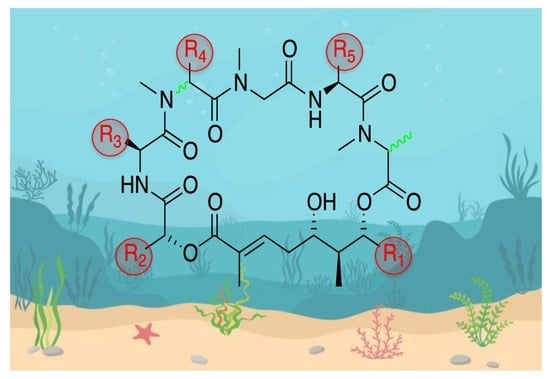
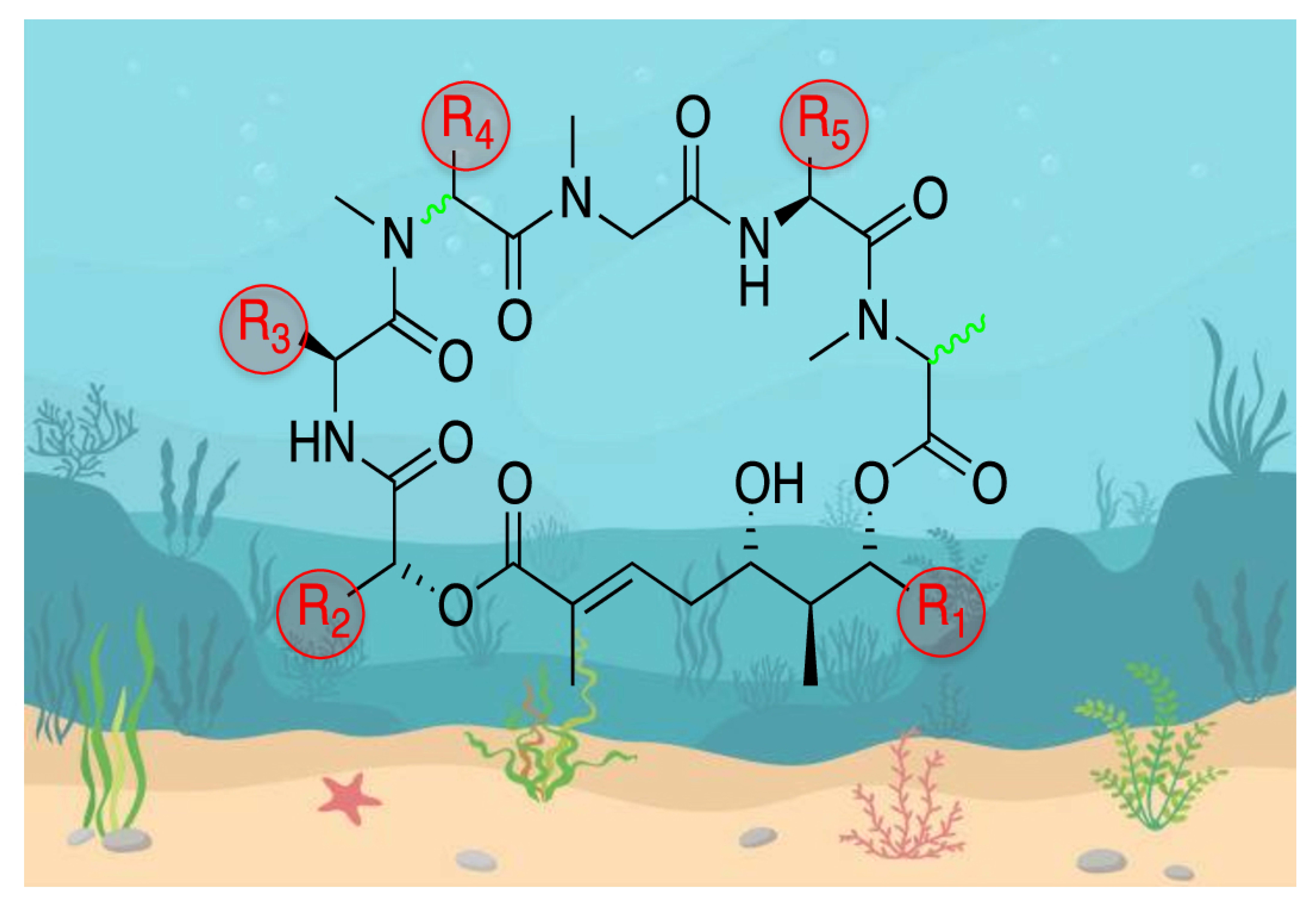
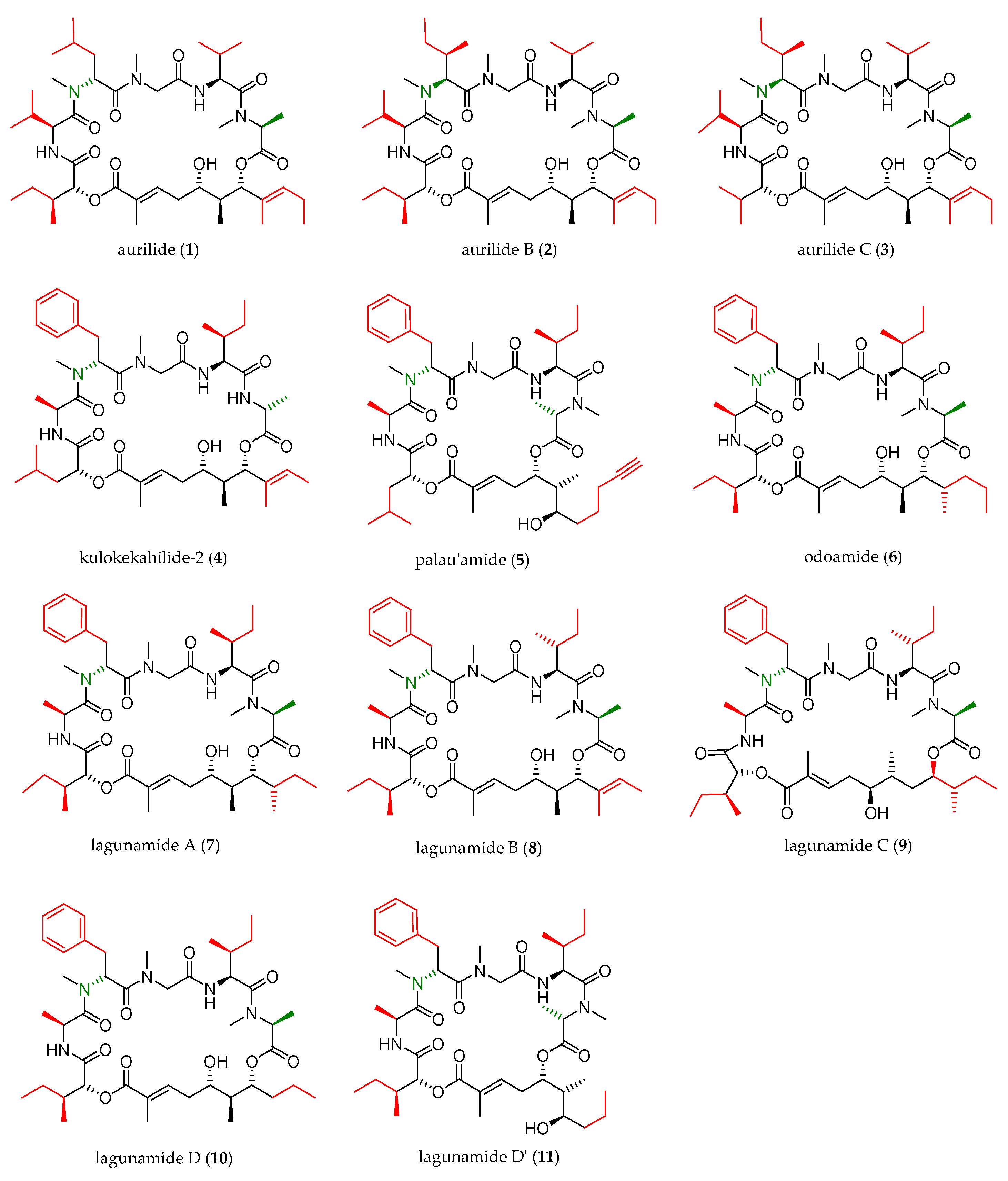
