4.2.2. Experimental Procedures and Characterization Data

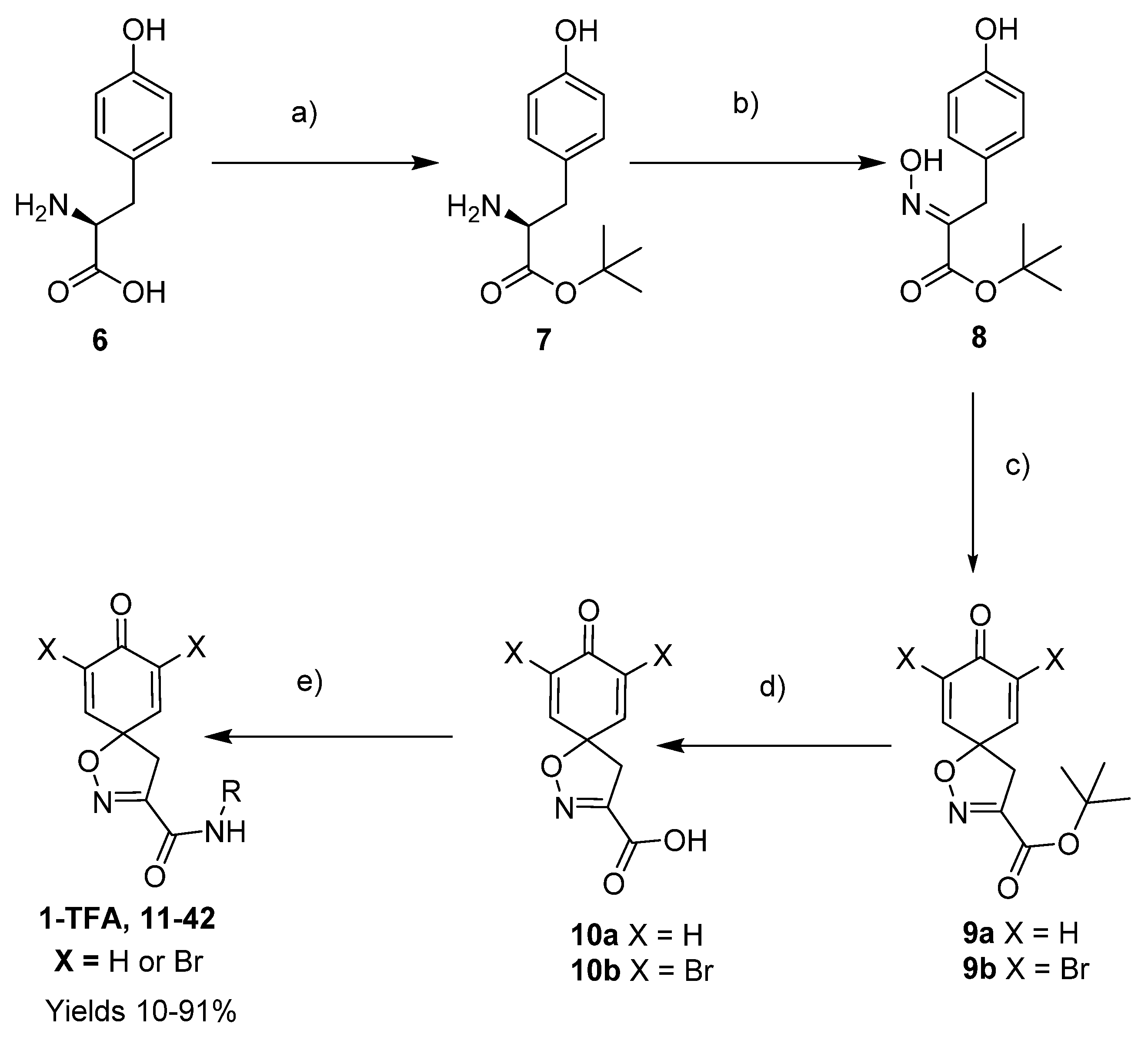
To a stirred suspension of
l-tyrosine
6 (25.0 g, 0.138 mol) in
tert-butyl acetate (100 mL) in an ice bath (0 °C), perchloric acid (15.7 mL, 0.276 mol, 2.0 equiv) was added dropwise. The reaction mixture was allowed to warm to room temperature and was stirred for 17 h. The mixture was washed with H
2O (300 mL) and a 1 M solution of HCl in H
2O (250 mL). The aqueous phase was diluted with H
2O (300 mL), followed by the addition of solid K
2CO
3 until the pH was 7. The resulting mixture was filtered, the filtrate was made alkaline (pH 9) by adding solid K
2CO
3, and then it was extracted with EtOAc (3 × 300 mL). The combined organic phases were washed with brine (300 mL), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo to give an off-white solid; crude yield: 29 g (86%). The crude product was purified with automated flash chromatography (DCM/MeOH, gradient: 0→10%) to give the product
7 as a white solid (25 g, 76%).
1H NMR (400 MHz, CDCl
3)
δ 7.01 (d,
J = 8.5 Hz, 2H), 6.65 (d,
J = 8.5 Hz, 2H), 3.60 (dd,
J = 7.7, 5.3 Hz, 1H), 3.00 (dd,
J = 13.8, 5.3 Hz, 1H), 2.77 (dd,
J = 13.8, 7.7 Hz, 1H), 1.45 (s, 9H).
13C NMR (101 MHz, CDCl
3)
δ 174.2, 155.5, 130.5, 128.2, 115.8, 81.8, 56.2, 40.0, 28.2.
1H NMR is in accordance with the literature [
24].

To a stirred solution of
tert-butyl L-tyrosinate
7 (1.20 g, 5.06 mmol) in EtOH (20 mL) in an ice bath (0 °C), Na
2WO
4·2H
2O (1.83 g, 10.4 mmol, 1.1 equiv), a 30% solution of H
2O
2 in H
2O (9 mL), and H
2O (14 mL) wereadded. The resulting mixture was stirred for 8 h and slowly allowed to reach room temperature. The mixture was diluted with EtOAc (25 mL) and washed with a 10% solution of Na
2SO
3 in H
2O (2 × 15 mL), H
2O (2 × 15 mL), and brine (15 mL). The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic phases were washed with brine (10 mL), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The crude product was purified with automated flash chromatography (
n-heptane/EtOAc gradient: 5→50%) to give the compound
8 as a white solid (1.00 g, 79%).
