
A Comparative In Vitro Evaluation of the Anti-Inflammatory Effects of a Tisochrysis lutea Extract and Fucoxanthin

 ,
, 
 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
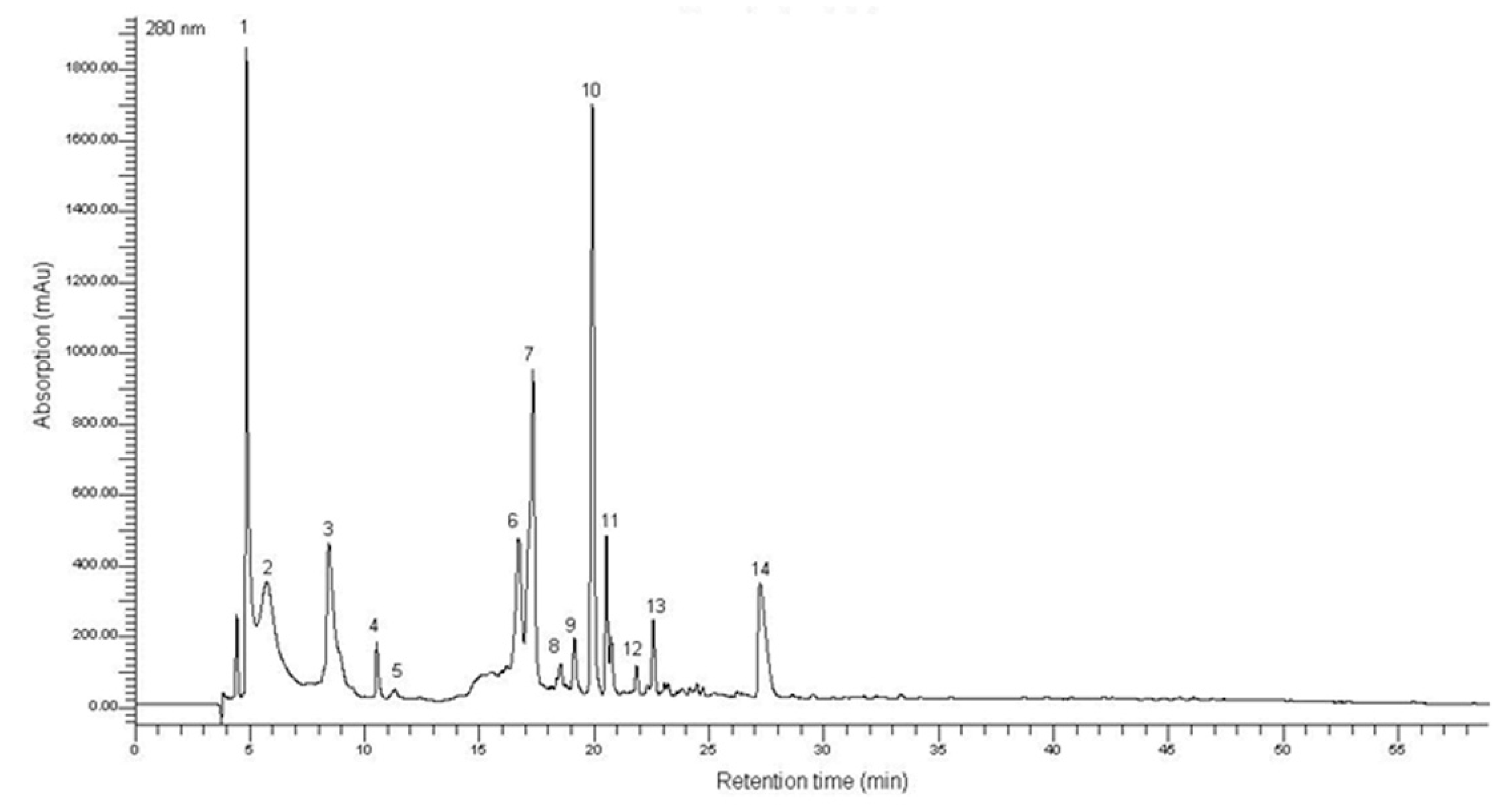
2.1. Characterization of the T. lutea F&M-M36 Methanolic Extract
2.2. Effects of T. lutea F&M-M36 Methanolic Extract and FX on RAW 264.7 Macrophages Viability
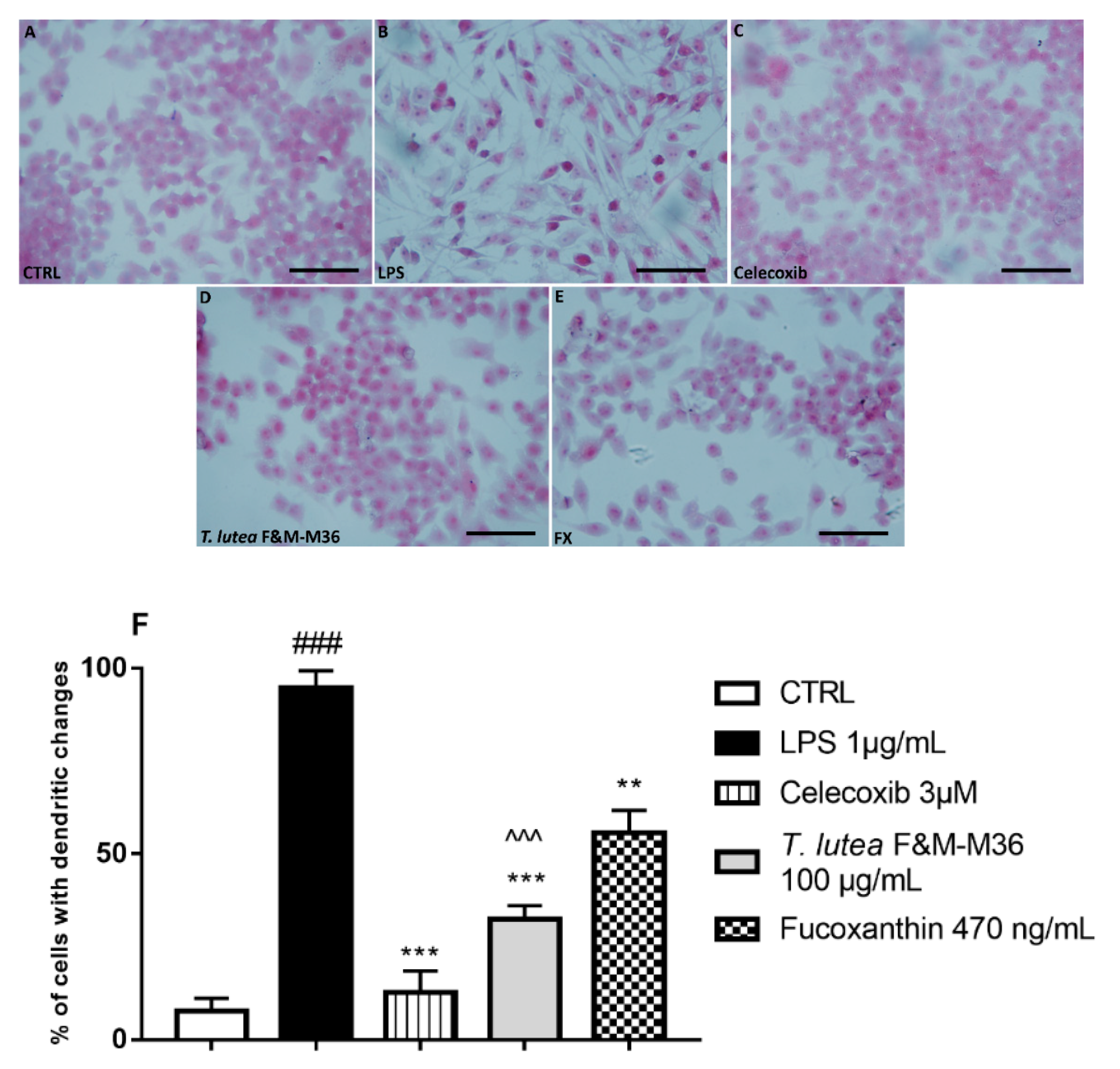
2.3. Comparative Effects of T. lutea F&M-M36 Extract and FX on Cell Morphology
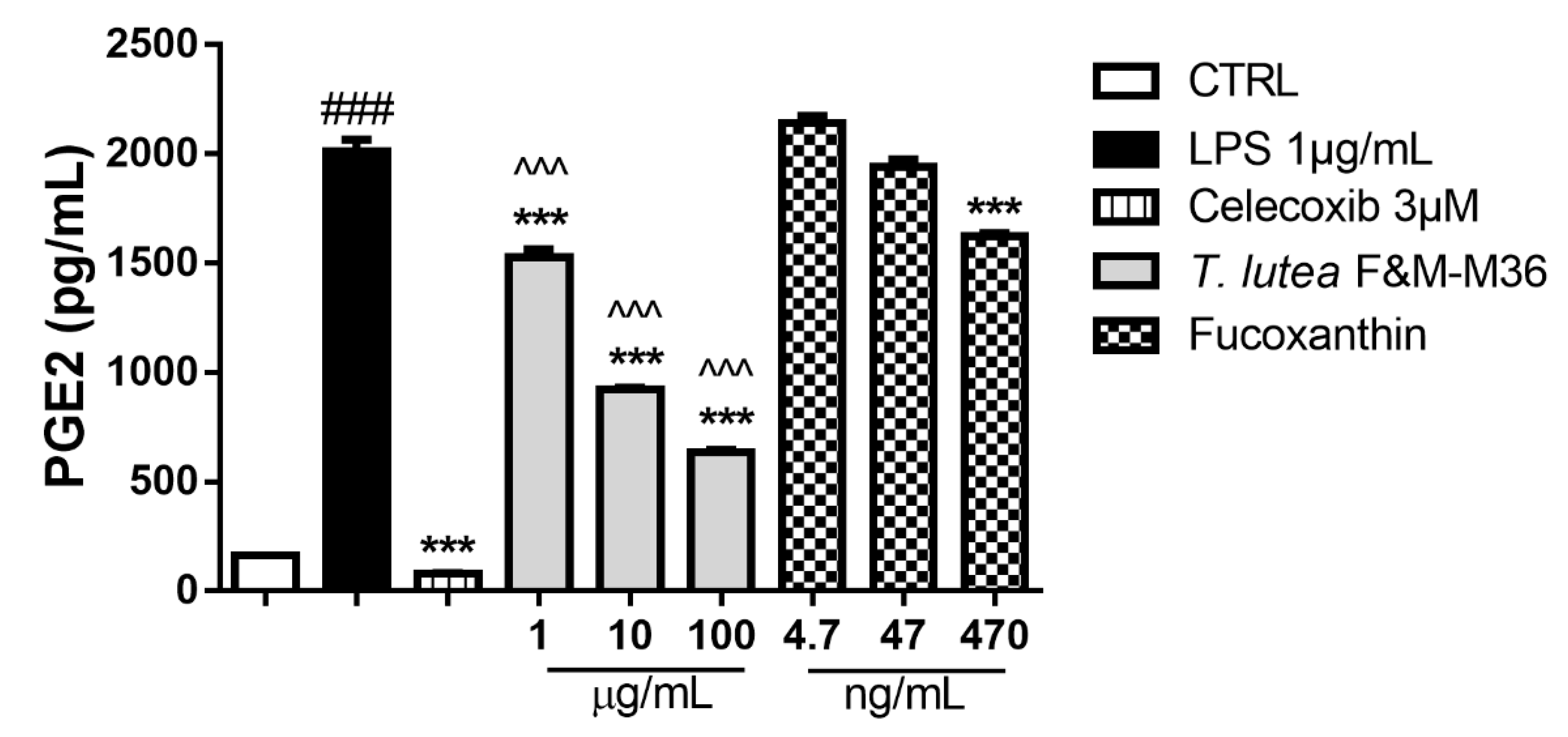
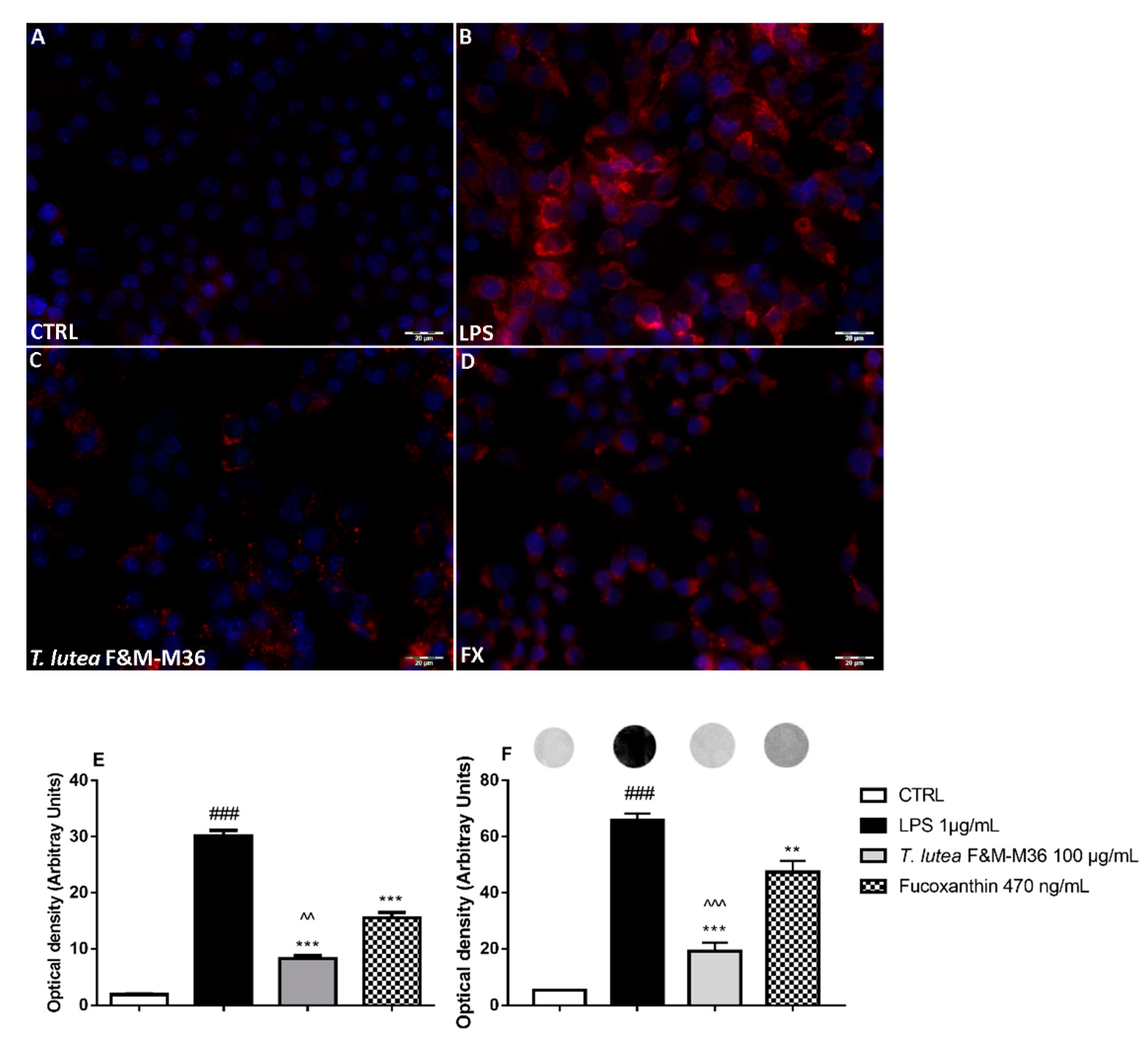
2.4. Comparative Effects of T. lutea F&M-M36 Extract and FX on PGE2 Production and COX-2 Protein Expression
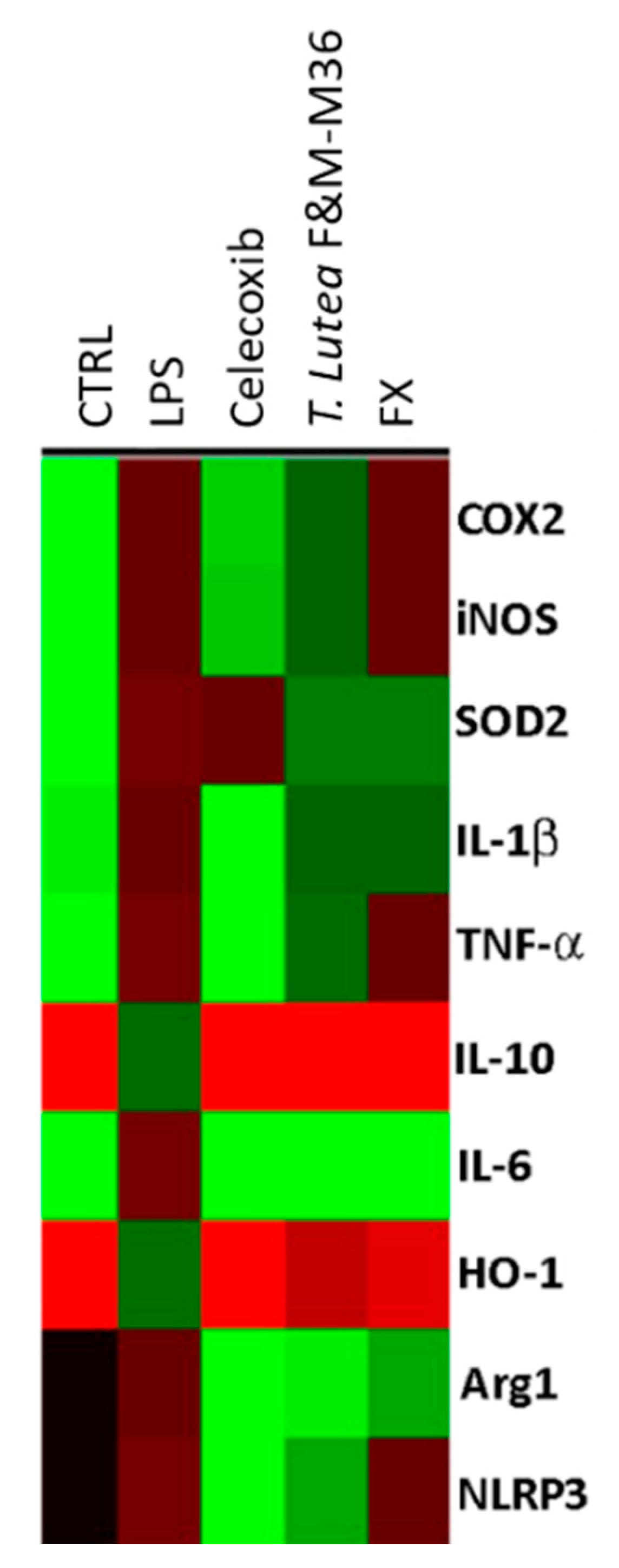
2.5. Comparative Effects of T. lutea F&M-M36 Extract and FX on the Expression of Pro-Inflammatory and Anti-Inflammatory Genes
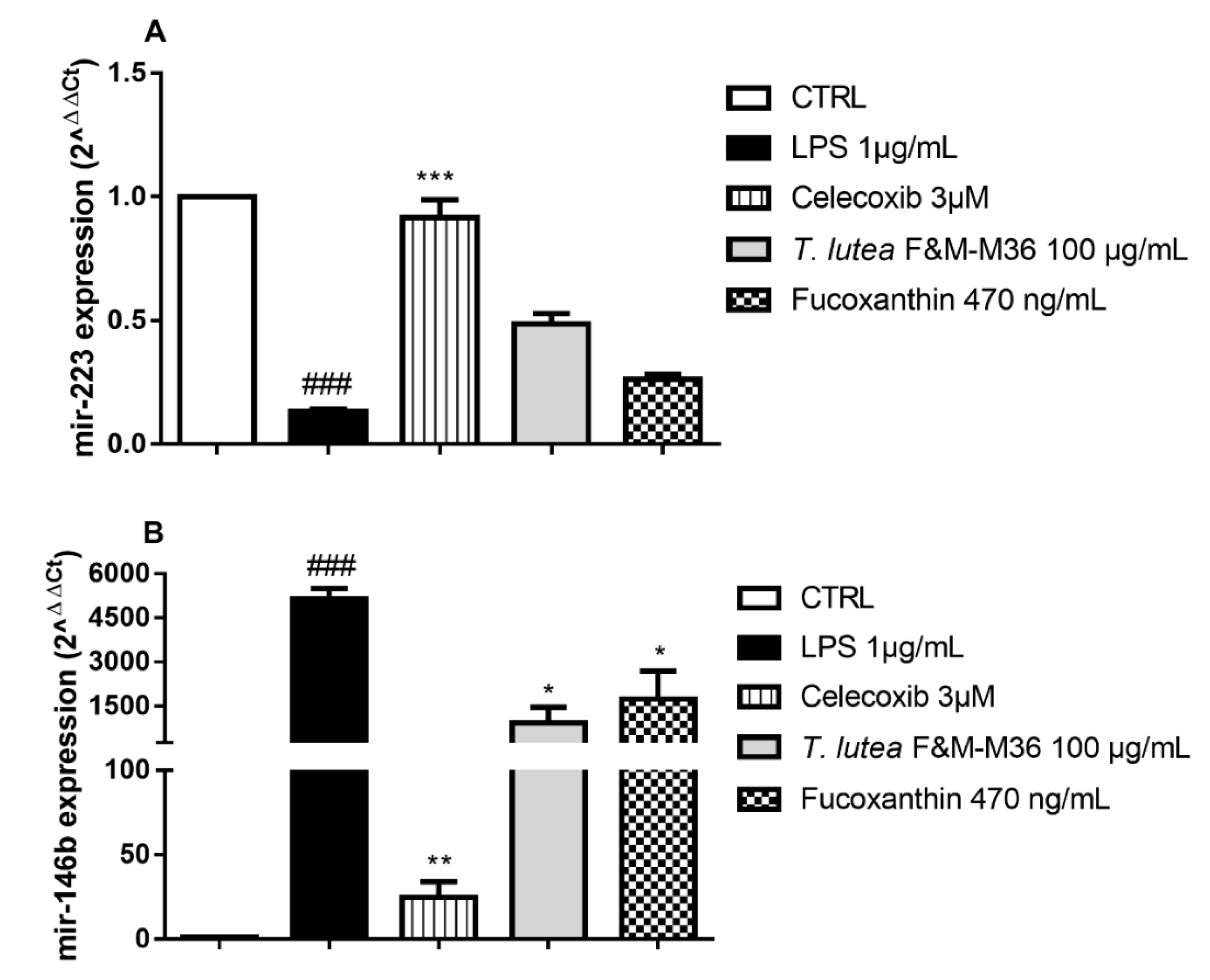
2.6. Comparative Effects of T. lutea F&M-M36 Extract and FX on mir-146b and mir-223 Expression
3. Discussion
4. Materials and Methods
4.1. Microalgal Biomass
4.2. Microalgal Extract Preparation
4.3. Sample Preparation and HPLC-DAD Analysis for Phenols Quantification and Characterization
4.4. Fucoxanthin Determination in the Methanolic Extract
4.5. In Vitro Model of Inflammation and Anti-Inflammatory Assay
4.6. Morphological Analysis: Hematoxylin and Eosin (H&E) Staining
4.7. PGE2 Determination
4.8. RT-PCR
4.9. Real-Time PCR for mir-146b and mir-223 Expression Analysis
4.10. Dot-Blotting for COX-2 Protein Expression
4.11. Immunocytochemistry for COX-2 Protein Expression
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bendif, E.; Probert, I.; Schroeder, D.C.; de Vargas, C. On the description of Tisochrysis lutea gen. nov., sp. nov. and Isochrysis nuda sp. nov. in the Isochrysidales, and the transfer of Dicrateria to the Prymnesiales (Haptophyta). J. Appl. Phycol. 2013, 25, 1763–1776. [Google Scholar] [CrossRef]
- Camacho-Rodríguez, J.; Cerón-García, M.C.; González-López, C.V.; López-Rosales, L.; Contreras-Gómez, A.; Molina-Grima, E. Use of continuous culture to develop an economical medium for the mass production of Isochrysis galbana for aquaculture. J. Appl. Phycol. 2020, 32, 851–863. [Google Scholar] [CrossRef]
