
Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
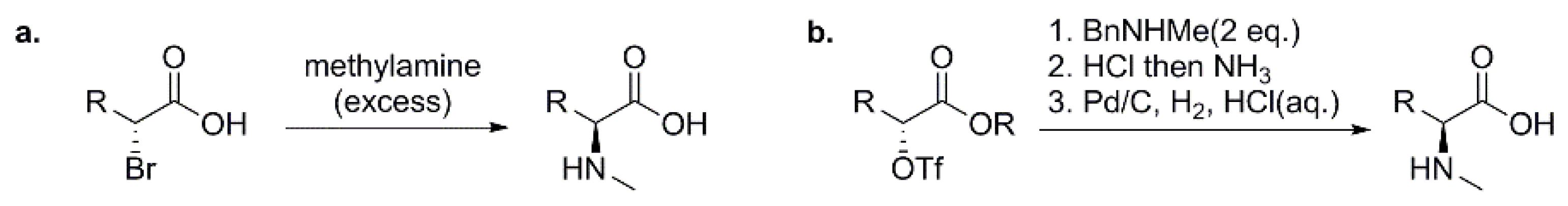{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cyclic Peptides in Drug Development
2.1. Peptides as Drugs
2.2. Membrane Permeability of Peptides
2.3. Advantages of Cyclic Peptides and Peptidomimetics on Cell Permeability
3. Backbone N-Methylation: Pivotal Roles to Improve the Permeability for Cyclic Peptides and Peptidomimetics
3.1. Chemical Synthesis of N-Methylated Cyclic Peptides
3.1.1. Preparation of N-Methyl Amino Acids (NMAAs) as Building Blocks for Solution-Phase Synthesis of Peptides
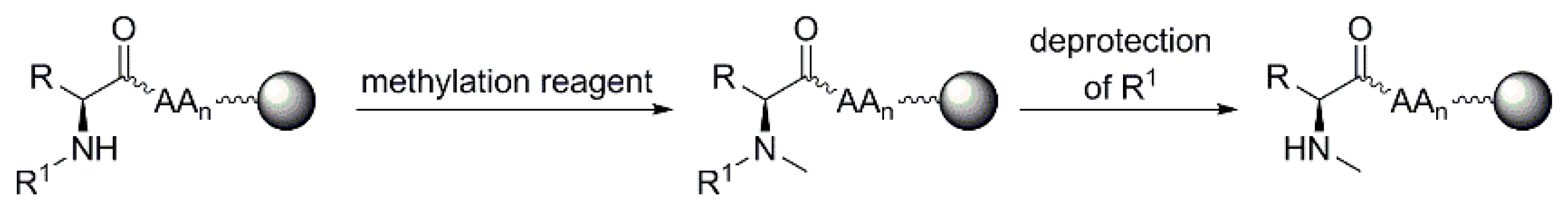
3.1.2. Regio-Specific N-Methylation for Solid-Phase Synthesis of Peptides
3.2. Backbone N-Methylation in the Discovery of Permeable Cyclic Peptide/Peptidomimetic
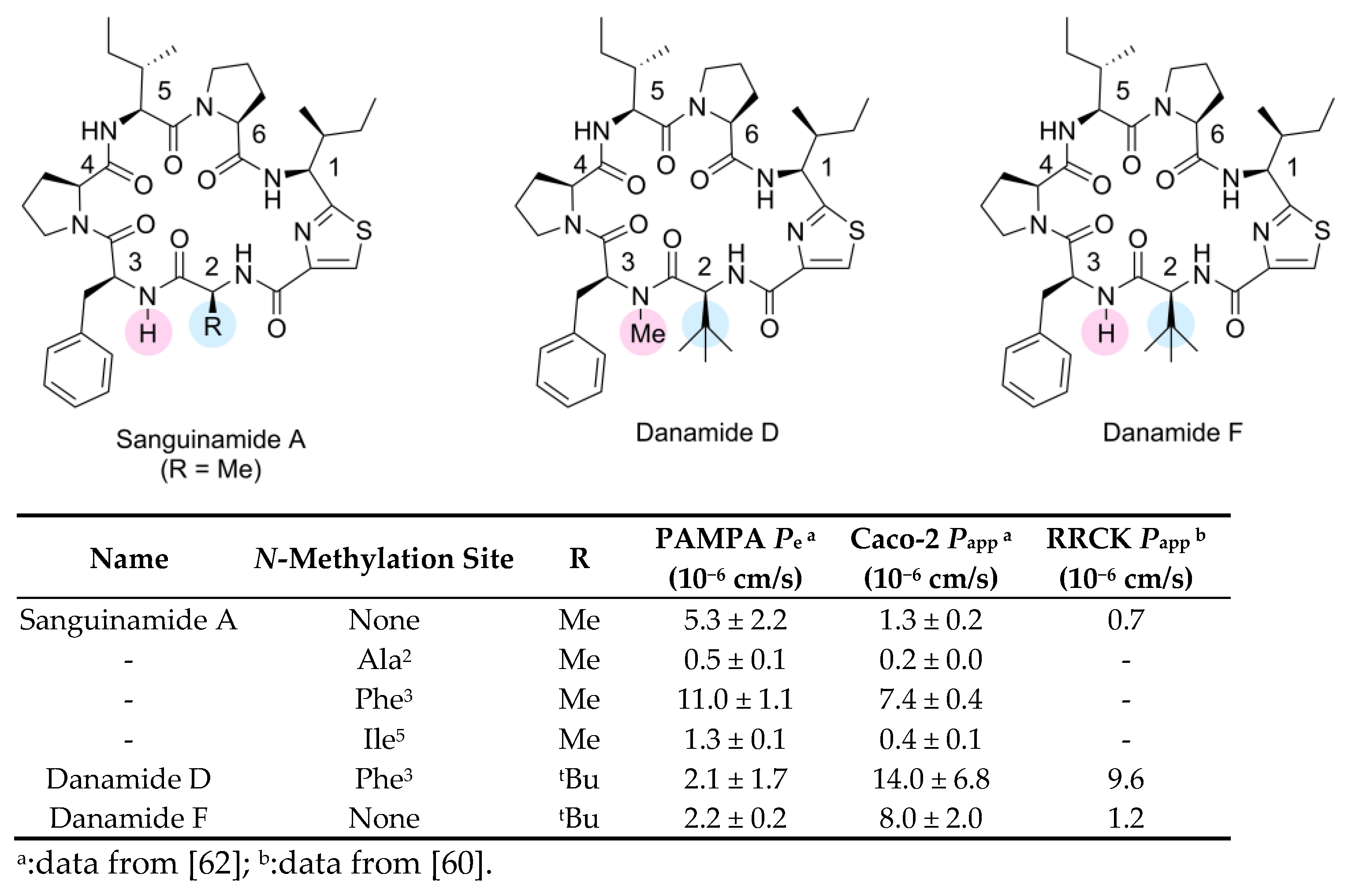
3.2.1. Studies on Methylated Analogs of Sanguinamide A
3.2.2. PAMPA Permeability of N-Methylated LB51 Analogs
3.2.3. Studies on N-Methylated Analogs of Cyclo(-Pro-Phe-D-Trp-Lys-Thr-Phe-)
3.2.4. Membrane Permeability of N-Methylated Poly Alanine Cyclic Pentapeptide/hexapeptide
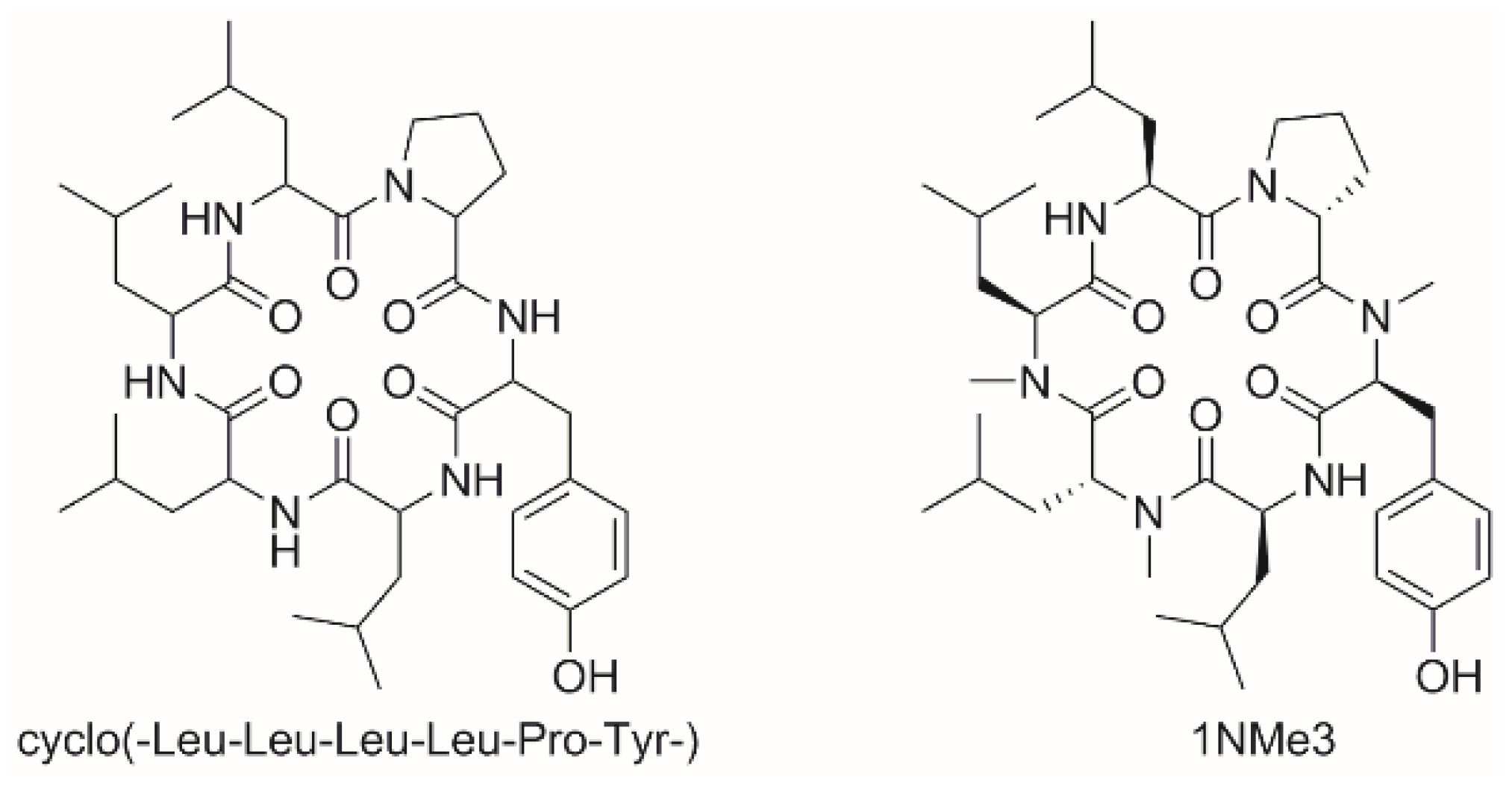
3.2.5. Studies on N-Methylated Analogs of Cyclo(-Leu-Leu-Leu-Leu-Pro-Tyr-)
3.2.6. Backbone N-Methylation on Modulators for Chemokine Receptor CXCR7
3.2.7. Influence of N-Methylation on Permeability of Semipeptide Macrocycles
3.2.8. Membrane Permeability of Hirsutellide A and Its Desmethyl Analog
3.2.9. Influence of Backbone N-Methylation on Permeability of Lariat Peptides
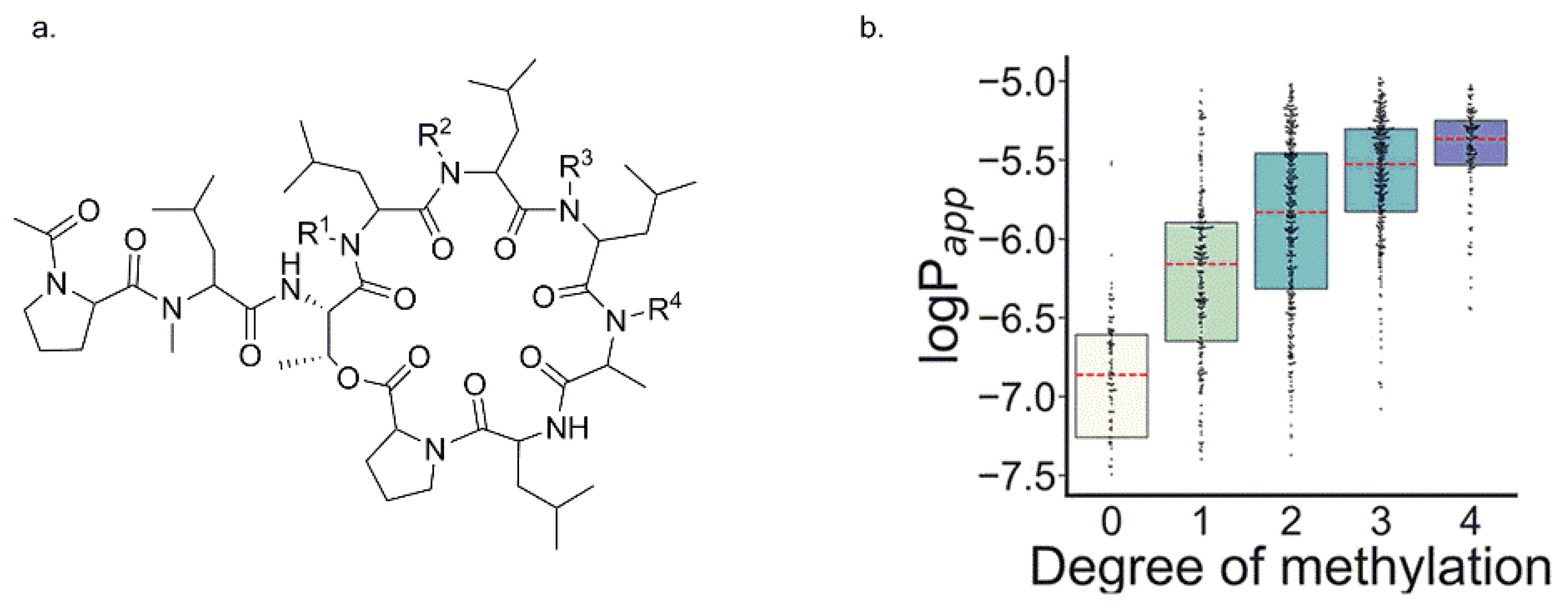
3.2.10. Backbone N-Methylation of Hexa-, Hepta- and Octo-Thioether-Containing Cyclic Peptides
4. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Buchwald, P. Small-molecule protein–protein interaction inhibitors: Therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 2010, 62, 724–731. [Google Scholar] [CrossRef]
- Qian, Z.; Dougherty, P.G.; Pei, D. Targeting intracellular protein-protein interactions with cell-permeable cyclic peptides. Curr. Opin. Chem. Biol. 2017, 38, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein–protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability challenges in peptide synthesis and purification: From R&D to production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar]