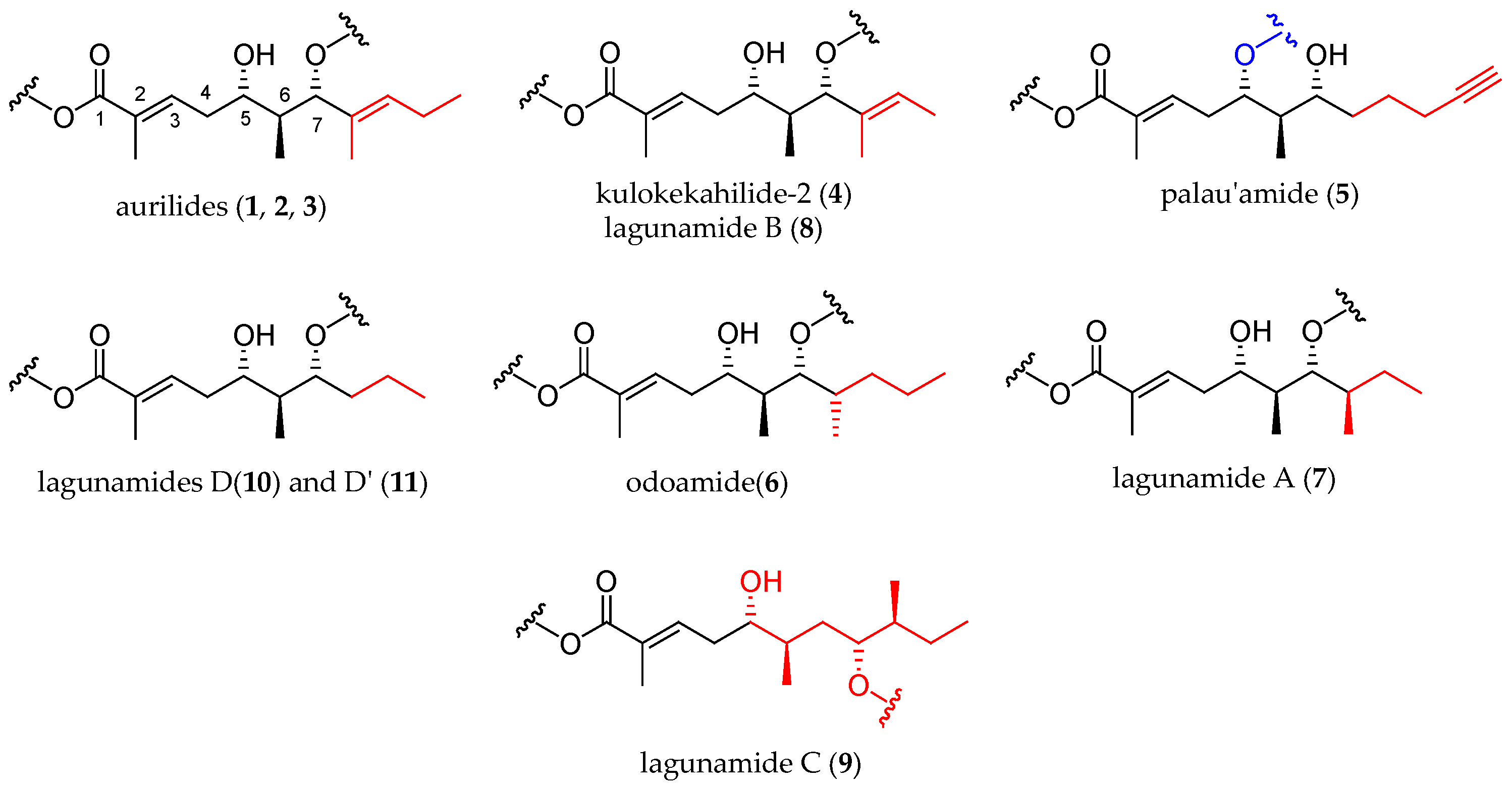
Figure 1.
Aurilide family members.
Figure 1.
Aurilide family members.
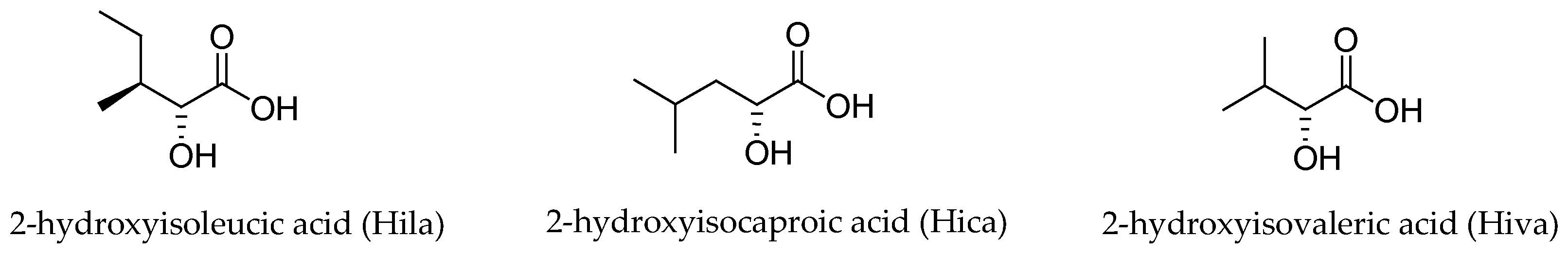
Figure 2.