- Custódio, L.; Soares, F.; Pereira, H.; Barreira, L.; Vizetto-Duarte, C.; Rodrigues, M.J.; Rauter, A.P.; Alberício, F.; Varela, J. Fatty acid composition and biological activities of Isochrysis galbana T-ISO, Tetraselmis sp. and Scenedesmus sp.: Possible application in the pharmaceutical and functional food industries. J. Appl. Phycol. 2014, 26, 151–161. [Google Scholar] [CrossRef]
- Delbrut, A.; Albina, P.; Lapierre, T.; Pradelles, R.; Dubreucq, E. Fucoxanthin and Polyunsaturated Fatty Acids Co-Extraction by a Green Process. Molecules 2018, 23, 874. [Google Scholar] [CrossRef] [PubMed]
- de los Reyes, C.; Ortega, M.J.; Rodríguez-Luna, A.; Talero, E.; Motilva, V.; Zubía, E. Molecular characterization and anti-inflammatory activity of galactosylglycerides and galactosylceramides from the microalga Isochrysis galbana. J. Agric. Food Chem. 2016, 64, 8783–8794. [Google Scholar] [CrossRef]
- Hwang, P.A.; Phan, N.N.; Lu, W.J.; Ngoc Hieu, B.T.; Lin, Y.C. Low-molecular-weight fucoidan and high-stability fucoxanthin from brown seaweed exert prebiotics and anti-inflammatory activities in Caco-2 cells. Food Nutr. Res. 2016, 60, 32033. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Cinci, L.; Niccolai, A.; Biondi, N.; Rodolfi, L.; D’Ottavio, M.; D’Ambrosio, M.; Lodovici, M.; Tredici, M.R.; Luceri, C. Preliminary data on the dietary safety, tolerability and effects on lipid metabolism of the marine microalga Tisochrysis lutea. Algal. Res. 2018, 34, 244–249. [Google Scholar] [CrossRef]
- Kim, S.M.; Kang, S.W.; Kwon, O.N.; Chung, D.; Pan, C.H. Fucoxanthin as a major carotenoid in Isochrysis aff. galbana: Characterization of extraction for commercial application. J. Korean Soc. Appl. Biol. Chem. 2012, 55, 477–483. [Google Scholar]
- Heo, S.J.; Yoon, W.J.; Kim, K.N.; Ahn, G.N.; Kang, S.M.; Kang, D.H.; Affan, A.; Oh, C.; Jung, W.K.; Jeon, Y.J. Evaluation of anti-inflammatory effect of fucoxanthin isolated from brown algae in lipopolysaccharide-stimulated RAW 264.7 macrophages. Food Chem. Toxicol. 2010, 48, 2045–2051. [Google Scholar] [CrossRef]