- Reese, H.R.; Shanahan, C.C.; Proulx, C.; Menegatti, S. Peptide science: A “rule model” for new generations of peptidomimetics. Acta Biomater. 2020, 102, 35–74. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive characterization of protein–protein interactions perturbed by disease mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef]
- Jwad, R.; Weissberger, D.; Hunter, L. Strategies for fine-tuning the conformations of cyclic peptides. Chem. Rev. 2020, 120, 9743–9789. [Google Scholar] [CrossRef]
- Viarengo-Baker, L.A.; Brown, L.E.; Rzepiela, A.A.; Whitty, A. Defining and navigating macrocycle chemical space. Chem. Sci. 2021, 12, 4309–4328. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell permeability beyond the rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Goetz, G.H.; Philippe, L.; Shapiro, M.J. EPSA: A novel supercritical fluid chromatography technique enabling the design of permeable cyclic peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.N.; Hill, T.A.; Fairlie, D.P. Connecting hydrophobic surfaces in cyclic peptides increases membrane permeability. Angew. Chem. Int. Ed. 2021, 60, 8385–8390. [Google Scholar] [CrossRef]
- Farley, K.A.; Che, Y.; Navarro-Vázquez, A.; Limberakis, C.; Anderson, D.; Yan, J.; Shapiro, M.; Shanmugasundaram, V.; Gil, R.R. Cyclic peptide design guided by residual dipolar couplings, j-couplings, and intramolecular hydrogen bond analysis. J. Org. Chem. 2019, 84, 4803–4813. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Schwochert, J.; Pye, C.R.; Asano, D.; Edmondson, Q.D.; Turmon, A.C.; Klein, V.G.; Ono, S.; Okada, O.; Lokey, R.S. Drug-Like Properties in Macrocycles above MW 1000: Backbone Rigidity versus Side-Chain Lipophilicity. Angew. Chem. Int. Ed. 2020, 59, 21571–21577. [Google Scholar] [CrossRef]
- Liras, S.; McClure, K.F. Permeability of Cyclic Peptide Macrocycles and Cyclotides and Their Potential as Therapeutics. ACS Med. Chem. Lett. 2019, 10, 1026–1032. [Google Scholar] [CrossRef]
- Stähelin, H.F. The history of cyclosporin A (Sandimmune®) revisited: Another point of view. Experientia 1996, 52, 5–13. [Google Scholar] [CrossRef]
- Graeb, C.; Arbogast, H.; Guba, M.; Jauch, K.W.; Land, W. Cyclosporine: 20 years of experience at the university of munich. Transplant. Proc. 2004, 36, S125–S129. [Google Scholar] [CrossRef] [PubMed]
- Loosli, H.-R.; Kessler, H.; Oschkinat, H.; Weber, H.-P.; Petcher, T.J.; Widmer, A. Peptide conformations. Part 31. The conformation of cyclosporin a in the crystal and in solution. Helv. Chim. Acta 1985, 68, 682–704. [Google Scholar] [CrossRef]