1H NMR (400 MHz, CD
3OD)
δ 7.05 (d,
J = 8.7 Hz, 2H), 6.67 (d,
J = 8.6 Hz, 2H), 3.79 (s, 2H), 1.43 (s, 9H).
13C NMR (101 MHz, CD
3OD)
δ 164.6, 156.9, 153.6, 131.0, 128.6, 116.1, 83.3, 30.3, 28.1.
1H NMR is in accordance with the literature [
10].

tert-Butyl (
E)-2-(hydroxyimino)-3-(4-hydroxyphenyl)propanoate
8 (0.40 g, 1.6 mmol) was dissolved in 2,2,2-trifluoroethanol (7.7 mL), followed by the addition of anhydrous pyridine (0.39 mL, 4.8 mmol, 2 equiv). The mixture was cooled in an ice bath (0 °C) for 5 min. Phenyliodine bis(trifluoroacetate) (0.76 g, 1.8 mmol, 1.1 equiv) was added to the cooled mixture, and stirring was continued for 1.5 h. The reaction was quenched with a 10% solution of Na
2S
2O
3 in H
2O (13 mL), and the resulting mixture extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with brine, dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The crude product was purified with automated flash chromatography (
n-heptane/EtOAc gradient: 12→100%) to give the compound
9a as a yellow oil (0.33 g, 84%).
1H NMR (400 MHz,
d6-acetone)
δ 7.12–7.05 (m, 2H), 6.24–6.18 (m, 2H), 3.48 (s, 2H), 1.52 (s, 9H).
13C NMR (101 MHz,
d6-acetone)
δ 184.9, 159.5, 153.7, 145.7, 129.2, 83.6, 83.6, 44.3, 28.1.
1H NMR is in accordance with the literature [
10].
To a solution of tert-butyl 8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxylate 9a (0.33 g, 1.3 mmol) in anhydrous DCM (10 mL), trifluoroacetic acid (5.0 mL) was added dropwise. The resulting mixture was stirred at room temperature for 3 h. The solvent was removed in vacuo to give the compound 10a as a light brown solid (0.24 g, 93%). 1H NMR (400 MHz, d6-acetone) δ 7.15–7.08 (m, 1H), 6.26–6.19 (m, 1H), 3.51 (s, 1H). 13C NMR (101 MHz, d6-acetone) δ 184.9, 161.0, 153.0, 145.7, 129.3, 116.5, 83.8.
To a solution of
tert-butyl (
E)-2-(hydroxyimino)-3-(4-hydroxyphenyl)propanoate
8 (0.59 g, 2.348 mmol) in anhydrous DMF (10 mL) in an ice bath (0 °C),
N-bromosuccinimide (1.35 g, 7.58 mmol, 3.25 equiv) in anhydrous DMF (5 mL) was added dropwise (15 min). The mixture was diluted with Et
2O (25 mL), and washed with H
2O (2 × 15 mL) and a 10% solution of Na
2S
2O
3 in H
2O (2 × 15 mL). The aqueous phase was back-extracted with Et
2O (2 × 30 mL). The combined organic phases were washed with brine (20 mL), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The crude product was purified with automated flash chromatography (isocratic DCM) to give the compound
9b as a white solid (0.58 g, 62%).
1H NMR (400 MHz, CDCl
3)
δ 7.32 (s, 2H), 3.42 (s, 2H), 1.57 (s, 9H).
13C NMR (101 MHz, CDCl
3)
δ 171.5, 158.2, 152.3, 144.1, 123.4, 85.8, 84.9, 43.2, 27.8.
1H NMR is in accordance with the literature [
10].
To a solution of
tert-butyl 7,9-dibromo-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxylate
9b (0.58 g, 1.4 mmol) in anhydrous DCM (10 mL), trifluoroacetic acid (5.0 mL) was added dropwise. The resulting mixture was stirred at room temperature for 1 h, after which the solvent was removed in vacuo. The product was purified via trituration with Et
2O to give the compound
10b as a white solid (0.50 g, 98%).
1H NMR (400 MHz,
d6-DMSO)
δ 7.81 (s, 2H), 3.52 (s, 2H).
1H NMR is in accordance with the literature [
22].
13C NMR (101 MHz,
d6-acetone)
δ 172.3, 160.7, 153.6, 146.8, 123.2, 87.3, 43.7. HRMS (ESI-): calculated 303.8609 (C
8H
4Br
2NO
2), found 303.8609. LC-MS: [M-CO
2]
− m/
z 304 (
tR = 2.12 min), >99%. Mp: 174–175 °C, (Lit. Mp: 167–168 °C) [
10].
