
Tedaniophorbasins A and B—Novel Fluorescent Pteridine Alkaloids Incorporating a Thiomorpholine from the Sponge Tedaniophorbas ceratosis

 , , and
, , and
Abstract
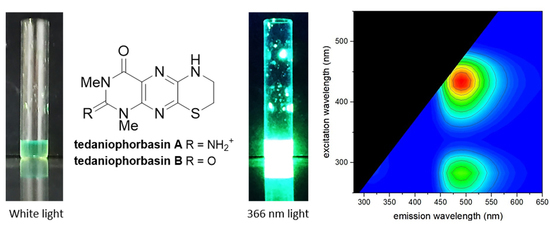
1. Introduction
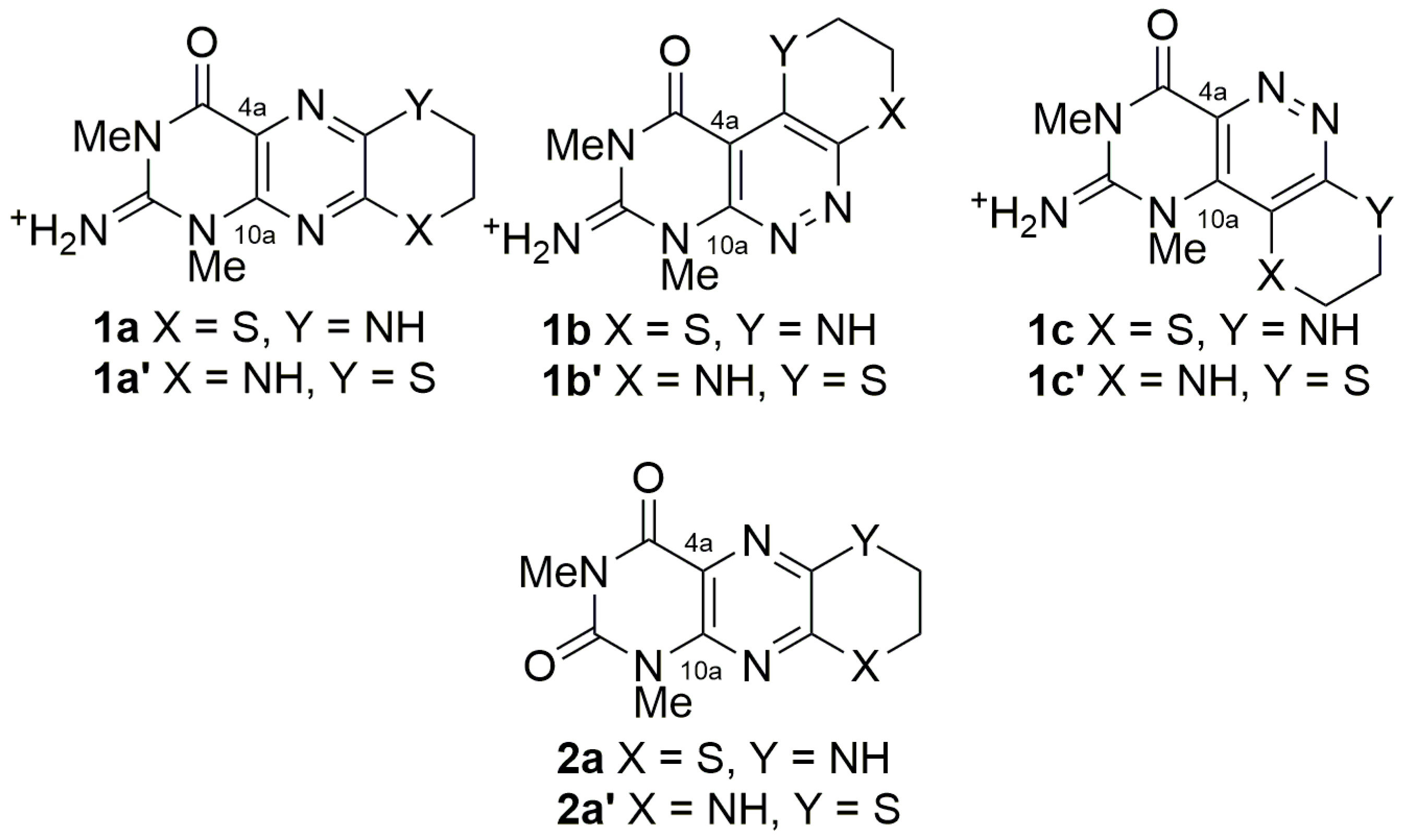
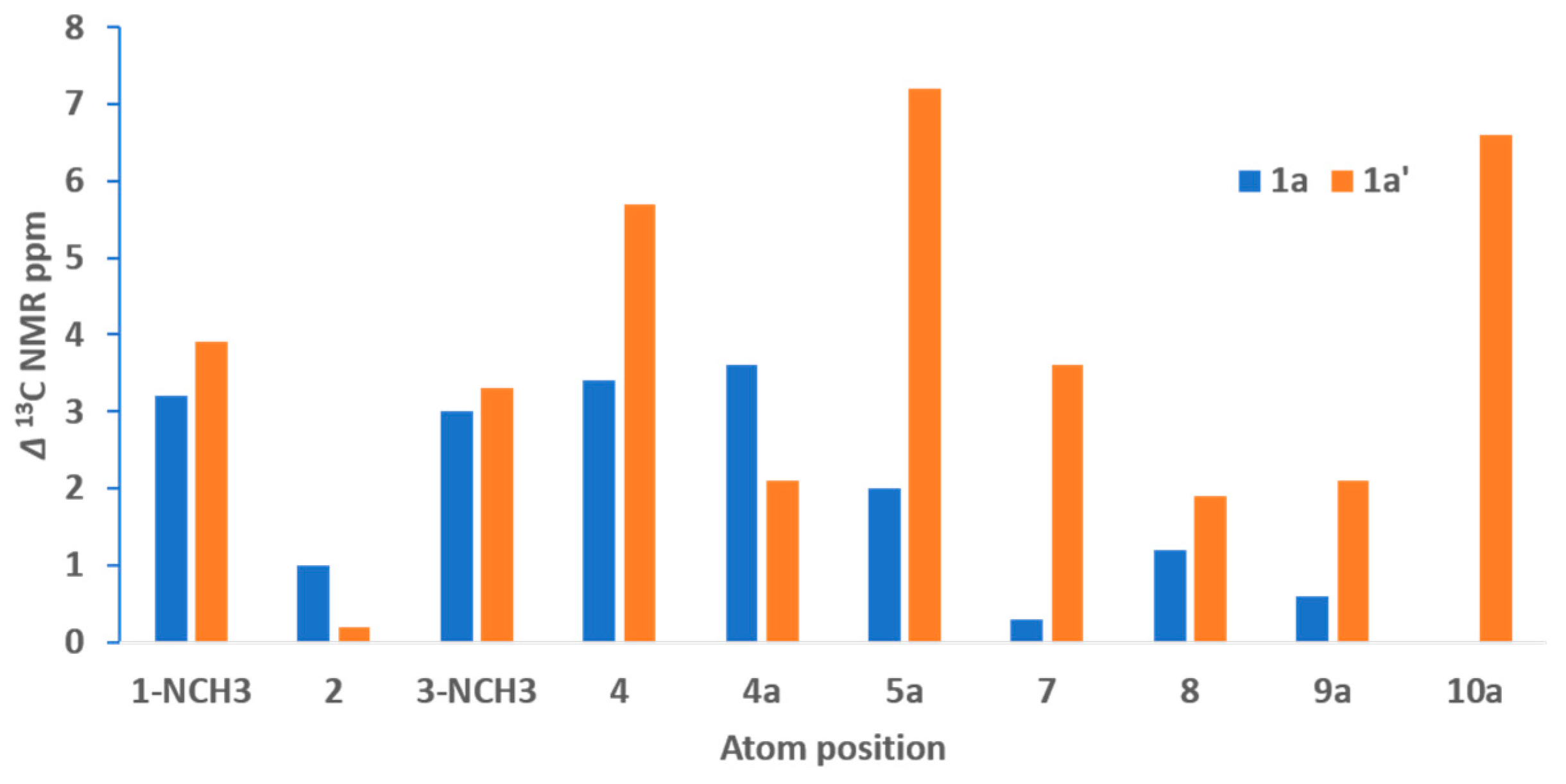
2. Results and Discussion
3. Materials and Methods
3.1. General Chemistry Experimental Procedures
3.2. Collections, Extraction, and Isolation
3.3. DFT 13C NMR Calculations
3.4. Biological Activity Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef]
- Pye, C.R.; Bertinb, M.J.; Lokeya, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef]
- Henkel, T.; Brunne, R.M.; Müller, H.; Reichel, F. Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem. Int. Ed. 1999, 38, 643–647. [Google Scholar] [CrossRef]
- Butler, A.J.; Rees, T.; Beesley, P.; Bax, N.J. Marine Biodiversity in the Australian Region. PLoS ONE 2010, 5, e11831. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef]
- Carroll, A.R.; Wild, S.J.; Duffy, S.; Avery, V.M. Kororamide A, a new tribrominated indole alkaloid from the Australian bryozoan Amathia tortuosa. Tetrahedron Lett. 2012, 53, 2873–2875. [Google Scholar] [CrossRef]
- Kleks, G.; Duffy, S.; Lucantoni, L.; Avery, V.M.; Carroll, A.R. Orthoscuticellines A–E, β-Carboline Alkaloids from the Bryozoan Orthoscuticella ventricosa Collected in Australia. J. Nat. Prod. 2020, 83, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K.; Robertson, L.P.; Rudolph, K.E.; Munn, A.L.; Carroll, A.R. Anti-prion Butenolides and Diphenylpropanones from the Australian Ascidian Polycarpa procera. J. Nat. Prod. 2019, 82, 2620–2626. [Google Scholar] [CrossRef]