- Wang, C.K.; Swedberg, J.E.; Harvey, P.J.; Kaas, Q.; Craik, D.J. Conformational flexibility is a determinant of permeability for cyclosporin. J. Phys. Chem. B 2018, 122, 2261–2276. [Google Scholar] [CrossRef]
- Witek, J.; Keller, B.G.; Blatter, M.; Meissner, A.; Wagner, T.; Riniker, S. Kinetic models of cyclosporin a in polar and apolar environments reveal multiple congruent conformational states. J. Chem. Inf. Model. 2016, 56, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, G.M.; Gangwar, S.; Okumu, F.W.; Siahaan, T.J.; Stella, V.J.; Borchardt, R.T. Esterase-sensitive cyclic prodrugs of peptides: Evaluation of an acyloxyalkoxy promoiety in a model hexapeptide. Pharm. Res. 1996, 13, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, S.; Pauletti, G.M.; Siahaan, T.J.; Stella, V.J.; Borchardt, R.T. Synthesis of a novel esterase-sensitive cyclic prodrug of a hexapeptide using an (acyloxy)alkoxy promoiety. J. Org. Chem. 1997, 62, 1356–1362. [Google Scholar] [CrossRef]
- Okumu, F.W.; Pauletti, G.M.; Vander Velde, D.G.; Siahaan, T.J.; Borchardt, R.T. Effect of restricted conformational flexibility on the permeation of model hexapeptides across Caco-2 cell monolayers. Pharm. Res. 1997, 14, 169–175. [Google Scholar] [CrossRef]
- Borchardt, R.T. Optimizing oral absorption of peptides using prodrug strategies. J. Control. Release 1999, 62, 231–238. [Google Scholar] [CrossRef]
- Gudmundsson, O.; Jois, S.; Velde, D.V.; Siahaan, T.; Borchardt, R.; Wang, B. The effect of conformation on the membrane permeation of coumarinic acid-and phenylpropionic acid-based cyclic prodrugs of opioid peptides. J. Pept. Res. 1999, 53, 383–392. [Google Scholar] [CrossRef]
- Hill, T.A.; Lohman, R.J.; Hoang, H.N.; Nielsen, D.S.; Scully, C.C.; Kok, W.M.; Liu, L.; Lucke, A.J.; Stoermer, M.J.; Schroeder, C.I.; et al. Cyclic Penta- and Hexaleucine Peptides without N-Methylation Are Orally Absorbed. ACS Med. Chem. Lett. 2014, 5, 1148–1151. [Google Scholar] [CrossRef]
- Pauletti, G.M.; Gangwar, S.; Wang, B.; Borchardt, R.T. Esterase-sensitive cyclic prodrugs of peptides: Evaluation of a phenylpropionic acid promoiety in a model hexapeptide. Pharm. Res. 1997, 14, 11–17. [Google Scholar] [CrossRef]
- Wang, B.; Wang, W.; Zhang, H.; Shan, D.; Nimkar, K.; Gudmundsson, O.; Gangwar, S.; Siahaan, T.; Borchardt, R. Synthesis and evaluation of the physicochemical properties of esterase-sensitive cyclic prodrugs of opioid peptides using coumarinic acid and phenylpropionic acid linkers. J. Pept. Res. 1999, 53, 370–382. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhu, L.J.; Xi, T.K.; Zhu, H.Y.; Chen, X.X.; Wu, M.; Sun, C.; Xu, C.; Fang, G.M.; Meng, X. Delivery of cell membrane impermeable peptides into living cells by using head-to-tail cyclized mitochondria-penetrating peptides. Org. Biomol. Chem. 2019, 17, 9693–9697. [Google Scholar] [CrossRef]