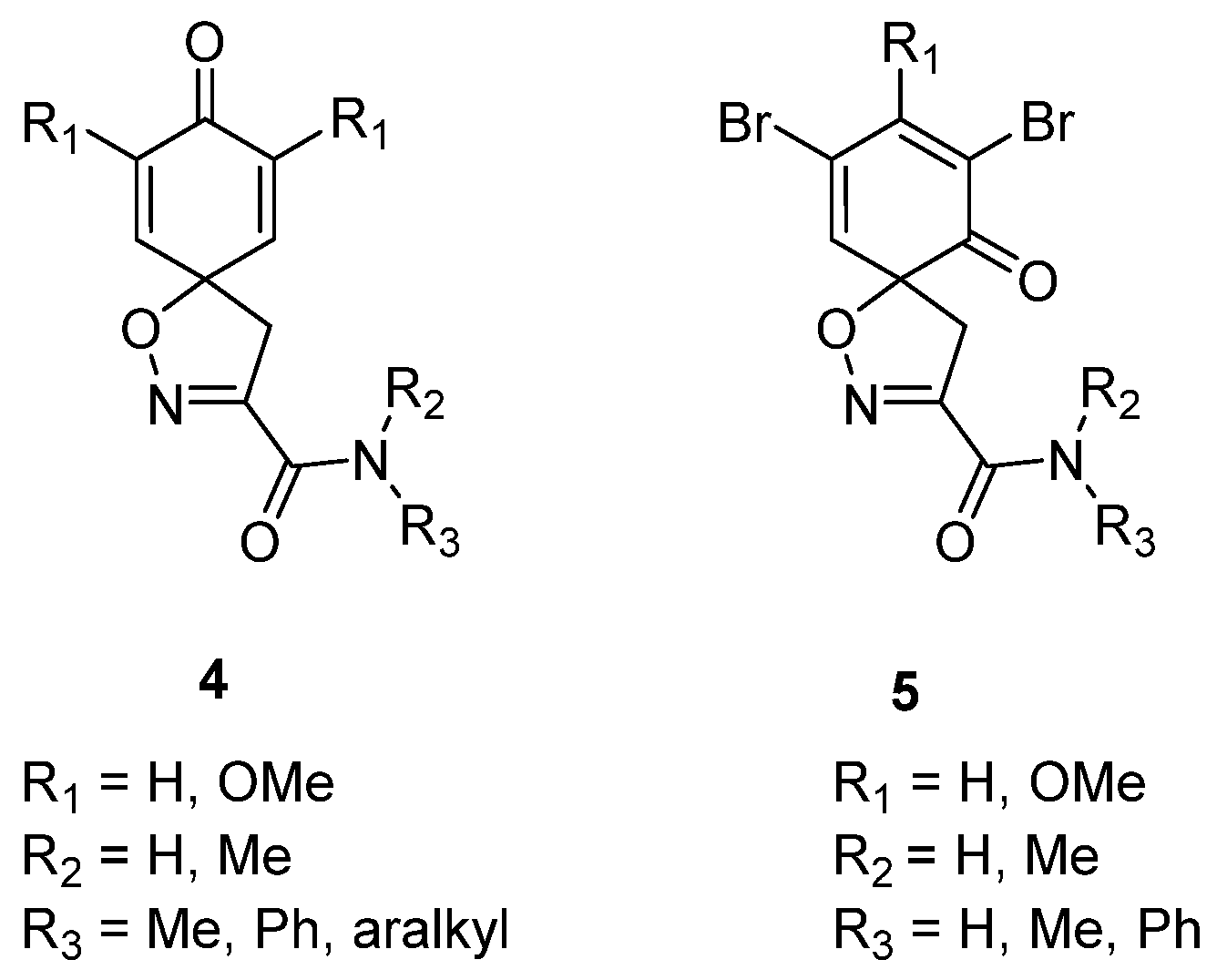
The spiro carboxylate core was mainly constructed using the synthetic method given below, followed by N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)-mediated coupling (General procedures for coupling A and B) to give the corresponding product. Some compounds were deprotected with trifluoroacetic acid to give the corresponding trifluoroacetate salts (General procedure C).
General procedure for EDC-mediated coupling (A). To a stirred solution of carboxylic acid 10a or 10b (0.36 mmol) in DCM (5 mL), 1-hydroxybenzotriazole (HOBt) hydrate (0.036 mmol, 0.1 equiv) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) hydrochloride (0.39 mmol, 1.1 equiv) at 0–5 °C were added and stirred for 15 min. After this, the amine/hydrazide (0.36 mmol, 1.0 equiv) was added. The reaction mixture was allowed to reach room temperature and was stirred for a further 8–16 h. The mixture was diluted with DCM (10 mL) and washed with 1 M hydrochloric acid (5 mL), a saturated solution of NaHCO3 in H2O (5 mL), and brine (5 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by automated flash chromatography (n-heptane/EtOAc gradient: 0→100%) to give the pure product.
General procedure for EDC-mediated coupling (B). Carboxylic acid 10a or 10b (0.3 mmol), amine (0.45 mmol, 1.5 equiv), HOBt hydrate (0.45 mmol, 1.5 equiv), and EDC·HCl (0.45 mmol, 1.5 equiv) were dissolved in anhydrous DCM (3 mL). The mixture was irradiated under microwave conditions at 60 °C for 2 h, after which it was diluted with DCM (10 mL). The solution was washed with a saturated solution of NH4Cl in H2O, water, and brine. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified with automated flash column chromatography (n-heptane/EtOAc-EtOH 3:1 (12→100%) to give the pure product.
General procedure for deprotection of the Boc groups (C). To a solution of clavatadine bis-Boc-derivative (0.28 mmol) in DCM (2 mL), TFA (1 mL) at 0–5 °C was added dropwise. The reaction mixture was allowed to reach room temperature. The resulting mixture was stirred for 3 h at room temperature. The solvent was removed in vacuo to give the crude product, which was triturated in Et2O to give a solid trifluoroacetate salt.
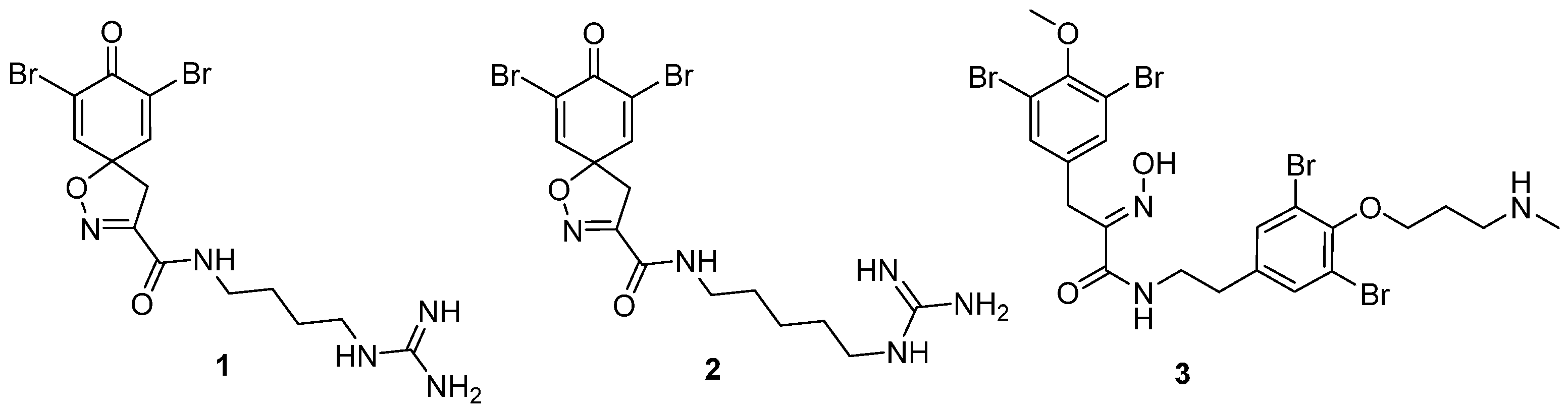
7,9-Dibromo-N-(4-guanidinobutyl)-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide TFA salt (1-TFA)
General procedures
A and
C were followed to give the clavatadine C TFA salt
12 as an off-white solid (0.10 g, 77%).
