
Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477

 , ,
, ,  ,
, 
Abstract
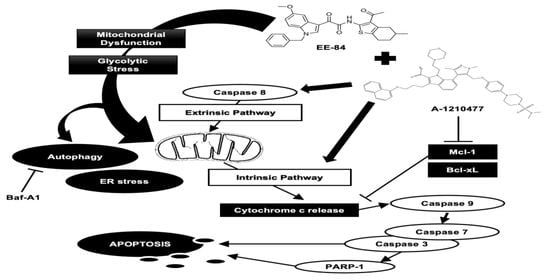
1. Introduction
2. Results
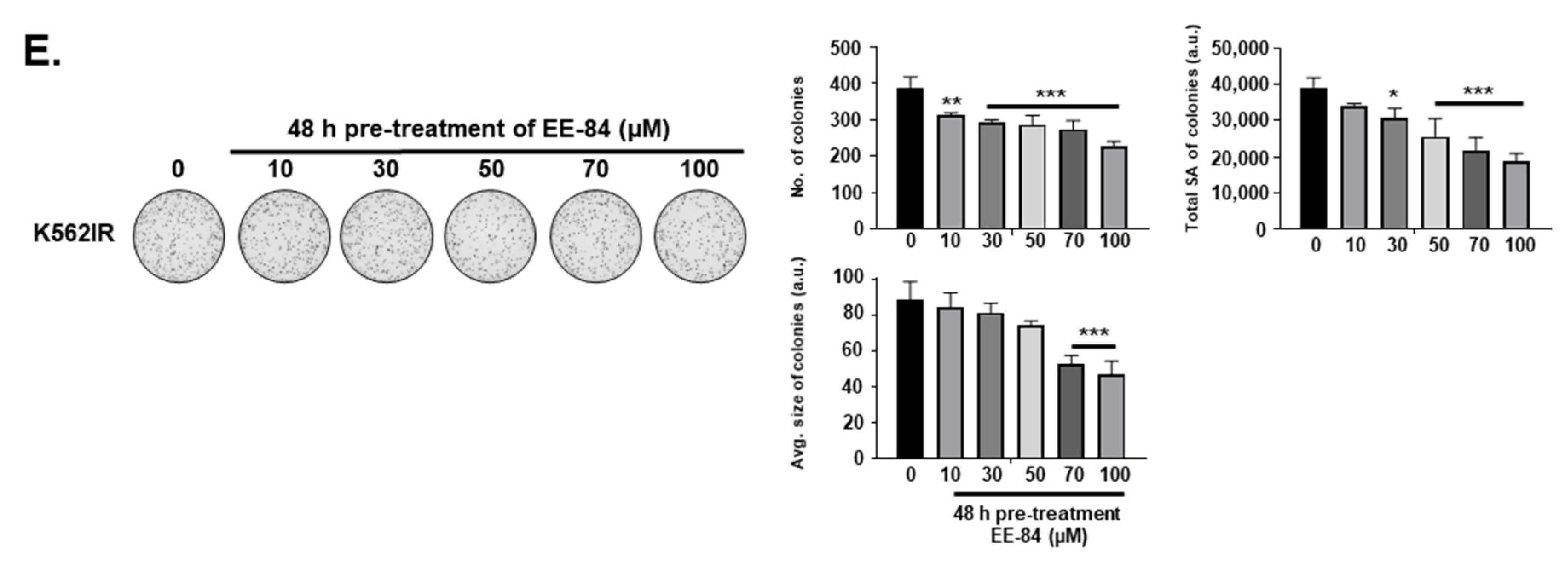
2.1. Aplysinopsin Analogs Display Cytostatic Activities in Myeloid Leukemia Cells
2.2. Drug-Likeness of Compounds EE-84 and EE-115
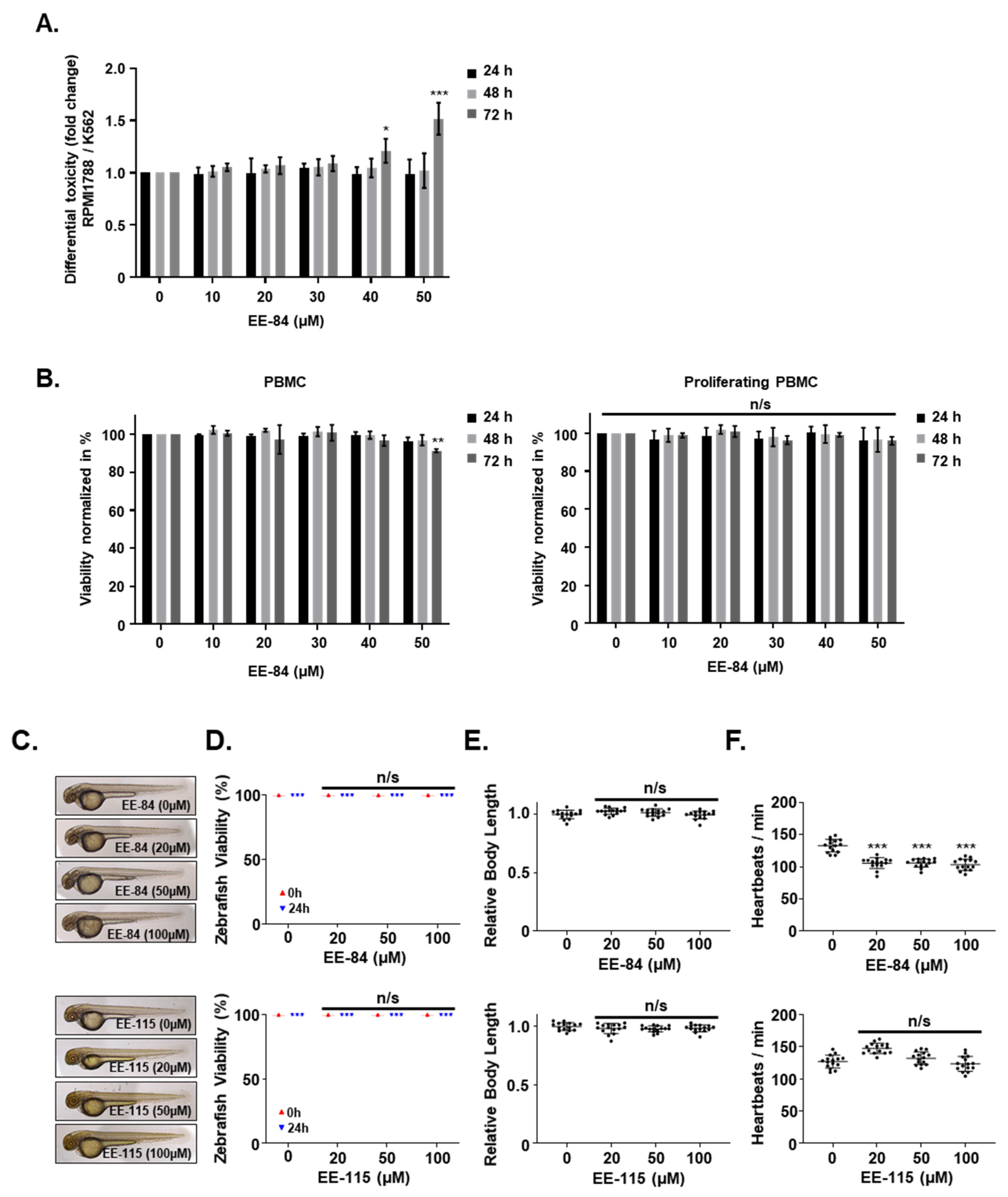
2.3. Effect of EE-84 and EE-115 on Healthy Cells and Zebrafish Embryos
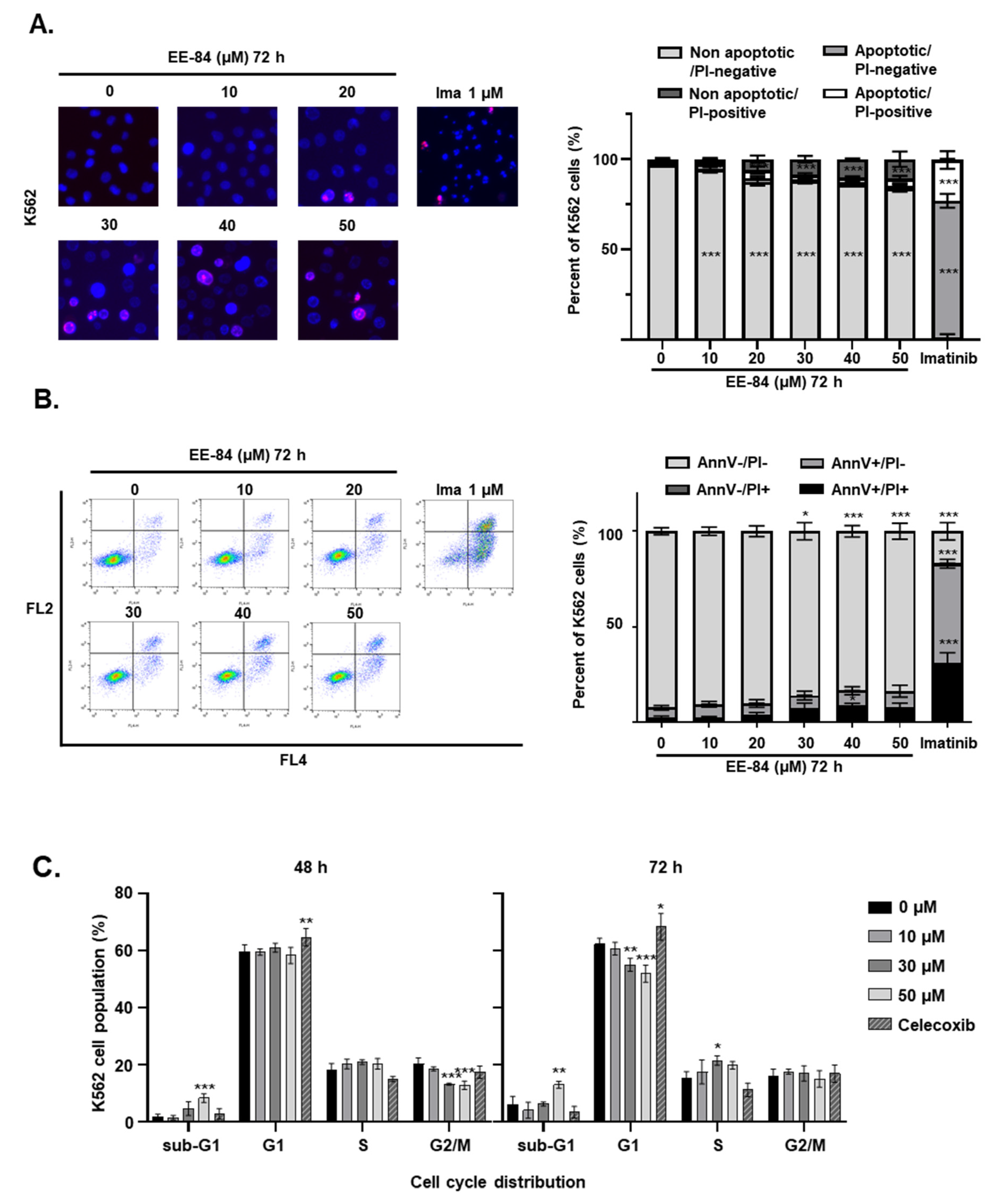
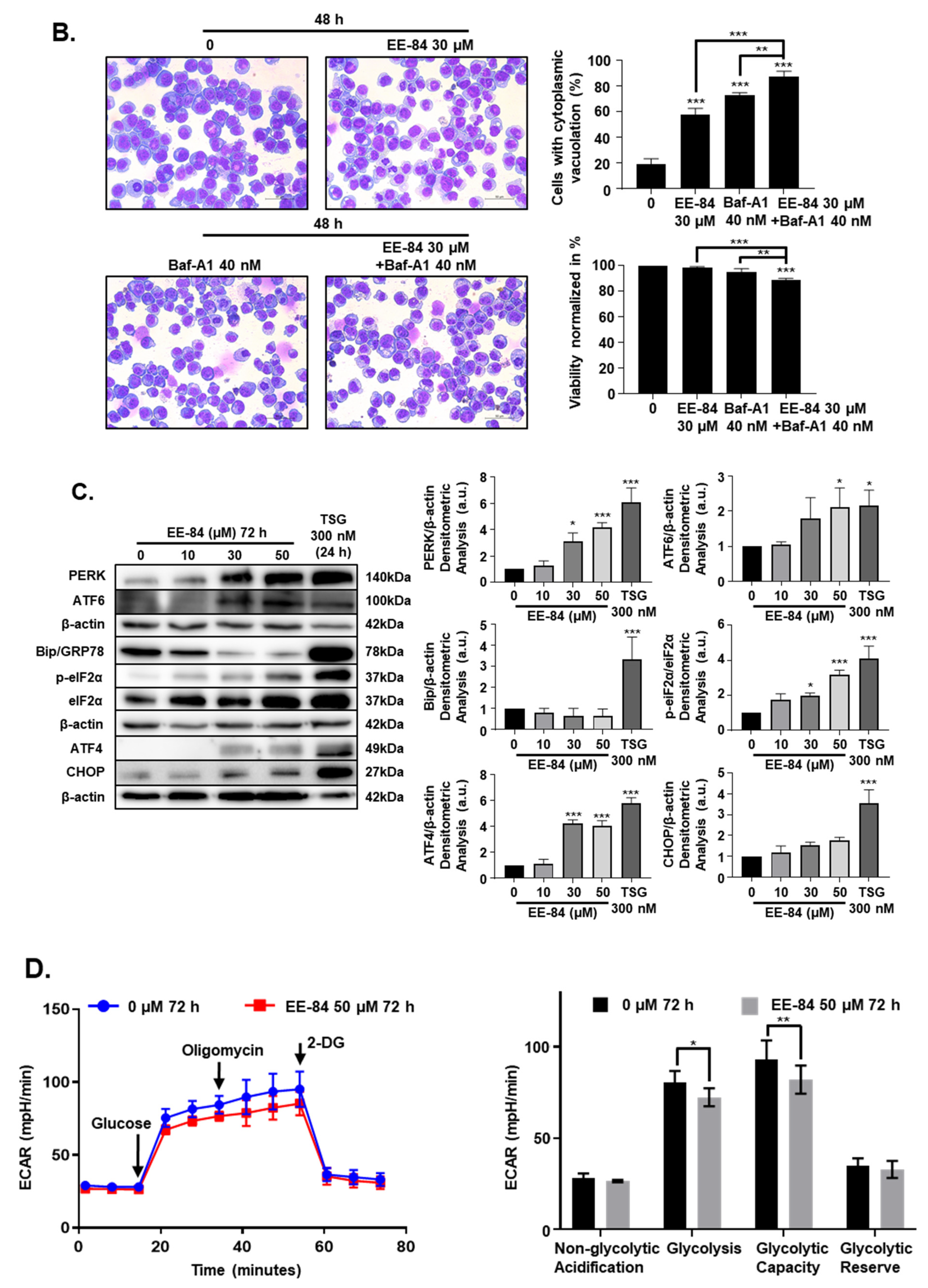
2.4. EE-84 Induced Morphological Changes and Cell Stress Responses in CML Cells
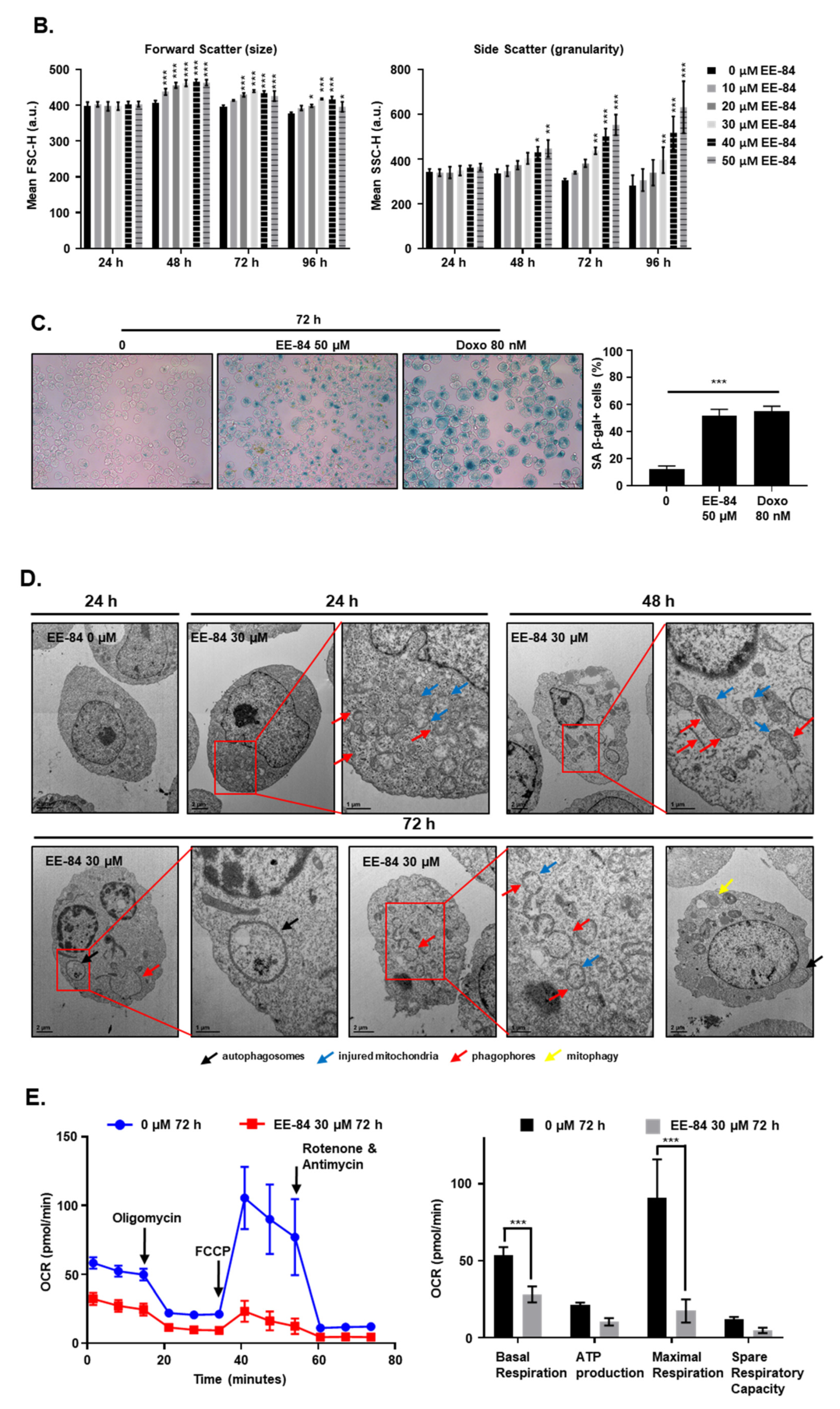
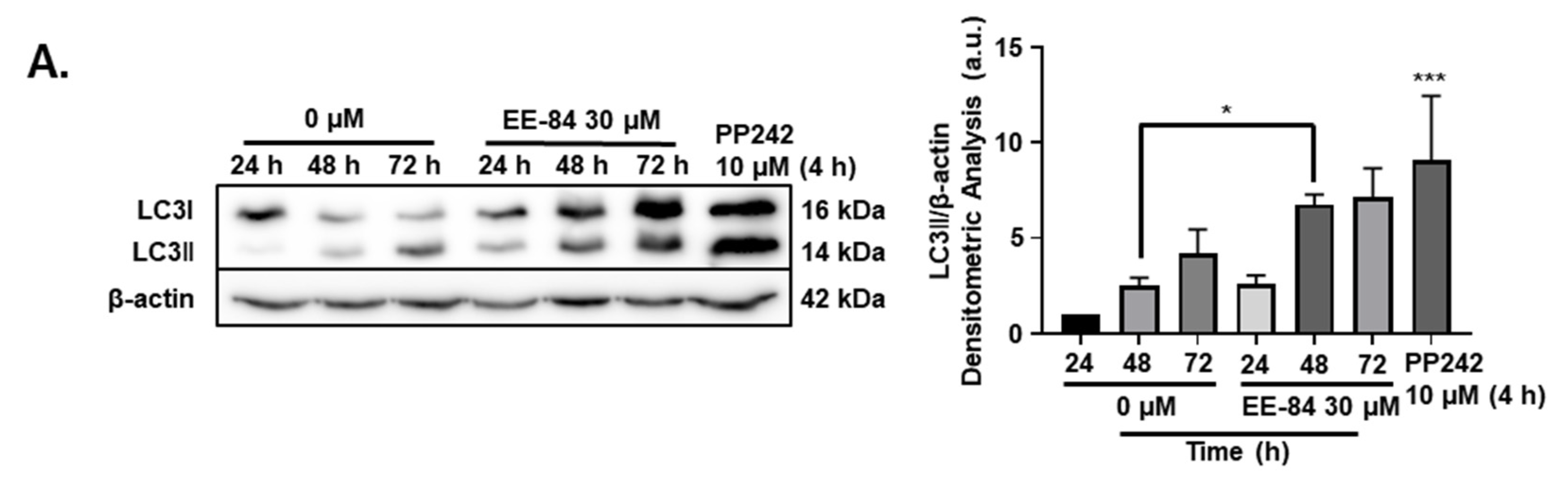
2.5. Autophagy and Endoplasmic Reticulum (ER) Stress Were Triggered by EE-84 as Cellular Stress Responses in K562 Cells
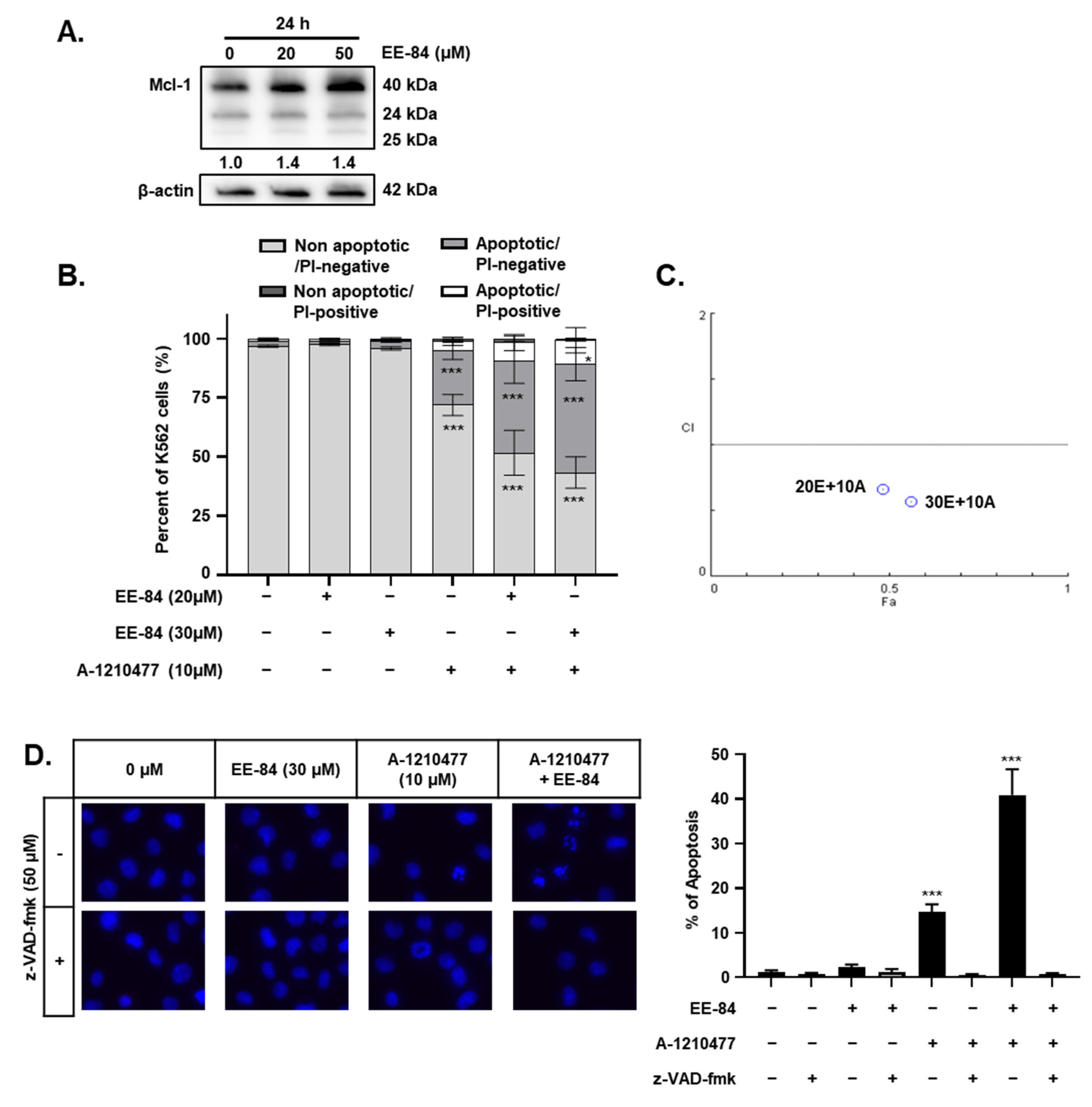
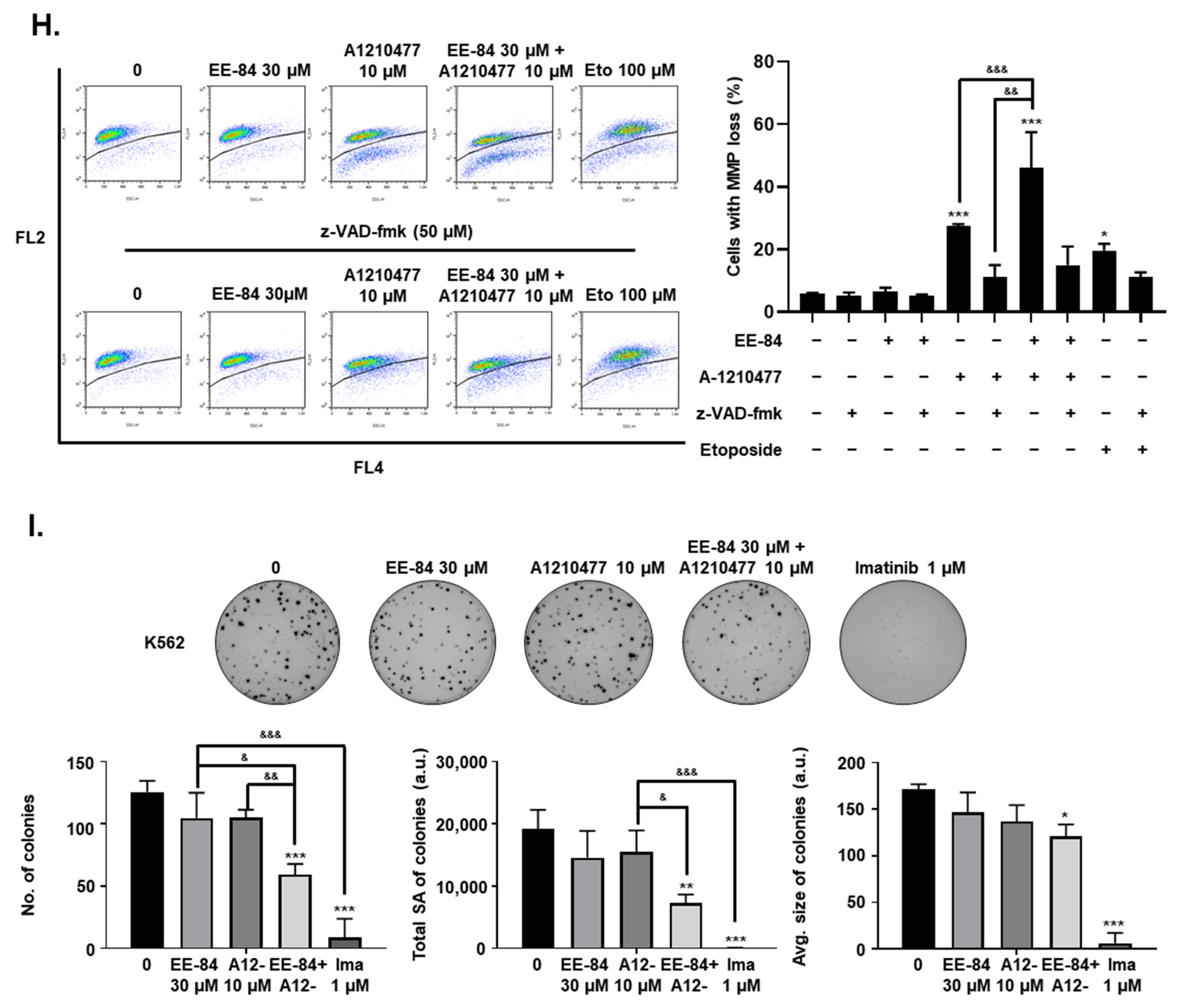
2.6. EE-84 Sensitized K562 Cells against Mcl-1 Inhibitor A-1210477 and Showed Synergistic Cytotoxicity in K562 and K562IR Cells
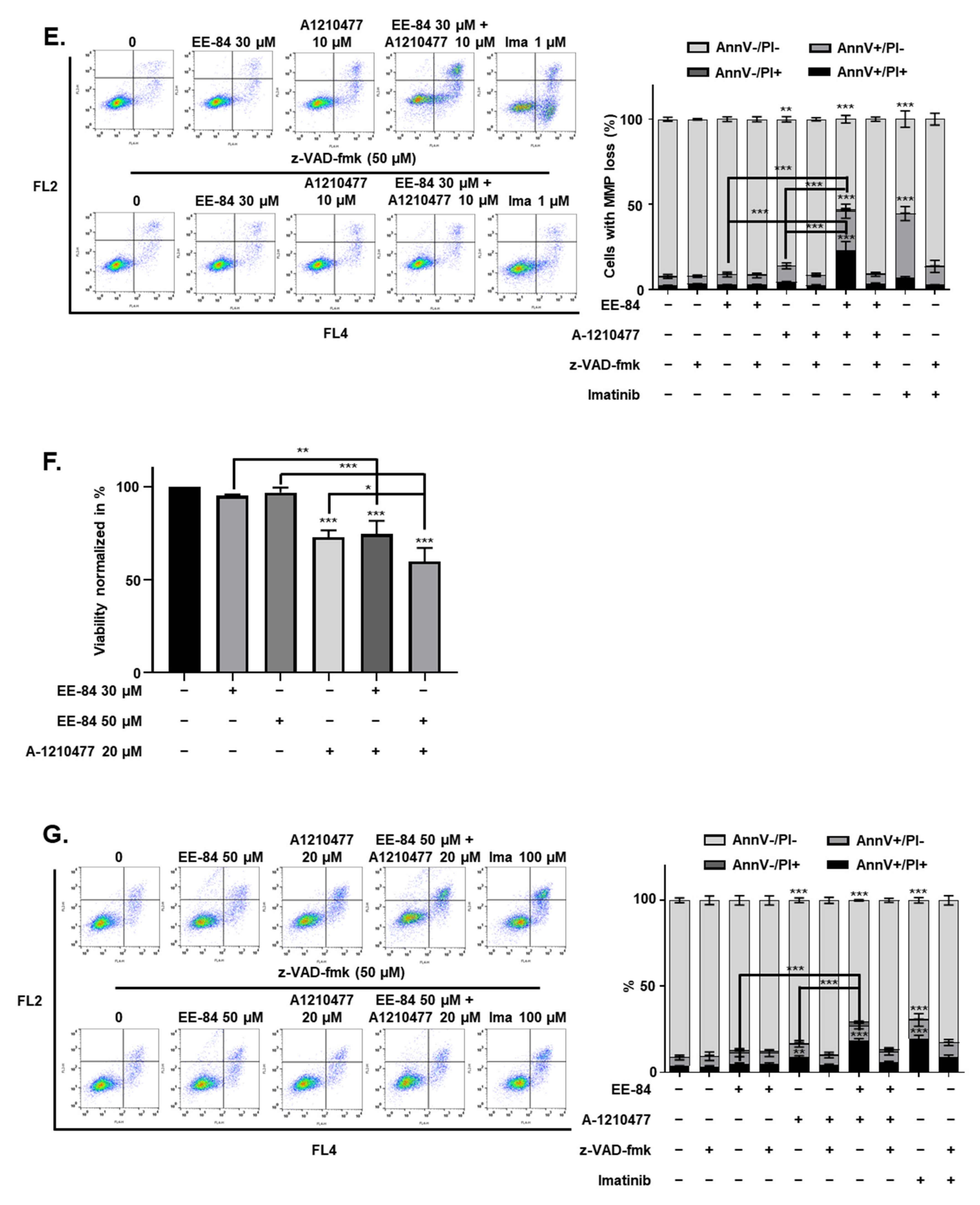
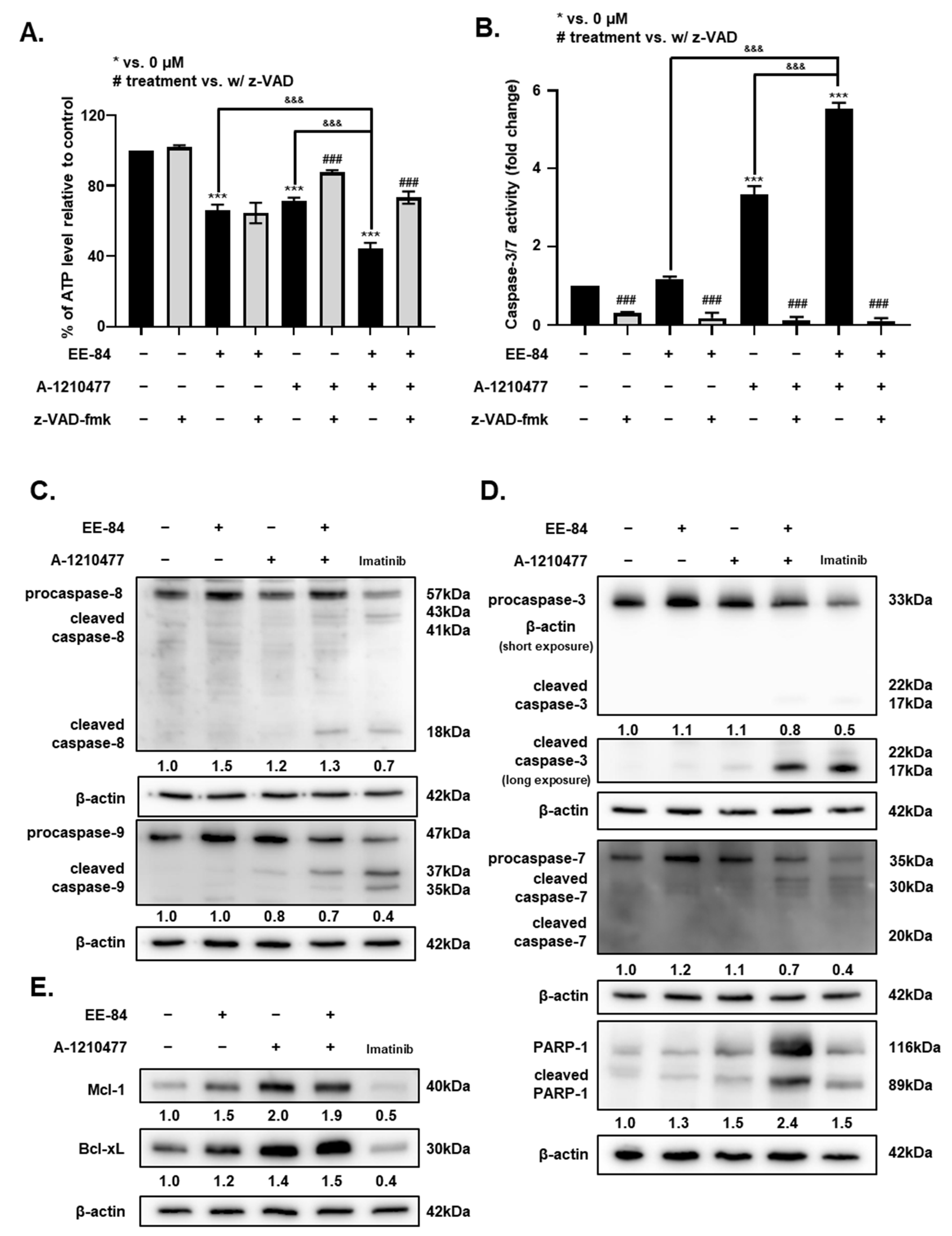
2.7. Synergistic Induction of Caspase-Dependent Apoptosis by Combination Treatment of EE-84 and A-1210477 in K562 Cells
3. Discussion
4. Materials and Methods
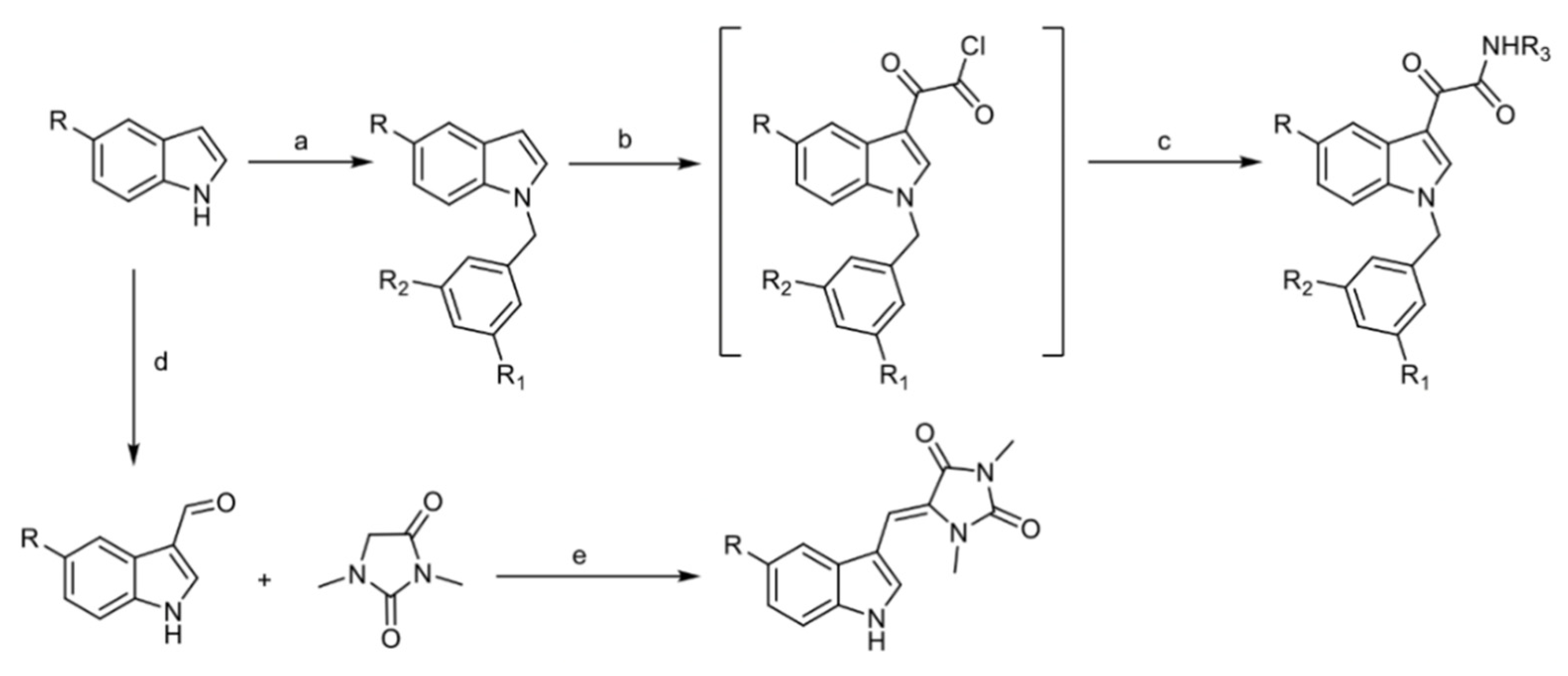
4.1. Chemistry
4.1.1. General Information
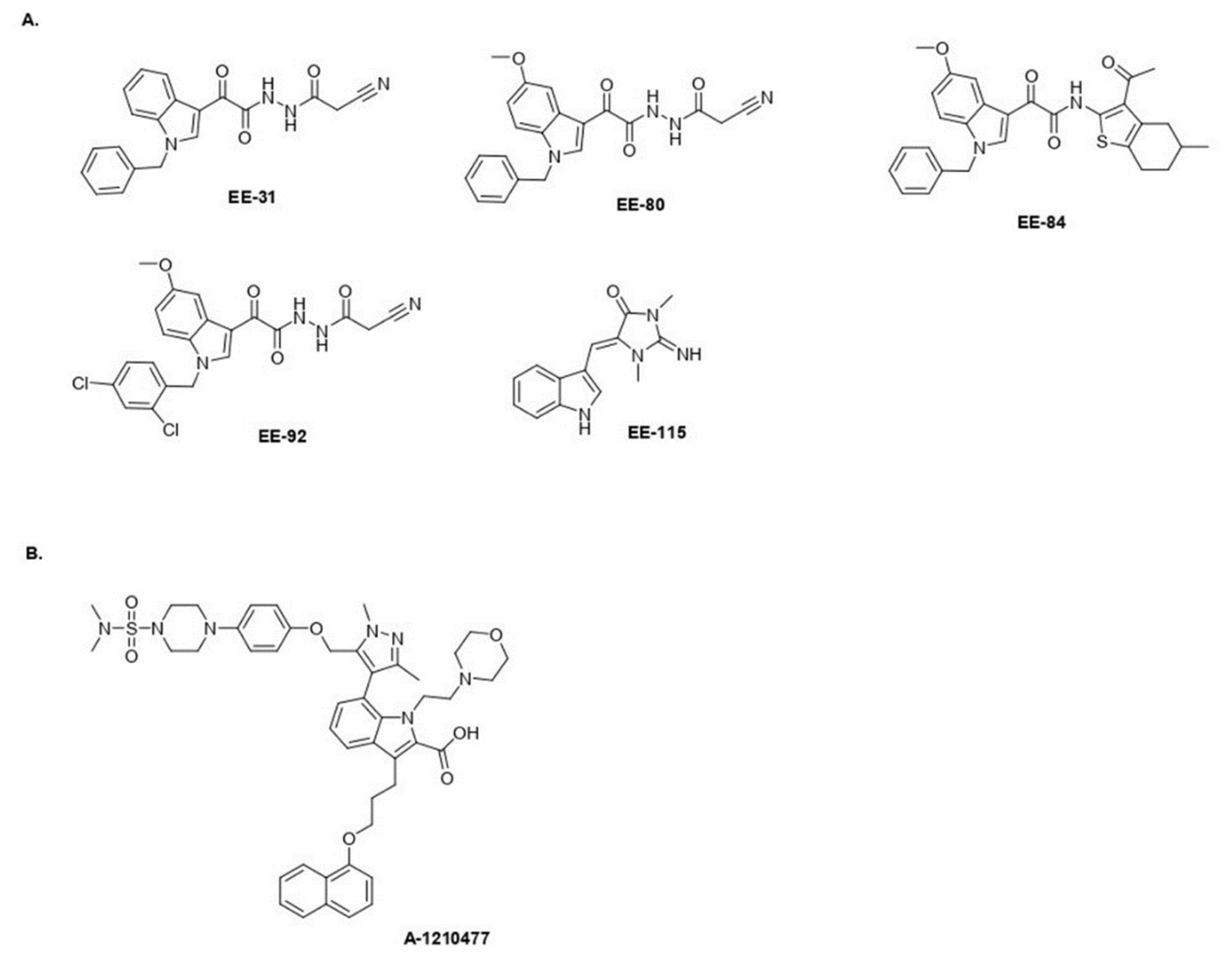
4.1.2. General Procedure for the Preparation of EE-31, EE-80, EE-84, EE-92
- 2-(1-Benzyl-1H-Indol-3-yl)-N′-(2-Cyanoacetyl)-2-Oxoacetohydrazide EE-31
- 2-(1-Benzyl-5-Methoxy-1H-Indol-3-yl)-N′-(2-Cyanoacetyl)-2-Oxoacetohydrazide EE-80
- (N-(3-Acetyl-4,5,6,7-Tetrahydro-5-Methylbenzo[b]Thiophen-2-yl)-2-(1-Benzyl-5-Methoxy-1H-Indol-3-yl)-2-Oxoacetamide EE-84
- N′-(2-Cyanoacetyl)-2-(1-(2,4-Dichlorobenzyl)-5-Methoxy-1H-Indol-3-yl)-2-Oxoacetohydrazide EE-92
4.1.3. General Procedure for the Preparation of EE-115
- (Z)-5-((1H-Indol-3-yl)Methylene)-1,3-Dimethylimidazolidine-2,4-Dione (Aplysinopsin) EE-115 [50]
4.2. Cell Lines and Cell Cultures