The three α-hydroxy acids present in the structure of the aurilide family members.
Figure 2.
The three α-hydroxy acids present in the structure of the aurilide family members.
Figure 3.
Polyketide moieties of the aurilide family members.
Figure 3.
Polyketide moieties of the aurilide family members.
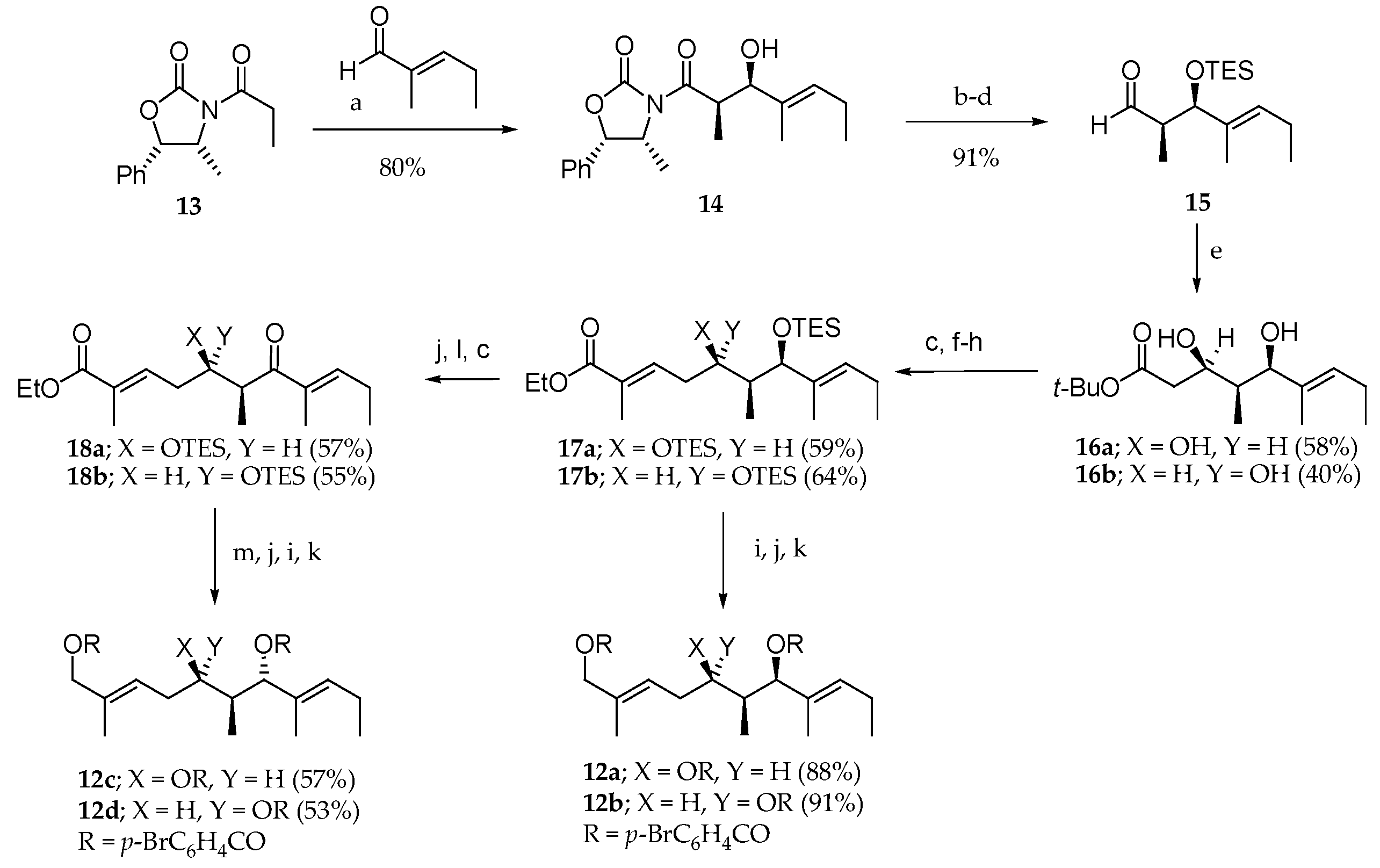
Scheme 1.
Synthesis of triols 12a–d. Reagents and conditions: (a) Bu2OTf, Et3N, CH2Cl2, −78 °C; (b) Me2AlN(Me)OMe, THF, toluene, 0 °C; (d) DiBAL-H, THF, hexane, −78 °C, 91% from 14; (e) LiCH2CO2tBu, THF, −78 °C; (f) DiBAL-H, CH2Cl2, hexane −23 °C; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to 0 °C; (h) (EtO)2P(O)CH(Me)CO2Et, NaH, DME, −20 °C; (i) DiBAL-H, CH2Cl2, hexane, −78 °C; (j) HF∙pyridine, pyridine, THF, 23 °C; (k) p-BrC6H4COCl, pyridine, 23 °C; (l) MnO2, CH2Cl2, 23 °C; (m) NaBH4, CeCl3∙7H2O, EtOH, −23 °C.
Scheme 1.
Synthesis of triols 12a–d. Reagents and conditions: (a) Bu2OTf, Et3N, CH2Cl2, −78 °C; (b) Me2AlN(Me)OMe, THF, toluene, 0 °C; (d) DiBAL-H, THF, hexane, −78 °C, 91% from 14; (e) LiCH2CO2tBu, THF, −78 °C; (f) DiBAL-H, CH2Cl2, hexane −23 °C; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to 0 °C; (h) (EtO)2P(O)CH(Me)CO2Et, NaH, DME, −20 °C; (i) DiBAL-H, CH2Cl2, hexane, −78 °C; (j) HF∙pyridine, pyridine, THF, 23 °C; (k) p-BrC6H4COCl, pyridine, 23 °C; (l) MnO2, CH2Cl2, 23 °C; (m) NaBH4, CeCl3∙7H2O, EtOH, −23 °C.