1H NMR (400 MHz,
d6-DMSO)
δ 8.65 (t,
J = 5.9 Hz, 1H), 7.80 (s, 2H), 7.59 (t,
J = 5.7 Hz, 1H), 3.55 (s, 2H), 3.23–3.06 (m, 4H), 1.48 (m, 4H).
13C NMR (101 MHz,
d6-DMSO)
δ 171.6, 158.2, 156.7, 155.0, 146.7, 121.6, 85.2, 43.2, 40.4, 38.4, 26.0, 25.9. Spectra are in accordance with the literature [
10]. HRMS (ESI
+): Calculated 463.9757 (C
14H
18Br
2N
5O
3), found 463.9755. LC-MS: [M + H]
+ m/
z 464 (
tR = 2.25 min), >91%. Mp: 115–118 °C, decomp. (Lit. Mp: 130–140 °C, decomp.) [
10].
General procedures A and C were followed to give the compound 11 as an off-white solid (0.11 g, 34%). 1H NMR (400 MHz, CD3OD) δ 7.04–7.00 (m, 2H), 6.26–6.22 (m, 2H), 3.44 (s, 2H), 3.36–3.34 (m, 2H), 3.23–3.20 (m, 2H), 1.64 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 186.4, 161.3, 158.6, 153.3, 146.7, 129.4, 83.6, 44.4, 42.0, 39.8, 27.5, 27.1. HRMS (ESI+): calculated 306.1566 (C14H20N5O3), found 306.1567. LC-MS: [M + H]+ m/z 306 (tR = 0.92 min), >98%. Mp: 170–173 °C.
General procedure A was followed to give the compound 12 as an off-white amorphous solid (0.011 g, 28%). 1H NMR (400 MHz, CD3OD) δ 7.05 (d, J = 10.1 Hz, 2H), 6.25 (d, J = 10.1 Hz, 2H), 3.50 (s, 2H), 3.28 (s, 3H), 3.06 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 185.1, 160.7, 153.4, 145.5, 128.7, 80.6, 45.2, 37.6, 34.7. HRMS (ESI+): calculated 221.0926 (C11H13N2O3), found 221.0927. LC-MS: [M + H]+ m/z 221 (tR = 1.85 min), >99%.
7,9-Dibromo-N,N-dimethyl-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (13)
General procedure A was followed to give the compound 13 as a white amorphous solid (0.12 g, 37%). 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 2H), 3.55 (s, 2H), 3.32 (s, 3H), 3.09 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 159.3, 153.7, 144.9, 123.8, 84.1, 45.8, 38.8, 36.5. HRMS (ESI+): calculated 378.9117 (C11H11Br2N2O3), found 378.9119.
General procedure A was followed to give the compound 14 as an off-white amorphous solid (0.061 g, 35%). 1H NMR (400 MHz, CDCl3) (1:1 mixture of rotamers) δ 7.33 (7.32) (s, 2H), 4.84 (4.66) (p, J = 6.8 Hz, 1H), 3.56 (3.55) (s, 2H), 3.10 (2.92) (s, 3H), 1.26 (1.19) (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.6, 159.0 (158.9), 154.2 (153.5), 144.98 (144.96), 123.71 (123.69), 84.0 (83.9), 49.6 (45.9), 46.03 (45.97), 30.1 (27.0), 20.9 (19.3). HRMS (ESI+): calculated 406.9430 (C13H15Br2N2O3), found 406.9426. LC-MS: [M + H]+ m/z 407 (tR = 4.31 min), >99%. Mp: 148–153 °C.
General procedure A was followed to give the compound 15 as an off-white amorphous solid (0.012 g, 12%). 1H NMR (400 MHz, CDCl3) δ 6.90–6.81 (m, 2H), 6.33–6.25 (m, 2H), 4.02–3.95 (m, 2H), 3.80–3.70 (m, 6H), 3.48 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.5, 158.9, 153.2, 144.1, 129.5, 80.9, 67.1, 66.8, 47.4, 46.2, 43.3. HRMS (ESI+): calculated 263.1032 (C13H15N2O4), found 263.1036. LC-MS: [M + H]+ m/z 442 (tR = 5.39 min), >99%.
General procedure A was followed to give the compound 16 as an off-white amorphous solid (0.024 g, 24%). 1H NMR (400 MHz, CDCl3): δ 6.92–6.83 (m, 2H), 6.33–6.24 (m, 2H), 4.23–4.14 (m, 1H), 4.08–4.00 (m, 2H), 3.72–3.60 (m, 1H), 3.53–3.42 (m, 3H), 2.02–1.90 (m, 2H), 1.69–1.59 (m, 2H), 1.57 (br s, 1H). 13C NMR (101 MHz, CDCl3) δ 184.5, 158.4, 153.2, 144.3, 129.4, 80.8, 66.7, 46.3, 39.9, 34.7, 33.8. HRMS (ESI+): calculated 277.1188 (C14H17N2O4), found 277.1187. LC-MS: [M + H]+ m/z 277 (tR = 1.61 min), >99%.
General procedure A was followed to give the compound 17 as an amorphous off-white solid (0.030 g, 17%). 1H NMR (400 MHz, CD3OD) δ 7.09–7.00 (m, 2H), 6.29–6.20 (m, 2H), 3.77–3.61 (m, 4H), 3.47 (s, 2H), 3.17 (s, 2H), 2.68–2.55 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 186.4, 171.6, 160.2, 154.3, 146.6, 129.5, 83.5, 67.7, 61.5, 54.7, 44.4. HRMS (ESI+): calculated 335.1356 (C15H19N4O5), found 335.1354. LC-MS: [M + H]+ m/z 335 (tR = 0.53 min), >96%.
General procedure A was followed to give the compound 18 as an off-white amorphous solid (0.098 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.36 (br s, 1H), 7.56 (d, J = 1.8 Hz, 2H), 7.17 (t, J = 1.8 Hz, 1H), 6.89–6.84 (m, 2H), 6.35–6.29 (m, 2H), 3.45 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 183.9, 156.3, 153.4, 143.1, 138.2, 135.4, 129.4, 125.0, 117.9, 83.6, 42.7. HRMS (ESI+): calculated 337.0147 (C15H11Cl2N2O3), found 337.0148. LC-MS: [M + H]+ m/z 337. (tR = 4.81 min), >91%.