- Heo, S.J.; Yoon, W.J.; Kim, K.N.; Oh, C.; Choi, Y.U.; Yoon, K.T.; Kang, D.H.; Qian, Z.J.; Choi, I.W.; Jung, W.K. Anti-inflammatory effect of fucoxanthin derivatives isolated from Sargassum siliquastrum in lipopolysaccharide-stimulated RAW 264.7 macrophage. Food Chem. Toxicol. 2012, 50, 3336–3342. [Google Scholar] [CrossRef]
- Kim, K.N.; Heo, S.J.; Yoon, W.J.; Kang, S.M.; Ahn, G.; Yi, T.H.; Jeon, Y.J. Fucoxanthin inhibits the inflammatory response by suppressing the activation of NF-jB and MAPKs in lipopolysaccharide-induced RAW 264.7 macrophages. Eur. J. Pharmacol. 2010, 649, 369–375. [Google Scholar] [CrossRef]
- Su, J.; Guo, K.; Huang, M.; Liu, Y.; Zhang, J.; Sun, L.; Li, D.; Pang, K.L.; Wang, G.; Chen, L.; et al. Fucoxanthin, a Marine Xanthophyll Isolated From Conticribra weissflogii ND-8: Preventive Anti-Inflammatory Effect in a Mouse Model of Sepsis. Front. Pharmacol. 2019, 10, 906. [Google Scholar] [CrossRef]
- Tan, C.P.; Hou, Y.H. First evidence for the anti-inflammatory activity of fucoxanthin in high-fat-diet-induced obesity in mice and the antioxidant functions in PC12 cells. Inflammation 2014, 37, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Cardoso, C.; Gomes, A.; Campos, A.M.; Falé, P.; Afonso, C.; Bandarra, N.M. Bioprospection of Isochrysis galbana and its potential as a nutraceutical. Food Funct. 2019, 10, 7333–7342. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Cinci, L.; D’Ambrosio, M.; Luceri, C. Pharmacological activities of an eye drop containing Matricaria chamomilla and Euphrasia officinalis extracts in UVB-induced oxidative stress and inflammation of human corneal cells. J. Photochem. Photobiol. B 2017, 173, 618–625. [Google Scholar] [CrossRef]
- Bigagli, E.; Cinci, L.; Paccosi, S.; Parenti, A.; D’Ambrosio, M.; Luceri, C. Nutritionally relevant concentrations of resveratrol and hydroxytyrosol mitigate oxidative burst of human granulocytes and monocytes and the production of pro-inflammatory mediators in LPS-stimulated RAW 264.7 macrophages. Int. Immunopharmacol. 