- Kleks, G.; Holland, D.C.; Kennedy, E.K.; Avery, V.M.; Carroll, A.R. Antiplasmodial Alkaloids from the Australian Bryozoan Amathia lamourouxi. J. Nat. Prod. 2020, 83, 3435–3444. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Prebble, D.W.; Xu, M.; Ekins, M.G.; Munn, A.L.; Mellick, G.D.; Carroll, A.R. Anti-prion and α-Synuclein Aggregation Inhibitory Sterols from the Sponge Lamellodysidea cf. chlorea. J. Nat. Prod. 2020, 83, 3751–3757. [Google Scholar] [CrossRef]
- Hayton, J.H.; Grant, G.D.; Carroll, A.R. Three New Spongian Diterpenes from the Marine Sponge Dendrilla rosea. Aust. J. Chem. 2019, 72, 964–968. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Tsukamoto, S.; Hirota, H.; Kato, H.; Fusetani, N. Urochordamines A and B: Larval Settlement/Metamorphosis-Promoting, Pteridine-Containing Physostigmine Alkaloids from the Tunicate Ciona savignyi. Tetrahedron Lett. 1993, 34, 4819–4822. [Google Scholar] [CrossRef]
- Murayama, S.; Nakao, Y.; Matsunaga, S. Asteropterin, an inhibitor of cathepsin B, from the marine sponge Asteropus simplex. Tetrahedron Lett. 2008, 49, 4186–4188. [Google Scholar] [CrossRef]
- Kutateladze, A.G.; Reddy, D.S. High-Throughput in Silico Structure Validation and Revision of Halogenated Natural Products Is Enabled by Parametric Corrections to DFT-Computed 13C NMR Chemical Shifts and Spin-Spin Coupling Constants. J. Org. Chem. 2017, 82, 3368–3381. [Google Scholar] [CrossRef]
- Abou-Hadeed, K.; Pfleiderer, W. Pteridines eVIII Reactions of 6, 7-Dichloro-I, 3-dimethyllumazine with Sulfur-Nucleophiles. Pteridines 1996, 7, 113–122. [Google Scholar] [CrossRef]