4.3. Compounds
4.4. Cell Proliferation and Viability
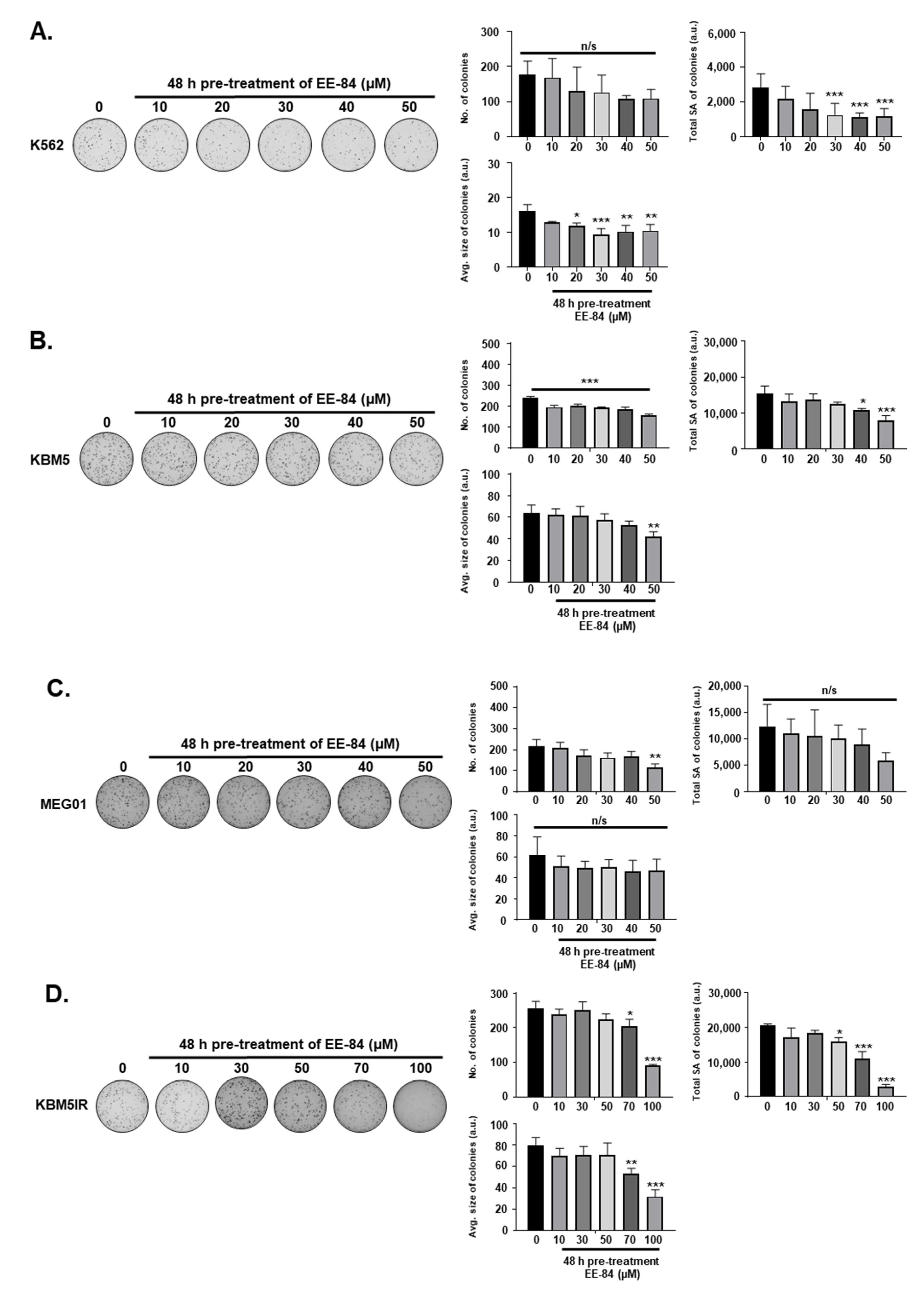
4.5. Colony Formation Assay
4.6. Quantification of Apoptosis and Necrosis
4.7. In Silico Drug Likeliness Properties
4.8. Zebrafish Toxicity
4.9. Cell Cycle Analysis
4.10. Cell Morphology/Wright-Giemsa Staining
4.11. SA-β-Gal Assay
4.12. Analysis of Cell Size and Complexity (Granularity)
4.13. Analysis of Mitochondrial Membrane Potential (MMP) Levels
4.14. Transmission Electron Microscopy (TEM)
4.15. Determination of the Oxygen Consumption Rate and Glycolysis Stress
4.16. Measurement of Intracellular ATP Content
4.17. Measurement of Caspase-3/7 Activity
4.18. Whole-Cell Extracts and Western Blotting
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| Bcr-Abl | breakpoint cluster region-Abelson |
| BH | Bcl-2 homology domain |
| BSA | bovine serum albumin |
| CCyR | complete cytogenetic response |
| CFA | colony formation assay |
| CML | chronic myeloid leukemia |
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| FDA | food and drug administration |
| FLT3 | FMS-like tyrosine kinase 3 |
| GI | growth inhibition |
| HPF | hours post fertilization |
| IC | inhibitory concentration |
| ITD | internal tandem duplication |
| LogP | octanol–water partition coefficient |
| MPER | mammalian protein extraction reagent |
| Mcl-1 | myeloid cell leukemia 1 |
| MMP | mitochondrial membrane potential |
| MTT | 3-(4,5-dimethylthiazol-2-1)-2,5,-diphenyltetrazolium bromide |
| PARP-1 | poly(ADP-ribose)polymerase |
| PBMC | peripheral blood mononuclear cell |
| PBS | phosphate buffered saline |
| PI | propidium iodide |
| PMSF | phenylmethylsulphonyl fluoride |
| PTU | phenylthiourea |
| RPMI | Roswell Park Memorial Institute |
| SA | surface area |
| SD | standard deviation |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TEM | transmission electron microscopy |
| TKD | tyrosine kinase domain |
| UPR | unfolded protein response |
References