2017, 43, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Luceri, C.; Scartabelli, T.; Dolara, P.; Casamenti, F.; Pellegrini-Giampietro, D.E.; Giovannelli, L. Long-term Neuroglial Cocultures as a Brain Aging Model: Hallmarks of Senescence, MicroRNA Expression Profiles, and Comparison with In Vivo Models. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 50–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castagnini, C.; Luceri, C.; Toti, S.; Bigagli, E.; Caderni, G.; Femia, A.P.; Giovannelli, L.; Lodovici, M.; Pitozzi, V.; Salvadori, M.; et al. Reduction of colonic inflammation in HLA-B27 transgenic rats by feeding Marie Ménard apples, rich in polyphenols. Br. J. Nutr. 2009, 102, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Luceri, C.; Bigagli, E.; Pitozzi, V.; Giovannelli, L. A nutrigenomics approach for the study of anti-aging interventions: Olive oil phenols and the modulation of gene and microRNA expression profiles in mouse brain. Eur. J. Nutr. 2017, 56, 865–877. [Google Scholar] [CrossRef]
- D’Ambrosio, M.; Bigagli, E.; Cinci, L.; Gori, A.; Brunetti, C.; Ferrini, F.; Luceri, C. Ethyl acetate extract from Cistus x incanus L. leaves enriched in myricetin and quercetin derivatives, inhibits inflammatory mediators and activates Nrf2/HO-1 pathway in LPS-stimulated RAW 264.7 macrophages. Z. Naturforsch. C J. Biosci. 2020, 76, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Ma, Q.; Cui, H.; Liu, G.; Zhao, X.; Li, W.; Piao, G. How Can Synergism of Traditional Medicines Benefit from Network Pharmacology? Molecules 2017, 22, 1135. [Google Scholar] [CrossRef]
- Dunkhunthod, B.; Talabnin, C.; Murphy, M.; Thumanu, K.; Sittisart, P.; Hengpratom, T.; Eumkeb, G. Intracellular ROS Scavenging and Anti-Inflammatory Activities of Oroxylum indicum Kurz (L.) Extract in LPS plus IFN-γ-Activated RAW264.7 Macrophages. Evid. Based. Complement. Alternat. Med. 2020, 27, 2020. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 26, 160. [Google Scholar] [CrossRef]