- Kakoi, H.; Tanino, H.; Okada, K.; Inoue, S. 6-Acyllumazines from the marine polychaete, Odontosyllis undecimdonta. Heterocycles 1995, 41, 789–797. [Google Scholar] [CrossRef]
- Rudolph, K.E.; Liberio, M.S.; Davis, R.A.; Carroll, A.R. Pteridine-, thymidine-, choline-and imidazole-derived alkaloids from the Australian ascidian, Leptoclinides durus. Org. Biomol. Chem. 2013, 11, 261–270. [Google Scholar] [CrossRef]
- You, M.; Liao, L.; Hong, S.H.; Park, W.; Kwon, D.I.; Lee, J.; Noh, M.; Oh, D.-C.; Oh, K.-B.; Shin, J. Lumazine Peptides from the Marine-Derived Fungus Aspergillus terreus. Mar. Drugs 2015, 13, 1290–1303. [Google Scholar] [CrossRef]
- Duffy, S.; Avery, V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [Google Scholar] [CrossRef]
- Sykes, M.L.; Avery, V.M. Development of an Alamar Blue™ Viability Assay in 384-Well Format for High Throughput Whole Cell Screening of Trypanosoma brucei brucei Bloodstream Form Strain 427. Am. J. Trop. Med. Hyg. 2009, 81, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Miniaturized three-dimensional cancer model for drug evaluation. Assay Drug Dev. Technol. 2013, 11, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A Guide to Small-Molecule Structure Assignment through Computation of (1H and 13C) NMR Chemical Shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds Using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| Position | δc, Type | δH, Mult. (J in Hz) | δc, Type | δH, Mult. (J in Hz) |
| 1-NCH3 | 31.3, CH3 | 3.66, s | 28.9, CH3 | 3.43, s |
| 2 | 151.0, C | - | 150.0, C | - |
| 2-NH2+ | - | 9.05, bs | - | - |