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Chang, J.-M.; Chen, C.-A.; Chen, H.-C. Autophagy modulates endoplasmic reticulum stress-induced cell death in podocytes: A protective role. Exp. Biol. Med. 2015, 240, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, P.K.; Kantarjian, H.; Cortes, J.; Cornelison, A.M.; Jabbour, E. Management of imatinib-resistant patients with chronic myeloid leukemia. Ther. Adv. Hematol. 2012, 4, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gong, W.; Yang, Z.Y.; Zhou, X.S.; Gong, C.; Zhang, T.R.; Wei, X.; Ma, D.; Ye, F.; Gao, Q.L. Quercetin induces protective autophagy and apoptosis through ER stress via the p-STAT3/Bcl-2 axis in ovarian cancer. Apoptosis 2017, 22, 544–557. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a Stress-Response and Quality-Control Mechanism: Implications for Cell Injury and Human Disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, L.; Jiang, X.; Li, S.; Huo, H.; Chen, H. DEV induce autophagy via the endoplasmic reticulum stress related unfolded protein response. PLoS ONE 2017, 12, e0189704. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.; Almendros, P.; Fresneda, P.M. Iminophosphorane-Mediated Imidazole Ring Formation—A New and General Entry to Aplysinopsin-Type Alkaloids of Marine Origin. Tetrahedron 1994, 50, 2241–2254. [Google Scholar] [CrossRef]
- Segraves, N.L.; Crews, P. Investigation of brominated tryptophan alkaloids from two thorectidae sponges: Thorectandra and Smenospongia. J. Nat. Prod. 2005, 68, 1484–1488. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Aplysinopsin, a new tryptophan derivative from a sponge. Tetrahedron Lett. 1977, 18, 61–64. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharmacol. 2015, 94, 1–11. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Newton, I.P.; Zhang, L.; Ji, P.; Li, Z. GRP78 is implicated in the modulation of tumor aerobic glycolysis by promoting autophagic degradation of IKKbeta. Cell Signal. 2015, 27, 1237–1245. [Google Scholar] [CrossRef]