- Maadane, A.; Merghoub, N.; Ainane, T.; Arroussi, H.E.; Bennhima, R.; Amzazi, S.; Bakri, Y.; Wahby, I. Antioxidant activity of some Moroccan marine microalgae:Pufa profiles, carotenoids and phenolic content. J. Biotechnol. 2015, 215, 13–19. [Google Scholar] [CrossRef]
- Robbins, R.J. Phenolic acids in foods: An overview of analytical methodology. J. Agric. Food Chem. 2003, 51, 2866–2887. [Google Scholar] [CrossRef]
- Stalikas, C.D. Extraction, separation, and detection methods for phenolic acids and flavonoids. J. Sep. Sci. 2007, 30, 3268–3295. [Google Scholar] [CrossRef]
- BenSaad, L.A.; Kim, K.H.; Quah, C.C.; Kim, W.R.; Shahimi, M. Anti-inflammatory potential of ellagic acid, gallic acid and punicalagin A&B isolated from Punica granatum. BMC Complement. Altern. Med. 2017, 14, 47. [Google Scholar]
- Luzardo-Ocampo, I.; Loarca-Piña, G.; Gonzalez de Mejia, E. Gallic and butyric acids modulated NLRP3 inflammasome markers in a co-culture model of intestinal inflammation. Food Chem. Toxicol. 2020, 146, 111835. [Google Scholar] [CrossRef]
- Luceri, C.; Guglielmi, F.; Lodovici, M.; Giannini, L.; Messerini, L.; Dolara, P. Plant phenolic 4-coumaric acid protects against intestinal inflammation in rats. Scand. J. Gastroenterol. 2004, 11, 1128–1133. [Google Scholar]
- Gutiérrez, S.; Svahn, S.L.; Johansson, M.E. Effects of Omega-3 Fatty Acids on Immune Cells. Int. J. Mol. Sci. 2019, 20, 5028. [Google Scholar] [CrossRef]
- Gallego, R.; Tardif, C.; Parreira, C.; Guerra, T.; Alves, M.J.; Ibáñez, E.; Herrero, M. Simultaneous extraction and purification of fucoxanthin from Tisochrysis lutea microalgae using compressed fluids. J. Sep. Sci. 2020, 43, 1967–1977. [Google Scholar] [CrossRef]
- Li, R.; Hong, P.; Zheng, X. β-carotene attenuates lipopolysaccharide-induced inflamma-tion via inhibition of the NF-κB, JAK2/STAT3 and JNK/p38 MAPK signaling pathways in macrophages. Anim. Sci. J. 2019, 90, 140–148. [Google Scholar] [CrossRef]
- Imamura, T.; Bando, N.; Yamanishi, R. Beta-carotene modulates the immunological function of RAW264, a murine macrophage cell line, by enhancing the level of intracellular glutathione. Biosci. Biotechnol. Biochem. 2006, 70, 2112–2120. [Google Scholar] [CrossRef][Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef]