- Price, D.A.; Eng, H.; Farley, K.A.; Goetz, G.H.; Huang, Y.; Jiao, Z.; Kalgutkar, A.S.; Kablaoui, N.M.; Khunte, B.; Liras, S.; et al. Comparative pharmacokinetic profile of cyclosporine (CsA) with a decapeptide and a linear analogue. Org. Biomol. Chem. 2017, 15, 2501–2506. [Google Scholar] [CrossRef]
- Masuda, Y.; Tanaka, R.; Ganesan, A.; Doi, T. Systematic Analysis of the Relationship among 3D Structure, Bioactivity, and Membrane Permeability of PF1171F, a Cyclic Hexapeptide with Paralyzing Effects on Silkworms. J. Org. Chem. 2017, 82, 11447–11463. [Google Scholar] [CrossRef]
- Ovadia, O.; Linde, Y.; Haskell-Luevano, C.; Dirain, M.L.; Sheynis, T.; Jelinek, R.; Gilon, C.; Hoffman, A. The effect of backbone cyclization on PK/PD properties of bioactive peptide-peptoid hybrids: The melanocortin agonist paradigm. Bioorg. Med. Chem. 2010, 18, 580–589. [Google Scholar] [CrossRef]
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef]
- Buckton, L.K.; Rahimi, M.N.; McAlpine, S.R. Cyclic peptides as drugs for intracellular targets: The next frontier in peptide therapeutic development. Chem. Eur. J. 2021, 27, 1487–1513. [Google Scholar] [CrossRef]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-methylation of peptides and proteins: An important element for modulating biological functions. Angew. Chem. Int. Ed. 2013, 52, 254–269. [Google Scholar] [CrossRef]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Rader, A.F.B.; Reichart, F.; Weinmuller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Aurelio, L.; Hughes, A.B. Synthesis of N-alkyl amino acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 245–289. [Google Scholar]
- Sharma, A.; Kumar, A.; Abdel Monaim, S.A.H.; Jad, Y.E.; El-Faham, A.; de la Torre, B.G.; Albericio, F. N-methylation in amino acids and peptides: Scope and limitations. Biopolymers 2018, 109, e23110. [Google Scholar] [CrossRef]
- Effenberger, F.; Burkard, U.; Willfahrt, J. Aminosäuren, 4. Enantioselektive Synthese N-substituierter α-Aminocarbonsäuren aus α-Hydroxycarbonsäuren. Liebigs Ann. Der Chem. 1986, 1986, 314–333. [Google Scholar] [CrossRef]