- Jiang, C.C.; Lucas, K.; Avery-Kiejda, K.A.; Wade, M.; Debock, C.E.; Thorne, R.F.; Allen, J.; Hersey, P.; Zhang, X.D. Up-regulation of Mcl-1 Is Critical for Survival of Human Melanoma Cells upon Endoplasmic Reticulum Stress. Cancer Res. 2008, 68, 6708–6717. [Google Scholar] [CrossRef]
- Hu, J.; Dang, N.; Menu, E.; De Bryune, E.; Xu, D.; Van Camp, B.; Van Valckenborgh, E.; Vanderkerken, K. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood 2012, 119, 826–837. [Google Scholar] [CrossRef]
- Folmer, F.; Jaspars, M.; Dicato, M.; Diederich, M. Marine cytotoxins: Callers for the various dances of death. Gastroenterol. Hepatol. 2009, 2, S34–S50. [Google Scholar]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Costa-Lotufo, L.V. Marine drugs for cancer: Surfacing biotechnological innovations from the oceans. Clincs 2018, 73, e482s. [Google Scholar] [CrossRef]
- Roberts, W.K.; Dekker, C.A. A convenient synthesis of arabinosylcytosine (cytosine arabinoside). J. Org. Chem. 1967, 32, 816–817. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, W.; Feeney, R.J. Contributions to the Study of Marine Products. Xxxii. The Nucleosides of Sponges. I.1. J. Org. Chem. 2002, 16, 981–987. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeak, K.H.; Schmitz, F.J. Aplysinopsin: Antineoplastic tryptophan derivative from the marine sponge Verongia spengelii. Lloydia 1977, 40, 479–481. [Google Scholar]
- Murata, M.; Miyagawa-Kohshima, K.; Nakanishi, K.; Naya, Y. Characterization of Compounds That Induce Symbiosis between Sea Anemone and Anemone Fish. Science 1986, 234, 585–587. [Google Scholar] [CrossRef]
- Okuda, R.K.; Klein, D.; Kinnel, R.B.; Li, M.; Scheuer, P.J. Marine natural products: The past twenty years and beyond. Pure Appl. Chem. 1982, 54, 1907–1914. [Google Scholar] [CrossRef]
- Bialonska, D.; Zjawiony, J.K. Aplysinopsins-Marine Indole Alkaloids: Chemistry, Bioactivity and Ecological Significance. Mar. Drugs 2009, 7, 166–183. [Google Scholar] [CrossRef]