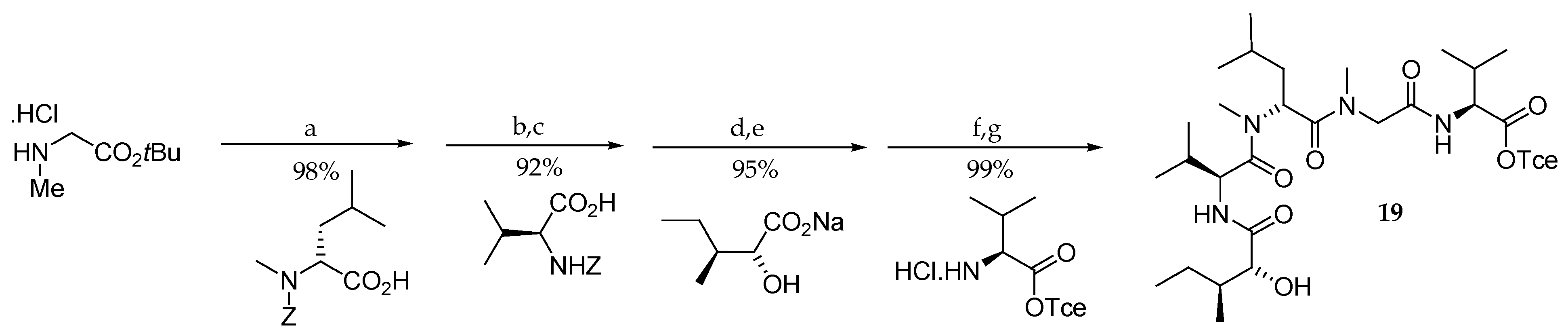
Scheme 2.
Synthesis of pentapeptide subunit 19. Reagents and conditions: (a) DEPC, Et3N, DMF, 23 °C; (b) H2, Pd/C, EtOH, 23 °C; (c) PyBOP, DIPEA, CH2Cl2; (d) H2, Pd/C, EtOH, 23 °C; (e) EDCI∙HCl, HOBt, DMF, 23 °C; (f) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (g) EDCI∙HCl, HOBt, Et3N, DMF, CH2Cl2, 23 °C. DEPC = diethylphosphoryl cyanide; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride. PyBOP = benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate; DIPEA = N,N-diisopropylethylamine; HOBt = 1-hydroxybenzotriazole; TMSOTf = trimethylsilyl trifluoromethanesulfonate; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride.
Scheme 2.
Synthesis of pentapeptide subunit 19. Reagents and conditions: (a) DEPC, Et3N, DMF, 23 °C; (b) H2, Pd/C, EtOH, 23 °C; (c) PyBOP, DIPEA, CH2Cl2; (d) H2, Pd/C, EtOH, 23 °C; (e) EDCI∙HCl, HOBt, DMF, 23 °C; (f) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (g) EDCI∙HCl, HOBt, Et3N, DMF, CH2Cl2, 23 °C. DEPC = diethylphosphoryl cyanide; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride. PyBOP = benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate; DIPEA = N,N-diisopropylethylamine; HOBt = 1-hydroxybenzotriazole; TMSOTf = trimethylsilyl trifluoromethanesulfonate; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride.
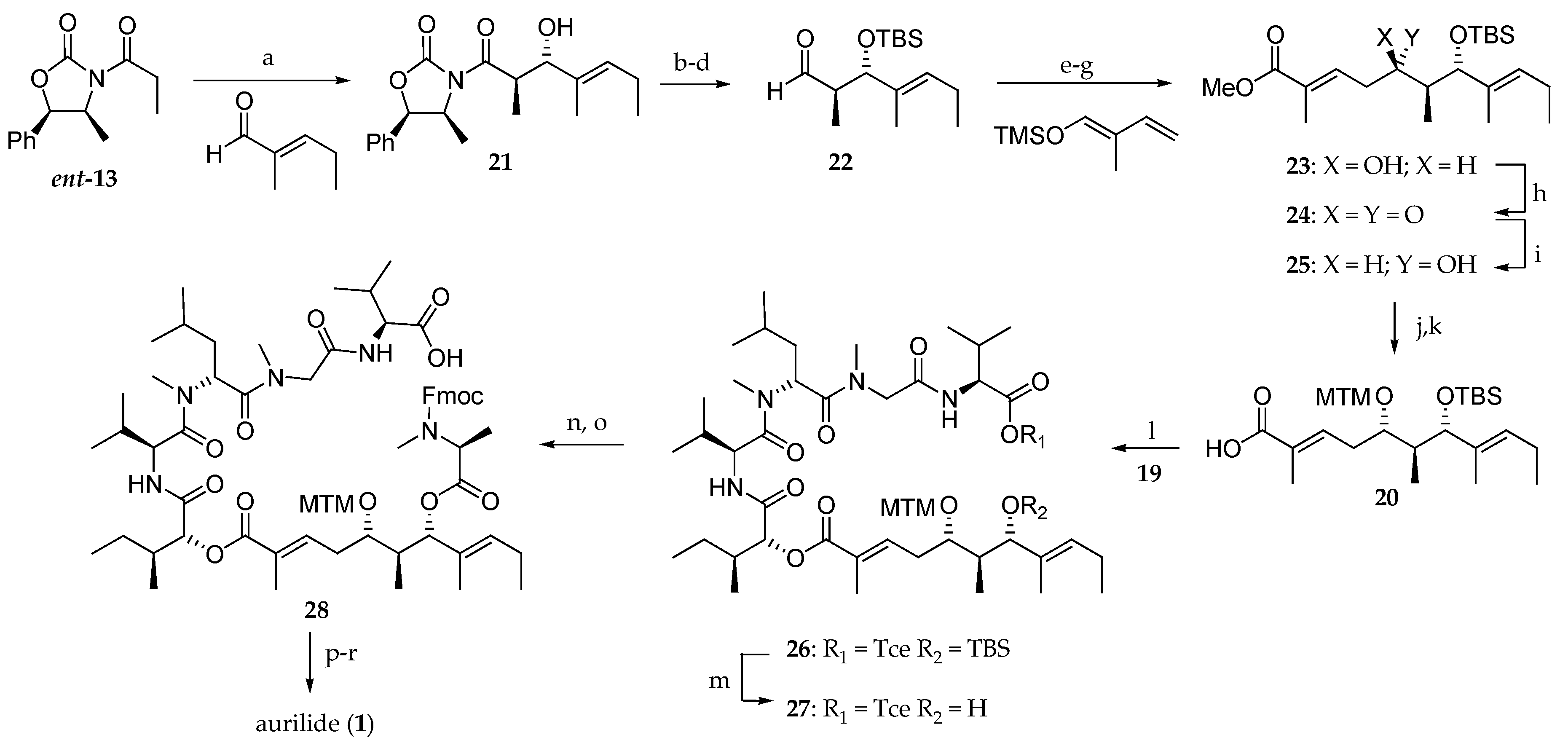
Scheme 3.