- Yang, L.; Chiu, K. Solid phase synthesis of Fmoc N-methyl amino acids: Application of the Fukuyama amine synthesis. Tetrahedron Lett. 1997, 38, 7307–7310. [Google Scholar] [CrossRef]
- Karamanos, N.; Stavropoulos, G.; Napoli, A.; Aksnesc, D.W.; Francis, G.W.; Sindona, G. Redox-alkylation of tosyl protected amino acid and peptide ester. Acta Chem. Scand. 1994, 994, 324–333. [Google Scholar]
- Abdel-Magid, A.F. Reduction of CN to CH–NH by Metal Hydrides. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 85–150. [Google Scholar]
- Ben-Ishai, D. Reaction of acylamino acids with paraformaldehyde. J. Am. Chem. Soc. 1957, 79, 5736–5738. [Google Scholar] [CrossRef]
- Auerbach, J.; Zamore, M.; Weinreb, S.M. N-Methylation of amides, lactams, and ureas. J. Org. Chem. 1976, 41, 725–726. [Google Scholar] [CrossRef]
- Freidinger, R.M.; Hinkle, J.S.; Perlow, D.S. Synthesis of 9-fluorenylmethyloxycarbonyl-protected N-alkyl amino acids by reduction of oxazolidinones. J. Org. Chem. 1983, 48, 77–81. [Google Scholar] [CrossRef]
- Aurelio, L.; Box, J.S.; Brownlee, R.T.C.; Hughes, A.B.; Sleebs, M.M. An efficient synthesis of n-methyl amino acids by way of intermediate 5-oxazolidinones. J. Org. Chem. 2003, 68, 2652–2667. [Google Scholar] [CrossRef]
- Aurelio, L.; Brownlee, R.T.C.; Hughes, A.B. A novel synthesis of N-methyl asparagine, arginine, histidine, and tryptophan. Org. Lett. 2002, 4, 3767–3769. [Google Scholar] [CrossRef]
- Reddy, G.V.; Rao, G.V.; Iyengar, D.S. A practical approach for the optically pure N-Methyl-α-amino acids. Tetrahedron Lett. 1998, 39, 1985–1986. [Google Scholar] [CrossRef]
- Fukuyama, T.; Jow, C.-K.; Cheung, M. 2- and 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar] [CrossRef]
- Miller, S.C.; Scanlan, T.S. Site-selective N-methylation of peptides on solid support. J. Am. Chem. Soc. 1997, 119, 2301–2302. [Google Scholar] [CrossRef]
- Miller, S.C.; Scanlan, T.S. oNBS−SPPS: A new method for solid-phase peptide synthesis. J. Am. Chem. Soc. 1998, 120, 2690–2691. [Google Scholar] [CrossRef]
- Biron, E.; Chatterjee, J.; Kessler, H. Optimized selective N-methylation of peptides on solid support. J. Pept. Sci. 2006, 12, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.A.; Hauksson, N.E.; Gipe, J.H.; Lokey, R.S. Selective, on-resin N-methylation of peptide N-trifluoroacetamides. Org. Lett. 2013, 15, 5012–5015. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Hoang, H.N.; Lohman, R.J.; Hill, T.A.; Lucke, A.J.; Craik, D.J.; Edmonds, D.J.; Griffith, D.A.; Rotter, C.J.; Ruggeri, R.B.; et al. Improving on nature: Making a cyclic heptapeptide orally bioavailable. Angew. Chem. Int. Ed. 2014, 53, 12059–12063. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.; Hoang, H.N.; Lohman, R.J.; Diness, F.; Fairlie, D.P. Total synthesis, structure, and oral absorption of a thiazole cyclic peptide, sanguinamide A. Org. Lett. 2012, 14, 5720–5723. [Google Scholar] [CrossRef] [PubMed]
- Bockus, A.T.; Schwochert, J.A.; Pye, C.R.; Townsend, C.E.; Sok, V.; Bednarek, M.A.; Lokey, R.S. Going Out on a Limb: Delineating The Effects of beta-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide A Analogues. J. Med. Chem. 2015, 58, 7409–7418. [Google Scholar] [CrossRef] [PubMed]
- Brinker, A.; Scheufler, C.; Von Der Mulbe, F.; Fleckenstein, B.; Herrmann, C.; Jung, G.; Moarefi, I.; Hartl, F.U. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 x Hop x Hsp90 complexes. J. Biol. Chem. 2002, 277, 19265–19275. [Google Scholar] [CrossRef] [PubMed]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain–peptide complexes. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Schmid, A.B.; Lagleder, S.; Grawert, M.A.; Rohl, A.; Hagn, F.; Wandinger, S.K.; Cox, M.B.; Demmer, O.; Richter, K.; Groll, M.; et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 2012, 31, 1506–1517. [Google Scholar] [CrossRef]
- Horibe, T.; Kohno, M.; Haramoto, M.; Ohara, K.; Kawakami, K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J. Transl. Med. 2011, 9, 8. [Google Scholar] [CrossRef]
- Buckton, L.K.; Wahyudi, H.; McAlpine, S.R. The first report of direct inhibitors that target the C-terminal MEEVD region on heat shock protein 90. Chem. Commun. 2016, 52, 501–504. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Seow, J.; Das, S.C.; Norton, R.S. Disordered epitopes as peptide vaccines. Pept. Sci. 2018, 110, e24067. [Google Scholar] [CrossRef]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; et al. Improving oral bioavailability of peptides by multiple N-methylation: Somatostatin analogues. Angew. Chem. Int. Ed. 2008, 47, 2595–2599. [Google Scholar] [CrossRef]
- Chatterjee, J.; Mierke, D.; Kessler, H. N-Methylated cyclic pentaalanine peptides as template structures. J. Am. Chem. Soc. 2006, 128, 15164–15172. [Google Scholar] [CrossRef]
- Ovadia, O.; Greenberg, S.; Chatterjee, J.; Laufer, B.; Opperer, F.; Kessler, H.; Gilon, C.; Hoffman, A. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol. Pharm. 2011, 8, 479–487. [Google Scholar] [CrossRef]