- Hu, J.F.; Schetz, J.A.; Kelly, M.; Peng, J.N.; Ang, K.K.; Flotow, H.; Leong, C.Y.; Ng, S.B.; Buss, A.D.; Wilkins, S.P.; et al. New antiinfective and human 5-HT2 receptor binding natural and semisynthetic compounds from the Jamaican sponge Smenospongia aurea. J. Nat. Prod. 2002, 65, 476–480. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Yoon, D.-S.; Lim, I.K.; Yoon, S.-H.; Chung, H.-Y.; Rojo, M.; Malka, F.; Jou, M.-J.; Martinou, J.-C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef]
- Keskin, D.; Sadri, S.; Eskazan, A.E. Dasatinib for the treatment of chronic myeloid leukemia: Patient selection and special considerations. Drug Des. Dev. Ther. 2016, 10, 3355–3361. [Google Scholar] [CrossRef]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Matulis, S.M.; Gupta, V.A.; Nooka, A.K.; Von Hollen, H.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia 2016, 30, 1086–1093. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27, S128–S136. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27, S149–S157. [Google Scholar] [CrossRef]
- Chonghaile, T.N. BH3 mimetics: Weapons of cancer cell destruction. Sci. Transl. Med. 2019, 11, eaaw5311. [Google Scholar] [CrossRef]
- Cerella, C.; Gaigneaux, A.; Mazumder, A.; Lee, J.-Y.; Saland, E.; Radogna, F.; Farge, T.; Vergez, F.; Récher, C.; Sarry, J.-E.; et al. Bcl-2 protein family expression pattern determines synergistic pro-apoptotic effects of BH3 mimetics with hemisynthetic cardiac glycoside UNBS1450 in acute myeloid leukemia. Leukemia 2016, 31, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Talhi, O.; Jang, D.; Cerella, C.; Gaigneaux, A.; Kim, K.-W.; Lee, J.W.; Dicato, M.; Bachari, K.; Han, B.W.; et al. Cytostatic hydroxycoumarin OT52 induces ER/Golgi stress and STAT3 inhibition triggering non-canonical cell death and synergy with BH3 mimetics in lung cancer. Cancer Lett. 2018, 416, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Klosa, J. Synthesis of various cyanoacetic acid hydrazide derivatives. VII. Synthesis of tuberculostatic agents. Arch. Pharm. Ber. Dtsch. Pharm. Ges. 