7,9-Dibromo-N-(3,5-dichlorophenyl)-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (19)
General procedure B was followed to give the compound 19 as a white amorphous solid (0.0045 g, 13%). 1H NMR (400 MHz, d6-acetone) δ 9.78 (br s, 1H), 7.93 (d, J = 1.9 Hz, 2H), 7.77 (s, 2H), 7.26 (t, J = 1.8 Hz, 1H), 3.76 (s, 2H). 13C NMR (101 MHz, d6-acetone) δ 172.2, 158.3, 155.8, 146.7, 141.3, 135.7, 124.7, 123.3, 119.1, 119.0, 87.4, 43.4. HRMS (ESI−): calculated 490.8200 (C15H9Br2Cl2N2O3), found 490.8198. LC-MS: [M-H]− m/z 491 (tR = 5.84 min), >99%.
General procedure A was followed to give the compound 20 as an off-white amorphous solid (0.025 g, 13%). 1H NMR (400 MHz, CDCl3) δ 8.23 (br s, 1H), 7.68 (d, J = 2.6 Hz, 1H), 7.43 (dd, J = 8.9, 2.6 Hz, 1H), 6.92 (d, J = 8.9 Hz, 1H), 6.90–6.85 (m, 2H), 6.34–6.28 (m, 2H), 3.91 (s, 3H), 3.46 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.3, 156.4, 154.0, 152.7, 143.7, 130.2, 129.6, 123.0, 122.5, 119.6, 112.4, 83.5, 56.5, 43.3. HRMS (ESI+): calculated 333.0642 (C16H14ClN2O4), found 333.0639. LC-MS: [M + H]+ m/z 333 (tR = 3.68 min), >99%.
7,9-Dibromo-N-(3-chloro-4-methoxyphenyl)-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (21)
General procedure B was followed to give the compound 21 as a white amorphous solid (0.037 g, 18%). 1H NMR (400 MHz, d6-DMSO) δ 10.59 (br s, 1H), 7.88 (d, J = 2.6 Hz, 1H), 7.85 (s, 2H), 7.68 (dd, J = 9.0, 2.6 Hz, 1H), 7.15 (d, J = 9.1 Hz, 1H), 3.83 (s, 3H), 3.65 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 156.8, 155.4, 151.2, 146.5, 131.6, 121.8, 121.8, 120.5, 120.2, 112.8, 85.6, 56.2, 43.0. HRMS (ESI+): calculated 486.8696 (C16H11Br2ClN2O4), found 486.8696. LC-MS: [M-H]− m/z 487 (tR = 4.95 min), >94%.
General procedure A was followed to give the compound 22 as a white amorphous solid (0.058 g, 56%). 1H NMR (400 MHz, CDCl3) δ 8.12 (br s, 1H), 7.65–7.59 (m, 1H), 7.39 (m, 1H), 7.23 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.15 (ddd, J = 8.0, 7.1, 1.0 Hz, 1H), 7.08 (d, J = 2.3 Hz, 1H), 6.84–6.78 (m, 2H), 6.67 (br s, 1H), 6.30–6.23 (m, 2H), 3.71 (q, J = 6.8 Hz, 2H), 3.36 (s, 2H), 3.06 (t, J = 6.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 184.4, 158.7, 153.8, 144.0, 136.5, 129.3, 127.2, 122.5, 122.2, 119.7, 118.8, 112.5, 111.4, 82.7, 43.7, 39.8. HRMS (ESI+): calculated 336.1348 (C19H18N3O3), found 336.1349. LC-MS: [M-H]− m/z 336 (tR = 4.95 min), >99%.
General procedure A was followed to give the compound 23 as an off-white amorphous solid (0.13 g, 67%). 1H NMR (400 MHz, CDCl3) δ 7.27–7.22 (m, 2H), 6.91–6.87 (m, 3H), 6.86–6.81 (m, 2H), 6.30–6.24 (m, 2H), 4.48 (d, J = 5.9 Hz, 2H), 3.81 (s, 3H), 3.41 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.4, 159.5, 158.5, 153.7, 143.9, 129.5, 129.4, 114.5, 114.4, 82.9, 55.4, 43.6, 43.3. HRMS (ESI+): calculated 313.1188 (C17H17N2O4), found 313.1191. LC-MS: [M + H]+ m/z 313 (tR = 3.06 min), >99%.
General procedure A was followed to give the compound 24 as an off-white amorphous solid (0.022 g, 20%). 1H NMR (400 MHz, d6-DMSO) δ 9.07 (t, J = 6.2 Hz, 1H), 7.82 (s, 2H), 7.28–7.19 (m, 2H), 6.93–6.84 (m, 2H), 4.29 (d, J = 6.2 Hz, 2H), 3.73 (s, 3H), 3.56 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 158.3, 158.1, 155.0, 146.7, 130.8, 128.8, 121.5, 113.6, 85.2, 55.0, 43.1, 41.7. HRMS (ESI+Na+): calculated 492.9199 (C17H15Br2N2O4Na), found 492.9197.
General procedure A was followed to give the compound 25 as an off-white amorphous solid (0.042 g, 41%). 1H NMR (400 MHz, CDCl3) δ 7.15–7.11 (m, 2H), 6.89–6.85 (m, 2H), 6.85–6.81 (m, 2H), 6.63 (app. t, 1H), 6.31–6.24 (m, 2H), 3.80 (s, 3H), 3.60 (q, J = 7.0 Hz, 2H), 3.38 (s, 2H), 2.83 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 184.4, 158.6, 158.6, 153.8, 143.9, 130.2, 129.7, 129.4, 114.3, 82.8, 55.4, 43.6, 41.0, 34.8. HRMS (ESI+): calculated 327.1345 (C18H19N2O4), found 327.1348. LC-MS: [M + H]+ m/z 327 (tR = 3.35 min), >99%.
General procedure A was followed to give the compound 26 as an off-white amorphous solid (0.047 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 2.5 Hz, 1H), 8.44 (dd, J = 4.7, 1.4 Hz, 1H), 8.40 (br s, 1H), 8.14 (m, 1H), 7.33 (dd, J = 8.3, 4.7 Hz, 1H), 6.91–6.86 (m, 2H), 6.35–6.30 (m, 2H), 3.47 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.2, 157.0, 153.8, 146.4, 143.5, 141.5, 133.7, 129.7, 127.2, 124.0, 83.8, 43.1. HRMS (ESI+): calculated 270.0879 (C14H12N3O3), found 270.0876. LC-MS: [M + H]+ m/z 270 (tR = 1.03 min), >92%.