First total synthesis of aurilide 1. Reagents and conditions: (a) Bu2BOTf, DIEA, Et2O, −100 to −78 °C, 67%; (b) MeNH(OMe)∙HCl, Me3Al, THF, 50 °C, 84%; (c) TBSCl, imidazole, DMF, 23 °C, 100%; (d) DiBAL-H, THF, −78 °C, 93%; (e) BF3∙OEt2, CH2Cl2, Et2O, −78 °C, 59%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, 23 °C; (g) CH2N2, Et2O, 0 °C, 100% (2 steps); (h) Dess–Martin periodinane, CH2Cl2, 23 °C, 99%; (i) NaBH4, MeOH, −23 °C, 82%; (j) DMSO, Ac2O, AcOH, 40 °C, 74%; (k) LiOH, MeOH, H2O, 30 °C, 92%; (l) EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 91%; (m) HF∙pyridine, pyridine THF, 40 °C, 100%; (n) Fmoc-N-Me-l-Ala-OH, EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 92%; (o) Zn, NH4OAc, THF, H2O, 23 °C, 91%; (p) Et2NH, MeCN, 23 °C; (q) Bop-Cl, Et3N, CH2Cl2, 23 °C, 32% (2 steps); (r) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 93%.
Scheme 3.
First total synthesis of aurilide 1. Reagents and conditions: (a) Bu2BOTf, DIEA, Et2O, −100 to −78 °C, 67%; (b) MeNH(OMe)∙HCl, Me3Al, THF, 50 °C, 84%; (c) TBSCl, imidazole, DMF, 23 °C, 100%; (d) DiBAL-H, THF, −78 °C, 93%; (e) BF3∙OEt2, CH2Cl2, Et2O, −78 °C, 59%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, 23 °C; (g) CH2N2, Et2O, 0 °C, 100% (2 steps); (h) Dess–Martin periodinane, CH2Cl2, 23 °C, 99%; (i) NaBH4, MeOH, −23 °C, 82%; (j) DMSO, Ac2O, AcOH, 40 °C, 74%; (k) LiOH, MeOH, H2O, 30 °C, 92%; (l) EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 91%; (m) HF∙pyridine, pyridine THF, 40 °C, 100%; (n) Fmoc-N-Me-l-Ala-OH, EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 92%; (o) Zn, NH4OAc, THF, H2O, 23 °C, 91%; (p) Et2NH, MeCN, 23 °C; (q) Bop-Cl, Et3N, CH2Cl2, 23 °C, 32% (2 steps); (r) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 93%.
Scheme 4.
Synthesis of the tetrapeptides library. Reaction conditions: (a) (i) 50% AcCl, CH2Cl2, r.t., 12 h, (ii) Fmoc-AAx-OH (0.1 M), DIEA (0.25 M), CH2Cl2, r.t., 12 h. This protocol was repeated twice; (b) 20% Piperidine, DMF, r.t., 30 min; (c) Fmoc-AAx-OH (0.1 M), coupling reagent (method A, Fmoc-AAx-OH, DIC, HOBt (1:1:1.2); method B, Fmoc-AAx-OH, HBTU, HOBt, DIEA (1:1:1:2); method C, Fmoc-AAx-OH, TFFH, DIEA (1:1:2); method D, Fmoc-AAx-OH, PyBrOP, DIEA (1:1:2), DMF, r.t., 22 h; (d) 1% TFA, CH2Cl2, r.t., 1 h.DIC = N,N-diisopropylcarbodiimide; HBTU = N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate; TFFH = tetramethylfluoroformamidinium hexafluorophosphate; PyBrOP = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate; TFA = trifluoroacetic acid.
Scheme 4.
Synthesis of the tetrapeptides library. Reaction conditions: (a) (i) 50% AcCl, CH2Cl2, r.t., 12 h, (ii) Fmoc-AAx-OH (0.1 M), DIEA (0.25 M), CH2Cl2, r.t., 12 h. This protocol was repeated twice; (b) 20% Piperidine, DMF, r.t., 30 min; (c) Fmoc-AAx-OH (0.1 M), coupling reagent (method A, Fmoc-AAx-OH, DIC, HOBt (1:1:1.2); method B, Fmoc-AAx-OH, HBTU, HOBt, DIEA (1:1:1:2); method C, Fmoc-AAx-OH, TFFH, DIEA (1:1:2); method D, Fmoc-AAx-OH, PyBrOP, DIEA (1:1:2), DMF, r.t., 22 h; (d) 1% TFA, CH2Cl2, r.t., 1 h.DIC = N,N-diisopropylcarbodiimide; HBTU = N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate; TFFH = tetramethylfluoroformamidinium hexafluorophosphate; PyBrOP = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate; TFA = trifluoroacetic acid.
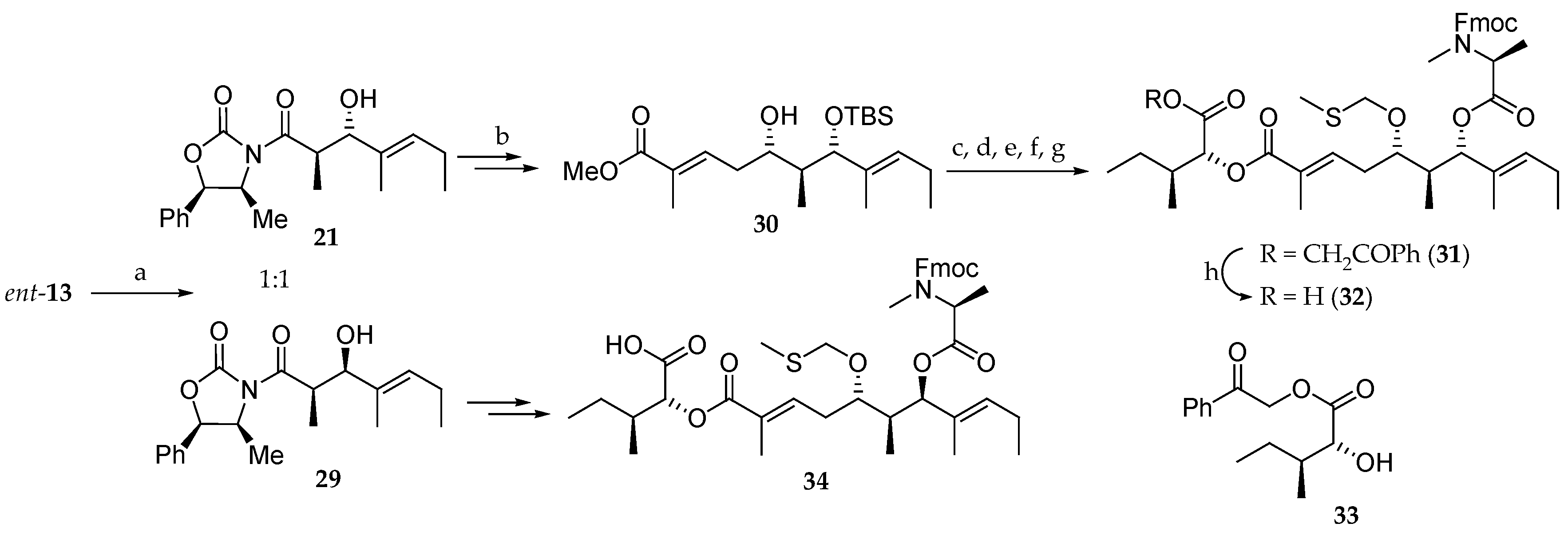
Scheme 5.