- Beck, J.G.; Chatterjee, J.; Laufer, B.; Kiran, M.U.; Frank, A.O.; Neubauer, S.; Ovadia, O.; Greenberg, S.; Gilon, C.; Hoffman, A.; et al. Intestinal Permeability of Cyclic Peptides: Common Key Backbone Motifs Identified. J. Am. Chem. Soc. 2012, 134, 12125–12133. [Google Scholar] [CrossRef] [PubMed]
- Marelli, U.K.; Bezencon, J.; Puig, E.; Ernst, B.; Kessler, H. Enantiomeric cyclic peptides with different Caco-2 permeability suggest carrier-mediated transport. Chem. Eur. J. 2015, 21, 8023–8027. [Google Scholar] [CrossRef]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.G.; Leung, S.S.; Kalgutkar, A.S.; et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Beaumont, K.; Jones, R.; Kalgutkar, A.S.; Zhang, L.; Atkinson, K.; Bai, G.; Brown, J.A.; Eng, H.; Goetz, G.H.; et al. Discovery of potent and orally bioavailable macrocyclic peptide-peptoid hybrid CXCR7 modulators. J. Med. Chem. 2017, 60, 9653–9663. [Google Scholar] [CrossRef]
- Le Roux, A.; Blaise, E.; Boudreault, P.L.; Comeau, C.; Doucet, A.; Giarrusso, M.; Collin, M.P.; Neubauer, T.; Kolling, F.; Goller, A.H.; et al. Structure-permeability relationship of semipeptidic macrocycles-understanding and optimizing passive permeability and efflux ratio. J. Med. Chem. 2020, 63, 6774–6783. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, L.; Duan, X.; Meng, Y.; Jiang, L.; Li, M.; Zhao, G.; Li, Y. Total synthesis of Hirsutellide A. Tetrahedron Lett. 2005, 46, 4377–4379. [Google Scholar] [CrossRef]
- Vongvanich, N.; Kittakoop, P.; Isaka, M.; Trakulnaleamsai, S.; Vimuttipong, S.; Tanticharoen, M.; Thebtaranonth, Y. Hirsutellide A, a new antimycobacterial cyclohexadepsipeptide from the entomopathogenic fungus hirsutella kobayasii. J. Nat. Prod. 2002, 65, 1346–1348. [Google Scholar] [CrossRef]
- Sahile, H.A.; Martínez-Martínez, M.S.; Dillenberger, M.; Becker, K.; Imming, P. Synthesis and Evaluation of Antimycobacterial and Antiplasmodial Activities of Hirsutellide A and Its Analogues. ACS Omega 2020, 5, 14451–14460. [Google Scholar] [CrossRef]
- Kelly, C.N.; Townsend, C.E.; Jain, A.N.; Naylor, M.R.; Pye, C.R.; Schwochert, J.; Lokey, R.S. Geometrically Diverse Lariat Peptide Scaffolds Reveal an Untapped Chemical Space of High Membrane Permeability. J. Am. Chem. Soc. 2021, 143, 705–714. [Google Scholar] [CrossRef]
- Golosov, A.A.; Flyer, A.N.; Amin, J.; Babu, C.; Gampe, C.; Li, J.; Liu, E.; Nakajima, K.; Nettleton, D.; Patel, T.J.; et al. Design of thioether cyclic peptide scaffolds with passive permeability and oral exposure. J. Med. Chem. 2021, 64, 2622–2633. [Google Scholar] [CrossRef]