1955, 288, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Ottoni, O.; Cruz, R.; Alves, R. Efficient and simple methods for the introduction of the sulfonyl, acyl and alkyl protecting groups on the nitrogen of indole and its derivatives. Tetrahedron 1998, 54, 13915–13928. [Google Scholar] [CrossRef]
- Cummings, D.F.; Canseco, D.C.; Sheth, P.; Johnson, J.E.; Schetz, J.A. Synthesis and structure–affinity relationships of novel small molecule natural product derivatives capable of discriminating between serotonin 5-HT1A, 5-HT2A, 5-HT2C receptor subtypes. Bioorg. Med. Chem. 2010, 18, 4783–4792. [Google Scholar] [CrossRef]
- James, P.N.; Snyder, H.R.; Boekelheide, V.; Knowles, R.N. Indole-3-Aldehyde. Org. Synth. 2003, 39, 30. [Google Scholar] [CrossRef]
- Kirsch, G.; Abdelwahab, A.B.; Hanna, A.G. Synthesis of Novel 3-Acetyl-2-aminothiophenes and Investigation of their Behaviour in the Reaction with Vilsmeier–Haack Reagent. Synthesis 2016, 48, 2881–2888. [Google Scholar] [CrossRef]
- Tymiak, A.A.; Rinehart, K.L.; Bakus, G.J. Constituents of Morphologically Similar Sponges-Aplysina and Smenospongia Species. Tetrahedron 1985, 41, 1039–1047. [Google Scholar] [CrossRef]
- Mazumder, A.; Lee, J.-Y.; Talhi, O.; Cerella, C.; Chateauvieux, S.; Gaigneaux, A.; Hong, C.R.; Kang, H.J.; Lee, Y.; Kim, K.-W.; et al. Hydroxycoumarin OT-55 kills CML cells alone or in synergy with imatinib or Synribo: Involvement of ER stress and DAMP release. Cancer Lett. 2018, 438, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Grandjenette, C.; Ghelfi, J.; Karius, T.; Foliguet, B.; Dicato, M.; Diederich, M. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 2011, 81, 364–378. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GI50 (μM) | ||||
|---|---|---|---|---|
| Compound | Cell Line | 24 h | 48 h | 72 h |
| EE-84 | KBM5 | >50 | 33.95 ± 1.89 | 28.14 ± 1.68 |
| MEG01 | >50 | 48.40 ± 5.02 | 28.89 ± 1.52 | |
| K562IR | >100 | 95.63 ± 16.56 | 55.09 ± 3.28 | |
| KBM5IR | >100 | 32.77 ± 2.25 | 29.15 ± 2.05 |