Synthesis of subunits
32 and
34. Reagents and conditions: (
a) (
E)-2-methyl-2-pentenal, Bu
2BOTf, DIEA, −100 °C in ether (
21 and
29 (1:1), 50%) and −78 °C in CH
2Cl
2; (
b) [
11,
12] (
c) Ac
2O, AcOH, DMSO, r.t., 27 h, 87%; (
d) LiOH, MeOH, H
2O, r.t., 46 h, 94%; (
e)
33, EDCI, DMAP, CH
2Cl
2, r.t., 96 h, 33%; (
f) HF∙pyridine, pyridine, THF, r.t., 3 h, 60%; (
g) Fmoc-
N-Me-Ala-OH, EDCI, DMAP, CH
2Cl
2, r.t., 20 h, 90%; (
h) Zn, AcOH, AcOEt, H
2O, 45 °C, 27 h, quant.
Scheme 5.
Synthesis of subunits
32 and
34. Reagents and conditions: (
a) (
E)-2-methyl-2-pentenal, Bu
2BOTf, DIEA, −100 °C in ether (
21 and
29 (1:1), 50%) and −78 °C in CH
2Cl
2; (
b) [
11,
12] (
c) Ac
2O, AcOH, DMSO, r.t., 27 h, 87%; (
d) LiOH, MeOH, H
2O, r.t., 46 h, 94%; (
e)
33, EDCI, DMAP, CH
2Cl
2, r.t., 96 h, 33%; (
f) HF∙pyridine, pyridine, THF, r.t., 3 h, 60%; (
g) Fmoc-
N-Me-Ala-OH, EDCI, DMAP, CH
2Cl
2, r.t., 20 h, 90%; (
h) Zn, AcOH, AcOEt, H
2O, 45 °C, 27 h, quant.
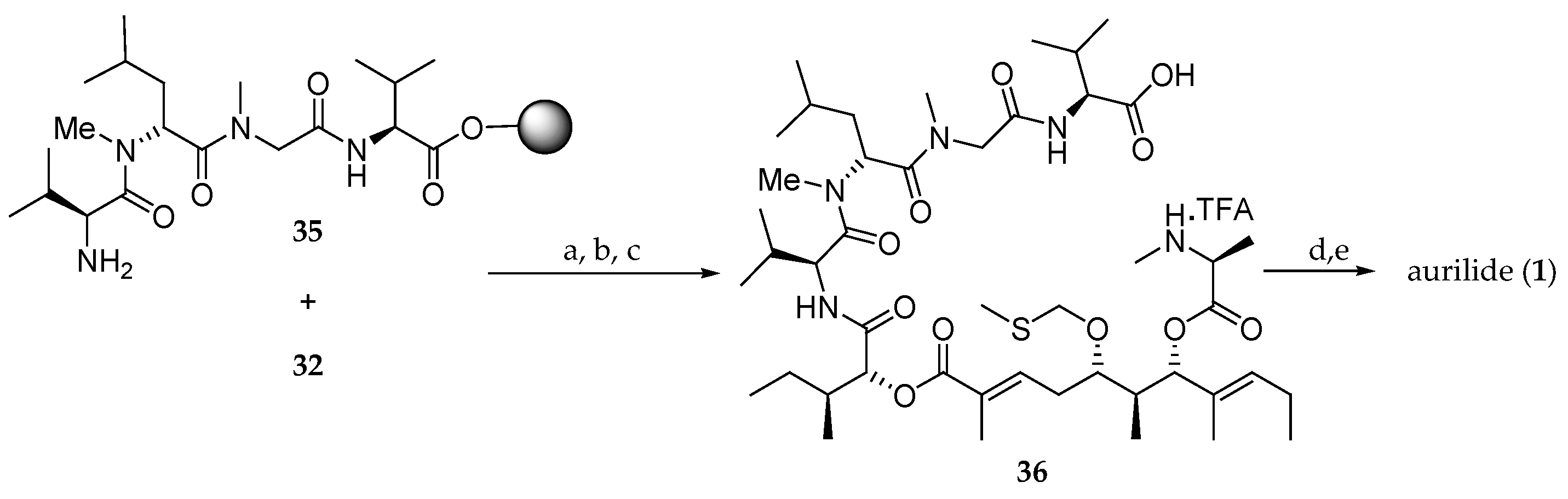
Scheme 6.
Completion of the synthesis of aurilide (1). Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
Scheme 6.
Completion of the synthesis of aurilide (1). Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
Scheme 7.
Synthesis of aurilide analogues 39. Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
Scheme 7.