- Zhang, N.; Fu, L.; Bu, Y.; Yao, Y.; Wang, Y. Downregulated expression of miR-223 promotes Toll-like receptor-activated inflammatory responses in macrophages by targeting RhoB. Mol. Immunol. 2017, 91, 42–48. [Google Scholar] [CrossRef]
- Yuan, X.; Berg, N.; Lee, J.W.; Le, T.T.; Neudecker, V.; Jing, N.; Eltzschig, H. MicroRNA miR-223 as regulator of innate immunity. J. Leukoc. Biol. 2018, 104, 515–524. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef]
- Montana, G.; Lampiasi, N. Substance P Induces HO-1 Expression in RAW 264.7 Cells Promoting Switch towards M2-Like Macrophages. PLoS ONE 2016, 11, e0167420. [Google Scholar] [CrossRef]
- Galvan-Pena, S.; O’Neill Luke, A.J. Metabolic reprogramming in macrophage polarization. Front. Immunol. 2014, 2, 420. [Google Scholar]
- Kim, M.B.; Kang, H.; Li, Y.; Park, Y.K.; Lee, J.Y. Fucoxanthin inhibits lipopolysaccharide-induced inflammation and oxidative stress by activating nuclear factor E2-related factor 2 via the phosphatidylinositol 3-kinase/AKT pathway in macrophages. Eur. J. Nutr. 2021, 60, 1–10. [Google Scholar]
- Mayer, C.; Richard, L.; Côme, M.; Ulmann, L.; Nazih, H.; Chénais, B.; Ouguerram, K.; Mimouni, V. The Marine Microalga, Tisochrysis lutea, Protects against Metabolic Disorders Associated with Metabolic Syndrome and Obesity. Nutrients 2021, 3, 430. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Sheedy, F.J.; McCoy, C.E. MicroRNAs: The fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 2011, 11, 163–175. [Google Scholar] [CrossRef]
- Batra, S.K.; Heier, C.R.; Diaz-Calderon, L.; Tully, C.B.; Fiorillo, A.A.; van den Anker, J.; Conklin, L.S. Serum miRNAs Are Pharmacodynamic Biomarkers Associated with Therapeutic Response in Pediatric Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2020, 26, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, Y.; Li, L.; Yu, Q.; Chao, K.; Zhou, G.; Qiu, Y.; Feng, R.; Huang, S.; He, Y.; et al. Circulating microRNA146b-5p is superior to C-reactive protein as a novel biomarker for monitoring inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49, 733–743. [Google Scholar] [CrossRef]