General procedure B was followed to give the compound 27 as a white amorphous solid (0.042 g, 34%). 1H NMR (400 MHz, d6-DMSO) δ 10.81 (br s, 1H), 8.93 (dd, J = 2.6, 0.8 Hz, 1H), 8.33 (dd, J = 4.7, 1.5 Hz, 1H), 8.14 (ddd, J = 8.4, 2.6, 1.5 Hz, 1H), 7.86 (s, 2H), 7.40 (ddd, J = 8.3, 4.7, 0.8 Hz, 1H), 3.66 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 157.4, 155.2, 146.4, 145.1, 142.0, 134.9, 127.4, 123.6, 121.8, 85.8, 42.9. HRMS (ESI+): calculated 425.9089 (C14H10Br2N3O3), found 425.9090. LC-MS: [M + H]+ m/z 426 (tR = 2.49 min), >99%.
General procedure A was followed to give the compound 28 as an off-white amorphous solid (0.040 g, 53%). 1H NMR (400 MHz, CDCl3) δ 9.01 (br s, 1H), 8.35 (ddd, J = 5.0, 1.9, 1.0 Hz, 1H), 8.18–8.16 (m, 1H), 7.76 (ddd, J = 8.4, 7.4, 1.9 Hz, 1H), 7.12 (ddd, J = 7.4, 5.0, 1.0 Hz, 1H), 6.92–6.85 (m, 2H), 6.35–6.28 (m, 2H), 3.46 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.3, 156.9, 153.7, 150.2, 148.4, 143.7, 138.7, 129.5, 120.7, 114.2, 83.6, 43.1. HRMS (ESI+): calculated 270.0879 (C14H12N3O3), found 270.0881. LC-MS: [M + H]+ m/z 270 (tR = 2.11 min), >99%.
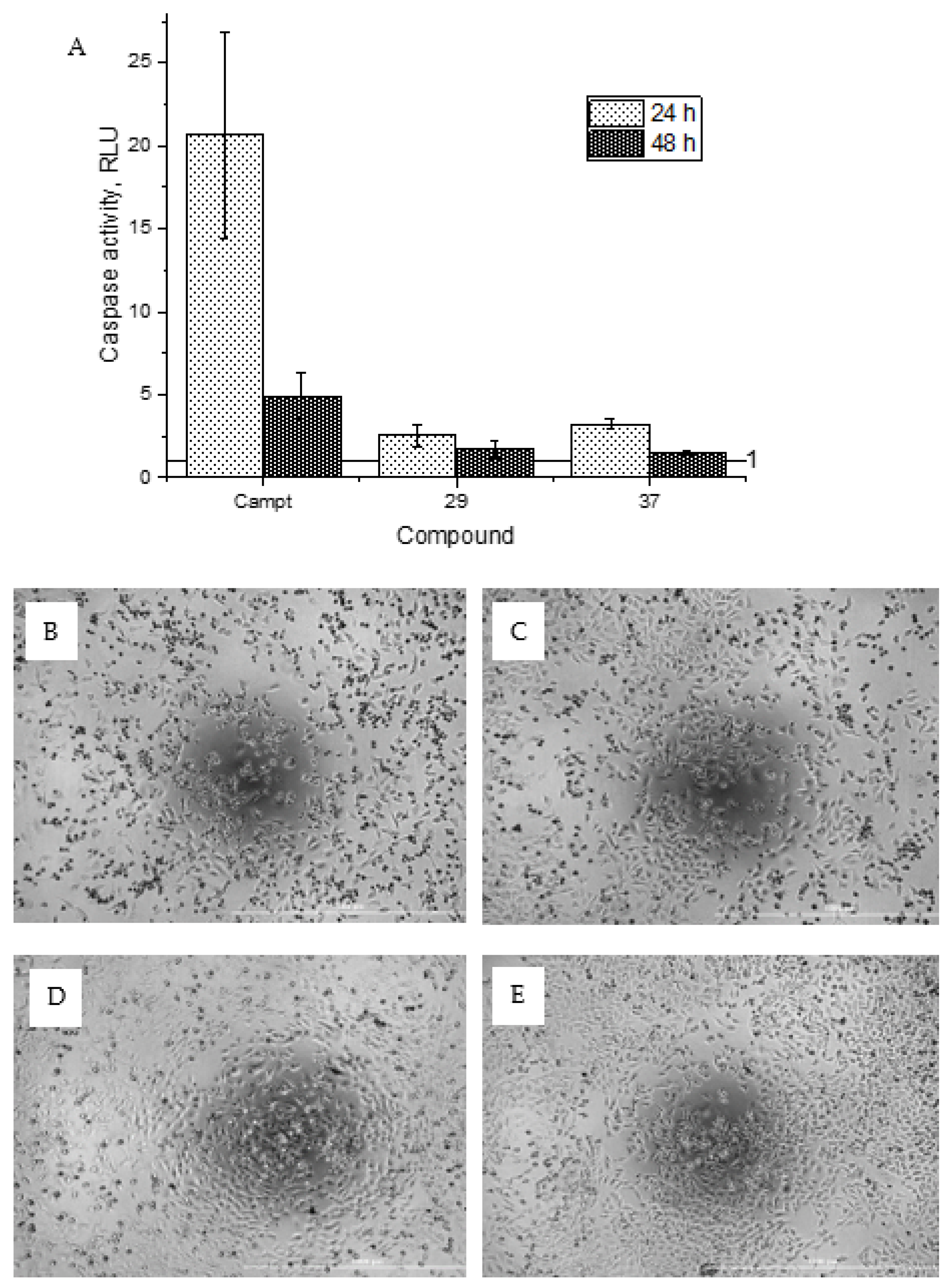
General procedure B was followed to give the compound 29 as a white amorphous solid (0.016 g, 13%). 1H NMR (400 MHz, d6-acetone) δ 9.22 (br s, 1H), 8.36 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.15 (dt, J = 8.3, 1.0 Hz, 1H), 7.85 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.80 (s, 2H), 7.19 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 3.80 (s, 2H). 13C NMR (101 MHz, d6-acetone) δ 172.3, 157.9, 155.9, 151.6, 150.7, 149.4, 146.7, 139.3, 123.3, 121.3, 114.5, 87.5, 43.3. HRMS (ESI+): calculated 427.9245 (C14H12Br2N3O3), found 427.9243. LC-MS: [M + H]+ m/z 426 (tR = 3.92 min), >95%.