| EE-84 (μM) | Ima (μM) | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 10 | 20 | 30 | 40 | 50 | 1 | |
| AnnV+/PI− (%) | 5.12 ± 1.19 | 7.05 ± 1.46 | 5.60 ± 1.98 | 6.65 ± 2.29 | 7.71 ± 2.10 | 8.22 ± 3.12 | 51.77 ± 2.29 |
| AnnV+/PI+ (%) | 2.68 ± 0.77 | 2.50 ± 0.62 | 4.40 ± 0.94 | 7.51 ± 2.70 | 8.97 ± 1.03 | 8.25 ± 1.95 | 31.27 ± 5.24 |
| Combined cell death (%) | 7.80 ± 1.73 | 9.55 ± 3.22 | 10.00 ± 0.85 | 14.16 ± 0.61 | 16.68 ± 0.89 | 16.46 ± 0.02 | 83.03 ± 14.50 |
| 48 h Treatment | EE-84 (μM) | Celecoxib (μM) | |||
| % | 0 | 10 | 30 | 50 | 40 |
| Sub-G1 | 1.78 ± 0.91 | 1.48 ± 0.81 | 4.65 ± 2.42 | 8.34 ± 1.51 | 2.79 ±1.90 |
| G1 | 59.81 ± 2.29 | 59.59 ± 1.12 | 61.11 ± 1.58 | 58.40 ± 2.81 | 64.77 ± 3.09 |
| S | 18.02 ± 2.49 | 20.30 ± 1.68 | 20.99 ± 0.83 | 20.41 ± 1.84 | 15.00 ± 0.93 |
| G2/M | 20.39 ± 2.12 | 18.64 ± 0.75 | 13.25 ± 0.32 | 12.86 ± 1.46 | 17.44 ± 2.13 |
| 72 h Treatment | EE-84 (μM) | Celecoxib (μM) | |||
| % | 0 | 10 | 30 | 50 | 40 |
| Sub-G1 | 6.07 ± 2.88 | 4.08 ± 2.80 | 6.33 ± 0.74 | 13.03 ± 1.19 | 3.41 ± 2.04 |
| G1 | 62.33 ± 2.13 | 60.83 ± 2.33 | 55.09 ± 2.26 | 52.03 ± 2.99 | 68.43 ± 4.67 |
| S | 15.50 ± 2.14 | 17.52 ± 4.24 | 21.55 ± 1.64 | 19.85 ± 1.33 | 11.20 ± 2.37 |
| G2/M | 16.10 ± 2.50 | 17.57 ± 0.90 | 17.03 ± 2.69 | 15.08 ± 2.89 | 16.96 ± 3.04 |
| EE-84 | EE-115 | |
|---|---|---|
| Mass | 500.00 | 254.00 |
| Hydrogen bond donor | 1 | 2 |
| Hydrogen bond acceptor | 5 | 4 |
| LogP | 5.91 | 1.85 |
| Molar reactivity | 142.51 | 74.54 |
| GI50 (µM) | |||
|---|---|---|---|
| Compound | 24 h | 48 h | 72 h |
| EE-84 | >50 | >50 | 40.10 ± 3.79 |
| EE-115 | >50 | 30.36 ± 1.95 | 27.72 ± 2.30 |
| EE-84 (µM) | A-1210477 (µM) | Fa | CI |
|---|---|---|---|
| 20 | 10 | 0.483 | 0.66 |
| 30 | 10 | 0.563 | 0.57 |
| Viability (24 h) | ||
|---|---|---|
| Treatment | Cell Line | % |
| EE-84 (30 µM) | K562 | 98.0 ± 1.0 |
| K562IR | 95.3 ± 0.6 | |
| EE-84 (50 µM) | K562 | 96.0 ± 0.7 |
| K562IR | 97.0 ± 2.7 | |
| A-1210477 (10 µM) | K562 | 79.3 ± 1.5 |