- Nuño, K.; Villarruel-López, A.; Puebla-Pérez, A.M.; Romero-Velarde, E.; Puebla-Mora, A.G.; Ascencio, F. Effects of the marine microalgae Isochrysis galbana and Nannochloropsis oculata in diabetic rats. J. Funct. Foods 2013, 5, 106–115. [Google Scholar] [CrossRef]
- Balakrishnan, J.; Dhavamani, S.; Sadasivam, S.G.; Arumugam, M.; Vellaikumar, S.; Ramalingam, J.; Shanmugam, K. Omega-3-rich Isochrysis sp. biomass enhances brain docosahexaenoic acid levels and improves serum lipid profile and antioxidant status in Wistar rats. J. Sci. Food Agric. 2019, 99, 6066–6075. [Google Scholar] [CrossRef]
- Guillard, R.R.L.; Ryther, J.H. Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt and Detonula confervacea Cleve. Can. J. Microbiol. 1962, 8, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Tredici, M.R.; Rodolfi, L.; Biondi, N.; Bassi, N.; Sampietro, G. Techno-economic analysis of microalgal biomass production in a 1-ha Green Wall Panel (GWP®) plant. Algal. Res. 2016, 19, 253–263. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | CTRL | LPS | Celecoxib | T. lutea F&M-M36 Extract | FX |
|---|---|---|---|---|---|
| COX-2 | 0.09 ± 0.01 | 0.86 ± 0.03 ### | 0.37 ± 0.03 *** | 0.81 ± 0.01 ^ | 0.91 ± 0.01 |
| iNOS | 0.02 ± 0.01 | 1.25 ± 0.03 ### | 0.51 ± 0.02 *** | 1.10 ± 0.06 | 1.20 ± 0.00 |
| SOD2 | 0.04 ± 0.02 | 0.64 ± 0.00 ### | 0.62 ± 0.01 | 0.44 ± 0.02 *** | 0.44 ± 0.01 *** |
| IL-1b | 0.36 ± 0.00 | 0.97 ± 0.02 ### | 0.31 ± 0.01 *** | 0.90 ± 0.01 | 0.92 ± 0.01 |
| TNF-a | 0.27 ± 0.03 | 0.76 ± 0.03 ### | 0.20 ± 0.02 *** | 0.69 ± 0.02 | 0.82 ± 0.01 |
| IL-10 | 0.62 ± 0.03 | 0.16 ± 0.00 ### | 0.71 ± 0.03 *** | 0.61 ± 0.06 *** | 0.62 ± 0.03 *** |
| IL-6 | 0.02 ± 0.01 | 0.74 ± 0.03 ### | 0.06 ± 0.01 *** | 0.04 ± 0.00 *** | 0.07 ± 0.01 *** |
| HO-1 | 1.04 ± 0.00 | 0.25 ± 0.00 ### | 0.95 ± 0.03 *** | 0.38 ± 0.01 *** | 0.46 ± 0.03 *** |
| Arg1 | 0.00 ± 0.00 | 1.71 ± 0.01 ### | 0.06 ± 0.00 *** | 0.61 ± 0.02 ***, ^^^ | 0.91 ± 0.02 *** |
| NLRP3 | 0.00 ± 0.00 | 0.74 ± 0.02 ### | 0.23 ± 0.02 *** | 0.37 ± 0.03 ***, ^^^ | 0.71 ± 0.05 |