7,9-Dibromo-8-oxo-N-[5-(trifluoromethyl)pyridin-2-yl]-1-oxa-2-azaspiro[4.5]-deca-2,6,9-triene-3-carboxamide (30)
General procedure A was followed to give the compound 30 as an off-white amorphous solid (0.012 g, 10%). 1H NMR (400 MHz, CDCl3) δ 9.14 (br s, 1H), 8.62 (d, J = 2.3 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 7.99 (dd, J = 8.7, 2.4 Hz, 1H), 7.34 (s, 2H), 3.56 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 171.3, 156.7, 153.6, 152.6, 146.0, 144.5 (q, JC,F = 40.4 Hz), 143.9, 136.2 (q, JC,F = 33.3 Hz), 124.3, 123.9, 113.6, 87.0, 42.4. HRMS (ESI+): calculated 495.8643 (C15H9Br2F3N3O3), found 495.8941. LC-MS: [M + H]+ m/z 496 and 498 (tR = 5.30 and 5.15 min), >97%.
7,9-Dibromo-8-oxo-N-[6-(trifluoromethyl)pyridin-3-yl]-1-oxa-2-azaspiro[4.5] deca-2,6,9-triene-3-carboxamide (31)
General procedure A was followed to give the compound 31 as an off-white amorphous solid (0.015 g, 13%). 1H NMR (400 MHz, d6-acetone) δ 10.09 (br s, 1H), 9.14 (d, J = 2.4 Hz, 1H), 8.55 (dd, J = 8.7, 2.5 Hz, 1H), 7.87 (d, J = 8.6 Hz, 1H), 7.78 (s, 2H), 3.80 (s, 2H). 13C NMR (101 MHz, d6-acetone) δ 172.3, 158.7 (m), 155.8 (m), 146.7, 143.4 (q, JC,F = 34.7 Hz), 142.6 (m), 138.5 (m), 128.4 (m), 123.4, 122.8 (q, JC,F = 272.6 Hz), 121.9 (q, JC,F = 2.9 Hz), 87.5, 43.4. HRMS (ESI+): calculated 495.8943 (C15H9Br2F3N3O3), found 495.8948. LC-MS: [M + H]+ m/z 496 and 498 (tR = 4.80 and 4.72 min), >91%.
General procedure A was followed to give the compound 32 as an off-white amorphous solid (0.076 g, 91%). 1H NMR (400 MHz, CDCl3) δ 8.59–8.54 (m, 1H), 7.71 (app. t, 1H), 7.65 (td, J = 7.7, 1.8 Hz, 1H), 7.21–7.19 (m, 1H), 7.19–7.16 (m, 1H), 6.87–6.81 (m, 2H), 6.29–6.23 (m, 2H), 3.81 (dt, J = 6.8, 5.9 Hz, 2H), 3.39 (s, 2H), 3.07 (t, J = 6.3 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 184.5, 159.0, 158.6, 153.9, 149.4, 144.1, 137.0, 129.3, 123.6, 121.9, 82.7, 43.8, 38.6, 36.5. HRMS (ESI+): calculated 298.1192 (C16H16N3O3), found 298.1191. LC-MS: [M + H]+ m/z 298 (tR = 0.74, salt and 1.15 min), >99%.
7,9-Dibromo-8-oxo-N-[2-(pyridin-2-yl)ethyl]-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (33)
General procedure B was followed to give the compound 33 as a white amorphous solid (0.062 g, 24%). 1H NMR (400 MHz, CDCl3) δ 8.59–8.54 (m, 1H), 7.79 (app. t, 1H), 7.64 (td, J = 7.7, 1.8 Hz, 1H), 7.30 (s, 2H), 7.21–7.15 (m, 2H), 3.81 (q, J = 6.0 Hz, 2H), 3.47 (s, 2H), 3.06 (t, J = 6.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 171.5, 159.1, 158.0, 154.1, 149.5, 144.6, 136.9, 123.8, 123.5, 122.0, 85.8, 43.4, 38.7, 36.4. HRMS (ESI+): calculated 453.9402 (C16H14Br2N3O3), found 453.9403. LC-MS: [M + H]+ m/z 454 (tR = 2.27 min), >90%.
7,9-Dibromo-8-oxo-N-[2-(pyridin-3-yl)ethyl]-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (34)
General procedure A was followed to give the compound 34 as an off-white solid (0.075 g, 39%). 1H NMR (400 MHz, d6-DMSO) δ 8.72 (t, J = 5.8 Hz, 1H), 8.44 (dq, J = 6.5, 2.6, 1.7 Hz, 2H), 7.80 (s, 2H), 7.66 (dt, J = 7.8, 2.0 Hz, 1H), 7.33 (ddd, J = 7.8, 4.8, 0.9 Hz, 1H), 3.54 (s, 2H), 3.48–3.38 (m, 2H), 2.83 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 158.2, 154.9, 149.8, 147.5, 146.7, 146.7, 136.1, 134.6, 123.4, 121.6, 85.2, 43.1, 31.7. HRMS (ESI+): calculated 492.9199 (C16H14Br2N3O3), found 492.9197. Mp: 202–205 °C.
General procedure B was followed to give the compound 35 as a white amorphous solid (0.13 g, 61%). 1H NMR (400 MHz, d6-acetone) δ 8.59 (dd, J = 2.3, 0.9 Hz, 1H), 8.48 (dd, J = 4.8, 1.7 Hz, 1H), 8.36 (br s, 1H), 7.80–7.75 (m, 1H), 7.32 (ddd, J = 7.9, 4.8, 0.9 Hz, 1H), 7.14–7.05 (m, 2H), 6.25–6.15 (m, 2H), 4.55 (d, J = 6.3 Hz, 2H), 3.50 (s, 2H). 13C NMR (101 MHz, d6-acetone) δ 184.9, 159.9, 155.1, 150.3, 149.4, 145.9, 136.1, 135.4, 129.2, 124.2, 83.2, 44.2, 41.2. HRMS (ESI+): calculated 284.1035 (C15H14N3O3), found 284.1032. LC-MS: [M + H]+ m/z 284 (tR = 0.68 min), >99%.