- Feldwisch, J.; Tolmachev, V. Engineering of affibody molecules for therapy and diagnostics. In Therapeutic Proteins: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; Volume 899, pp. 103–126. [Google Scholar]
- Frejd, F.Y.; Kim, K.-T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Karlström, A.E.; Frejd, F.Y.; Nygren, P.-Å.; Löfblom, J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting nanobodies’ singular traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody technology: A versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Širochmanová, I.; Čomor, Ľ.; Káňová, E.; Jiménez-Munguía, I.; Tkáčová, Z.; Bhide, M. Permeability of the blood-brain barrier and transport of nanobodies across the blood-brain barrier. Folia Vet. 2018, 62, 59–66. [Google Scholar] [CrossRef][Green Version]
- Herce, H.D.; Schumacher, D.; Schneider, A.F.L.; Ludwig, A.K.; Mann, F.A.; Fillies, M.; Kasper, M.-A.; Reinke, S.; Krause, E.; Leonhardt, H.; et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat. Chem. 2017, 9, 762–771. [Google Scholar] [CrossRef]
- Schumacher, D.; Helma, J.; Schneider, A.F.L.; Leonhardt, H.; Hackenberger, C.P.R. Nanobodies: Chemical functionalization strategies and intracellular applications. Angew. Chem. Int. Ed. 2018, 57, 2314–2333. [Google Scholar] [CrossRef]
- Bruce, V.J.; Lopez-Islas, M.; McNaughton, B.R. Resurfaced cell-penetrating nanobodies: A potentially general scaffold for intracellularly targeted protein discovery. Protein Sci. 2016, 25, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Mix, K.A.; Lomax, J.E.; Raines, R.T. Cytosolic delivery of proteins by bioreversible esterification. J. Am. Chem. Soc. 2017, 139, 14396–14398. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, W.; Xu, Z. Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Mar. Drugs 2021, 19, 311. https://doi.org/10.3390/md19060311
Li Y, Li W, Xu Z. Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Marine Drugs. 2021; 19(6):311. https://doi.org/10.3390/md19060311
Chicago/Turabian StyleLi, Yang, Wang Li, and Zhengshuang Xu. 2021. "Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool" Marine Drugs 19, no. 6: 311. https://doi.org/10.3390/md19060311
APA StyleLi, Y., Li, W., & Xu, Z. (2021). Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Marine Drugs, 19(6), 311. https://doi.org/10.3390/md19060311