| Gene | Primer Forward | Primer Reverse | Base Pair |
|---|---|---|---|
| RPLP-1 | ATCTACTCCGCCCTCATCCT | CAGATGAGGCTCCCAATGTT | 155 |
| COX-2 | TCCTCCTGGAACATGGACTC | CCCCAAAGATAGCATCTGGA | 321 |
| iNOS | CCCCAAAGATAGCATCTGGA | CCCCAAAGATAGCATCTGGA | 305 |
| SOD2 | ACCCAAAGGAGAGTTGCTGGA | ATGTGGCCGTGAGTGAGGTT | 354 |
| HO-1 | GGCTGCCCTGGAGCAGGACGT | AGGTCACCCAGGTAGCGG | 165 |
| TNFα | TAGCCCACGTCGTAGCAAAC | ACCCTGAGCCATAATCCCCT | 566 |
| NLRP3 | TGGGTTCTGGTCAGACACGAG | GGGGCTTAGGTCCACACAGAA | 176 |
| ARG1 | CATTGGCTTGCGAGACGTAG | CGGCCTTTTCTTCCTTCCCAG | 151 |
| IL-1β | CAGGCAGGCAGTATCACTCA | AGGCCACAGGTATTTTGTCG | 350 |
| IL-10 | AGGCGCTGTCATCGATTTCTC | AGGAAGAACCCCTCCCATCA | 489 |
| IL-6 | TCCTCTCTGCAAGAGACTTCC | TCCTCTCTGCAAGAGACTTCC | 513 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigagli, E.; D’Ambrosio, M.; Cinci, L.; Niccolai, A.; Biondi, N.; Rodolfi, L.; Dos Santos Nascimiento, L.B.; Tredici, M.R.; Luceri, C. A Comparative In Vitro Evaluation of the Anti-Inflammatory Effects of a Tisochrysis lutea Extract and Fucoxanthin. Mar. Drugs 2021, 19, 334. https://doi.org/10.3390/md19060334
Bigagli E, D’Ambrosio M, Cinci L, Niccolai A, Biondi N, Rodolfi L, Dos Santos Nascimiento LB, Tredici MR, Luceri C. A Comparative In Vitro Evaluation of the Anti-Inflammatory Effects of a Tisochrysis lutea Extract and Fucoxanthin. Marine Drugs. 2021; 19(6):334. https://doi.org/10.3390/md19060334
Chicago/Turabian StyleBigagli, Elisabetta, Mario D’Ambrosio, Lorenzo Cinci, Alberto Niccolai, Natascia Biondi, Liliana Rodolfi, Luana Beatriz Dos Santos Nascimiento, Mario R. Tredici, and Cristina Luceri. 2021. "A Comparative In Vitro Evaluation of the Anti-Inflammatory Effects of a Tisochrysis lutea Extract and Fucoxanthin" Marine Drugs 19, no. 6: 334. https://doi.org/10.3390/md19060334
APA StyleBigagli, E., D’Ambrosio, M., Cinci, L., Niccolai, A., Biondi, N., Rodolfi, L., Dos Santos Nascimiento, L. B., Tredici, M. R., & Luceri, C. (2021). A Comparative In Vitro Evaluation of the Anti-Inflammatory Effects of a Tisochrysis lutea Extract and Fucoxanthin. Marine Drugs, 19(6), 334. https://doi.org/10.3390/md19060334