General procedure B was followed to give 36 as a white amorphous solid (0.068 g, 55%). 1H NMR (400 MHz, d6-DMSO) δ 8.53 (dd, J = 2.3, 0.9 Hz, 1H), 8.47 (dd, J = 4.8, 1.7 Hz, 1H), 7.83 (s, 2H), 7.74–7.68 (m, 1H), 7.36 (ddd, J = 7.8, 4.8, 0.9 Hz, 1H), 4.39 (s, 2H), 3.57 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 158.4, 158.4, 154.9, 154.8, 148.9, 148.2, 146.7, 135.3, 134.3, 134.3, 123.4, 121.6, 85.4, 43.0, 40.0. HRMS (ESI+): calculated 439.9245 (C15H12Br2N3O3), found 439.9244. LC-MS: [M + H]+ m/z 440 (tR = 2.08 min), >94%.
General procedure A was followed to give the compound 37 as an off-white solid (0.025 g, 16%). 1H NMR (400 MHz, d6-DMSO) δ 10.71 (br s, 1H), 10.65 (br s, 1H), 8.75–8.67 (m, 1H), 8.10–8.00 (m, 2H), 7.69–7.63 (m, 1H), 7.22–7.13 (m, 2H), 6.31–6.22 (m, 2H), 3.54 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 184.5, 163.0, 157.8, 153.2, 149.1, 148.7, 145.6, 137.9, 128.2, 127.1, 122.5, 81.6, 43.3. HRMS (ESI+): calculated 313.0937 (C15H13N4O4), found 313.0937. LC-MS: [M + H]+ m/z 313 (tR = 1.51 min), >93%. Mp: 191–193 °C.
General procedure A was followed to give the compound 38 as an off-white solid (0.1 g, 50%). 1H NMR (400 MHz, d6-DMSO) δ 10.74 (s, 1H), 10.65 (s, 1H), 8.70 (dt, J = 4.7, 1.4 Hz, 1H), 8.08–7.99 (m, 2H), 7.88 (s, 2H), 7.71–7.63 (m, 1H), 3.63 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.7, 163.0, 157.5, 153.6, 149.0, 148.6, 146.7, 137.9, 127.1, 122.5, 121.7, 85.2, 43.5. HRMS (ESI+): calculated 470.9128 (C15H11Br2N4O4), found 470.9129. LC-MS: [M + H]+ m/z 471 and 473 (tR = 3.14 and 3.11 min), >98%. Mp: 199–202 °C.
General procedure A was followed to give the compound 39 as an off-white solid (0.025 g, 14%). 1H NMR (400 MHz, d6-DMSO) δ 10.73 (br s, 2H), 9.06–9.01 (m, 1H), 8.78 (dd, J = 4.9, 1.7 Hz, 1H), 8.23 (ddd, J = 8.0, 2.3, 1.7 Hz, 1H), 7.88 (s, 2H), 7.62–7.53 (m, 1H), 3.64 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 164.1, 157.7, 153.5, 152.6, 148.4, 146.6, 135.2, 128.0, 123.7, 121.77, 85.2, 43.0. HRMS (ESI+): calculated 470.9128 (C15H11Br2N4O4), found 470.9127. LC-MS: [M + H]+ m/z 471 and 473 (tR = 2.48 and 2.37 min), >99%. Mp: 163–166 °C.
General procedure A was followed to give the compound 40 as an off-white solid (0.021 g, 18%). 1H NMR (400 MHz, CDCl3) δ 8.25 (br s, 1H), 7.74–7.68 (m, 2H), 7.40–7.33 (m, 2H), 6.91–6.87 (m, 2H), 6.67 (s, 1H), 6.36–6.31 (m, 2H), 3.86 (s, 3H), 3.48 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.0, 156.2, 152.9, 149.2, 143.1, 134.9, 133.7, 131.5, 129.6, 128.8, 126.6, 96.9, 83.8, 42.8, 35.8. HRMS (ESI+): calculated 383.0911 (C19H16ClN4O3), found 383.0911. LC-MS: [M + H]+ m/z 383 and 385 (tR = 4.21 and 4.14min), >99%.
7,9-Dibromo-N-[3-(4-chlorophenyl)-1-methyl-1H-pyrazol-5-yl]-8-oxo-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (41)
General procedure A was followed to give the compound 41 as an off-white solid (0.075 g, 35%). 1H NMR (400 MHz, d6-DMSO) δ 10.74 (br s, 1H), 7.88 (s, 2H), 7.85–7.76 (m, 2H), 7.50–7.42 (m, 2H), 3.74 (s, 3H), 3.65 (s, 2H). 13C NMR (101 MHz, d6-DMSO) δ 171.6, 157.6, 154.6, 147.1, 146.4, 136.4, 132.0, 132.0, 128.7, 126.5, 121.9, 98.3, 85.9, 42.8, 36.0. Mp: 251–254 °C. HRMS (ESI+): calculated 538.9121 (C19H14Br2ClN4O3), found 538.9120. LC-MS: [M + H]+ m/z 541 (tR = 5.29 min), >99%.
7,9-Dibromo-8-oxo-N-(4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)-1-oxa-2-azaspiro[4.5]deca-2,6,9-triene-3-carboxamide (42)
General procedure A was followed to give the compound 42 as a pale yellow solid (0.085 g, 40%). 1H NMR (400 MHz, d6-acetone) δ 10.92 (br s, 1H), 7.78 (s, 2H), 3.79 (s, 2H), 2.72–2.69 (m, 2H), 2.62–2.59 (m, 2H), 1.84 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, d6-acetone) δ 172.29, 157.75, 155.14, 155.10, 146.75, 145.21, 123.69, 123.34, 87.35, 43.40, 29.84, 26.92, 24.02, 23.67, 23.33. HRMS (ESI+): calculated 487.9102 (C16H14Br2N3O3S), found 487.9109. LC-MS: [M + H]+ m/z 488 and 490 (tR = 4.92 and 5.03 min), >99%. Mp: 185–190 °C.


 ,
, 











































{kind=link}
{kind=link}
{kind=link}
{kind=link}

