Synthesis of aurilide analogues 39. Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
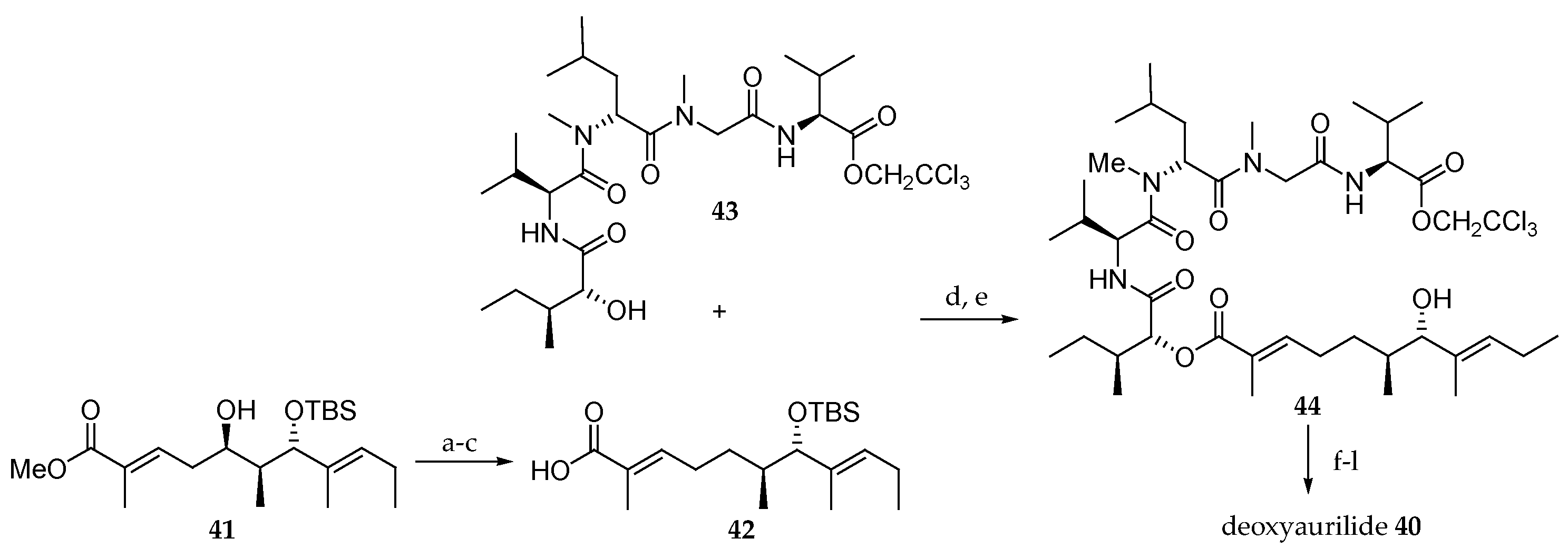
Scheme 8.
Synthesis of deoxyaurilide 40. Reagents and conditions: (a) o-NO2C6H4SeCN, PBu3, THF, r.t., 83%; (b) H2, Lindlar’s cat., quinoline, EtOAc, r.t., 62%; (c) 5M aq LiOH, MeOH, r.t., 95%; (d) EDCI∙HCl, DMAP, CH2Cl2; (e) HF∙pyridine, pyridine, r.t., 36% in 2 steps from 43; (f) Fmoc-N-Me-l-Ala, EDCl∙HCl, DMAP, CH2Cl2, r.t., 51%; (g) Zn, aq NH4OAc, r.t.; (h) Et2NH, CH3CN, r.t.; (i) EDCl∙HCl, HOAt, CH2Cl2, DMF, r.t., 25% (3 steps).
Scheme 8.
Synthesis of deoxyaurilide 40. Reagents and conditions: (a) o-NO2C6H4SeCN, PBu3, THF, r.t., 83%; (b) H2, Lindlar’s cat., quinoline, EtOAc, r.t., 62%; (c) 5M aq LiOH, MeOH, r.t., 95%; (d) EDCI∙HCl, DMAP, CH2Cl2; (e) HF∙pyridine, pyridine, r.t., 36% in 2 steps from 43; (f) Fmoc-N-Me-l-Ala, EDCl∙HCl, DMAP, CH2Cl2, r.t., 51%; (g) Zn, aq NH4OAc, r.t.; (h) Et2NH, CH3CN, r.t.; (i) EDCl∙HCl, HOAt, CH2Cl2, DMF, r.t., 25% (3 steps).
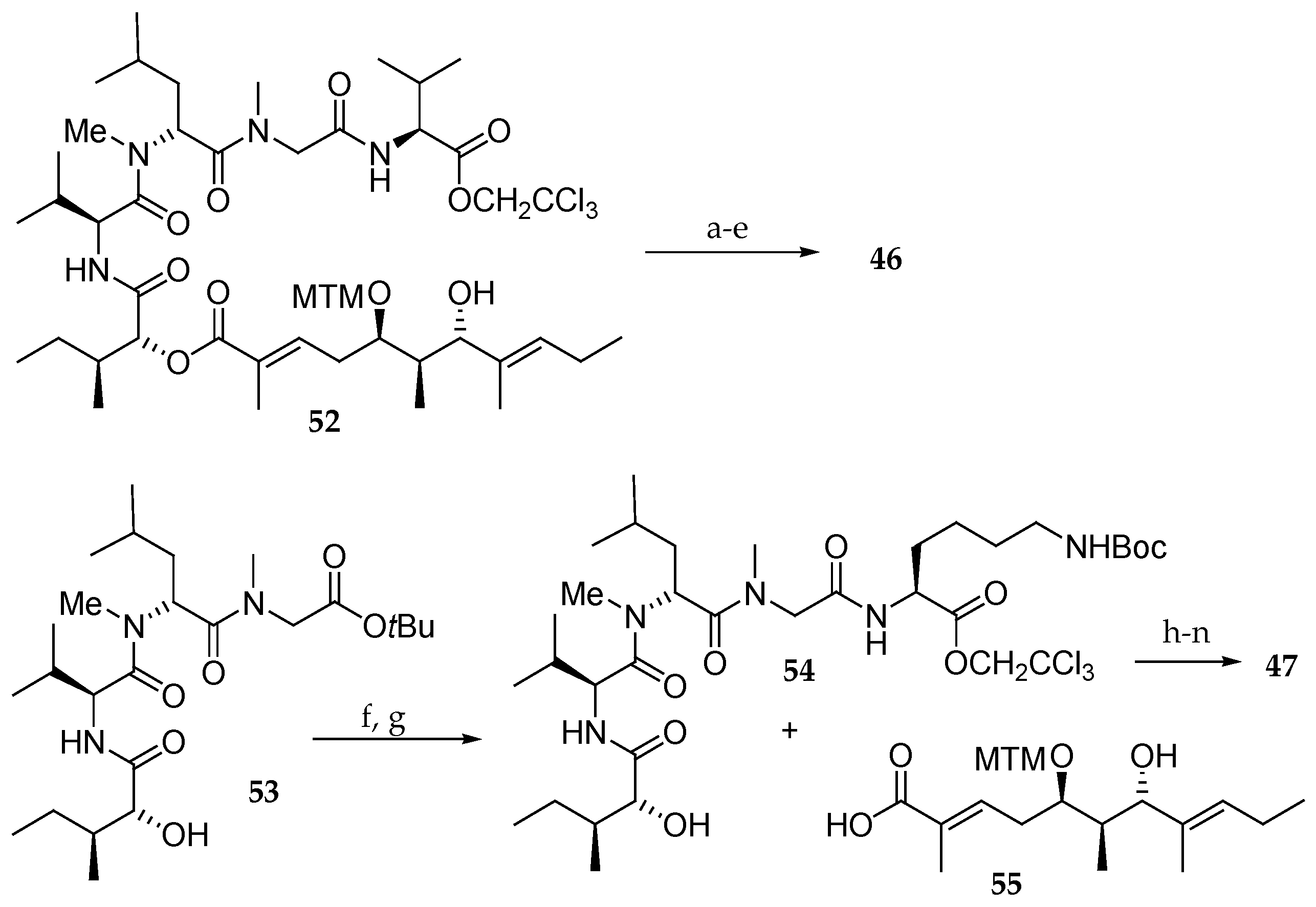
Scheme 9.