| 3-NCH3 | 30.3, CH3 | 3.46, s | 28.2, CH3 | 3.27, s |
| 4 | 157.4, C | - | 159.9, C | - |
| 4a | 120.6, C | - | 123.6, C | - |
| 5a | 148.0 b, C | - | 146.6, C | - |
| 6 | - | 8.15, t (2.7) | - | 7.64, t (2.7) |
| 7 | 40.6, CH2 | 3.65, m | 40.7, CH2 | 3.61, m |
| 8 | 26.3, CH2 | 3.34, m | 26.5, CH2 | 3.29, m |
| 9a | 147.9 b, C | - | 146.0, C | - |
| 10a | 137.6, C | - | 139.8, C | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiranrat, A.; Holland, D.C.; Mahabusarakam, W.; Hooper, J.N.A.; Avery, V.M.; Carroll, A.R. Tedaniophorbasins A and B—Novel Fluorescent Pteridine Alkaloids Incorporating a Thiomorpholine from the Sponge Tedaniophorbas ceratosis. Mar. Drugs 2021, 19, 95. https://doi.org/10.3390/md19020095
Hiranrat A, Holland DC, Mahabusarakam W, Hooper JNA, Avery VM, Carroll AR. Tedaniophorbasins A and B—Novel Fluorescent Pteridine Alkaloids Incorporating a Thiomorpholine from the Sponge Tedaniophorbas ceratosis. Marine Drugs. 2021; 19(2):95. https://doi.org/10.3390/md19020095
Chicago/Turabian StyleHiranrat, Asadhawut, Darren C. Holland, Wilawan Mahabusarakam, John N. A. Hooper, Vicky M. Avery, and Anthony R. Carroll. 2021. "Tedaniophorbasins A and B—Novel Fluorescent Pteridine Alkaloids Incorporating a Thiomorpholine from the Sponge Tedaniophorbas ceratosis" Marine Drugs 19, no. 2: 95. https://doi.org/10.3390/md19020095
APA StyleHiranrat, A., Holland, D. C., Mahabusarakam, W., Hooper, J. N. A., Avery, V. M., & Carroll, A. R. (2021). Tedaniophorbasins A and B—Novel Fluorescent Pteridine Alkaloids Incorporating a Thiomorpholine from the Sponge Tedaniophorbas ceratosis. Marine Drugs, 19(2), 95. https://doi.org/10.3390/md19020095