| A-1210477 (20 µM) | K562IR | 73.0 ± 3.6 |
| EE-84 (30 µM) + A-1210477 (10 µM) | K562 | 69.0 ± 3.0 |
| EE-84 (30 µM) + A-1210477 (20 µM) | K562IR | 74.7 ± 7.1 |
| EE-84 (50 µM) + A-1210477 (20 µM) | K562IR | 60.0 ± 7.2 |
| 0 | EE-84 30 μM | A-121047710 μM | EE-84 30 μM +A-121047710 μM | Ima 1 μM | |
| AnnV+/PI− (%) | 5.36 ± 1.14 | 6.00 ± 1.43 | 9.31 ± 1.78 | 23.03 ± 4.08 | 37.77 ± 4.07 |
| AnnV+/PI+ (%) | 2.53 ± 0.28 | 2.98 ± 0.23 | 4.73 ± 0.17 | 23.07 ± 5.32 | 6.97 ± 0.88 |
| Combined cell death (%) | 7.90 ± 2.00 | 8.98 ± 2.14 | 14.04 ± 3.24 | 46.10 ± 0.02 | 44.74 ± 21.78 |
| z-VAD-fmk (50 μM) | |||||
| 0 | EE-84 30 μM | A-121047710 μM | EE-84 30 μM +A-121047710 μM | Ima 1 μM | |
| AnnV+/PI− (%) | 4.46 ± 0.68 | 5.33 ± 1.35 | 6.04 ± 0.99 | 5.24 ± 1.15 | 10.86 ± 3.57 |
| AnnV+/PI+ (%) | 3.61 ± 0.13 | 3.13 ± 0.21 | 2.81 ± 0.21 | 3.84 ± 0.28 | 2.93 ± 0.15 |
| Combined cell death (%) | 8.07 ± 0.60 | 8.46 ± 1.55 | 8.85 ± 2.28 | 9.08 ± 0.99 | 13.79 ± 5.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Kim, S.; El-Sawy, E.R.; Cerella, C.; Orlikova-Boyer, B.; Kirsch, G.; Christov, C.; Dicato, M.; Diederich, M. Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477. Mar. Drugs 2021, 19, 285. https://doi.org/10.3390/md19060285
Song S, Kim S, El-Sawy ER, Cerella C, Orlikova-Boyer B, Kirsch G, Christov C, Dicato M, Diederich M. Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477. Marine Drugs. 2021; 19(6):285. https://doi.org/10.3390/md19060285
Chicago/Turabian StyleSong, Sungmi, Sua Kim, Eslam R. El-Sawy, Claudia Cerella, Barbora Orlikova-Boyer, Gilbert Kirsch, Christo Christov, Mario Dicato, and Marc Diederich. 2021. "Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477" Marine Drugs 19, no. 6: 285. https://doi.org/10.3390/md19060285
APA StyleSong, S., Kim, S., El-Sawy, E. R., Cerella, C., Orlikova-Boyer, B., Kirsch, G., Christov, C., Dicato, M., & Diederich, M. (2021). Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477. Marine Drugs, 19(6), 285. https://doi.org/10.3390/md19060285