Synthesis of aurilide analogues 46 and 47. Reagents and conditions: (a) ε-Boc-Fmoc-N-Me-l-Lys, EDCI∙HCl, DMAP, CH2Cl2, 88%; (b) Zn, NH4OAc aq, r.t.; (c) Et2NH, CH3CN, r.t.; (d) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (e) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 17% in 4 steps; (f) TMSOTf, 2.6-lutidine, 0 °C; (g) ε-Boc-l-Lys-OCH2CCl3, EDCI∙HCl, HOBt, DMF, CH2Cl2, r.t., 54% in 2 steps; (h) EDCI∙HCl, DMAP, CH2Cl2, r.t.; (i) HF∙pyridine, pyridine, r.t., 32% in 2 steps from 55; (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, r.t., 66%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (n) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 70% in 4 steps.
Scheme 9.
Synthesis of aurilide analogues 46 and 47. Reagents and conditions: (a) ε-Boc-Fmoc-N-Me-l-Lys, EDCI∙HCl, DMAP, CH2Cl2, 88%; (b) Zn, NH4OAc aq, r.t.; (c) Et2NH, CH3CN, r.t.; (d) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (e) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 17% in 4 steps; (f) TMSOTf, 2.6-lutidine, 0 °C; (g) ε-Boc-l-Lys-OCH2CCl3, EDCI∙HCl, HOBt, DMF, CH2Cl2, r.t., 54% in 2 steps; (h) EDCI∙HCl, DMAP, CH2Cl2, r.t.; (i) HF∙pyridine, pyridine, r.t., 32% in 2 steps from 55; (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, r.t., 66%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (n) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 70% in 4 steps.
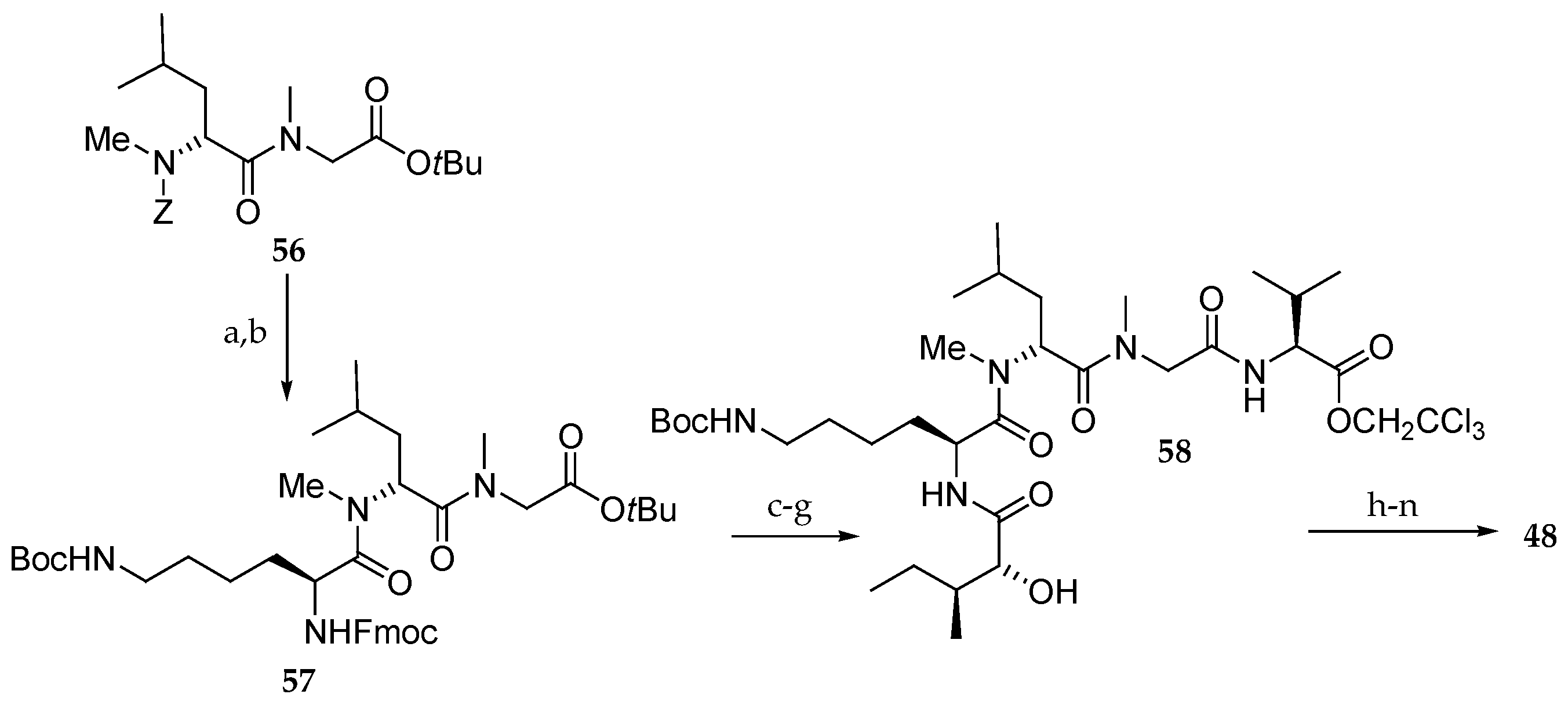
Scheme 10.
Synthesis of aurilide analogue 48. Reagents and conditions: (a) H2, Pd/C, EtOH, r.t.; (b) ε-Boc-α-Fmoc-l-Lys, PyBOP, DIPEA, CH2Cl2, r.t., 100% (2 steps); (c) Et2NH, CH3CN, r.t.; (d) sodium salt of allo-d-isoleucic acid, EDCI∙HCl, HOBt, DMF, r.t., 98% (2 steps); (e) TMSOTf, 2,6-lutidine, 0 °C; (f) (Boc)2O, 1 M aq NaOH, r.t.; (g) l-Val-OCH2CCl3, HOBt, Et3N, DMF, CH2Cl2, r.t., 76% (3 steps); (h) 55, EDCI∙HCl, DMAP, CH2Cl2, r.t., 74%; (i) HF∙pyridine, pyridine, 40 °C, 78%, (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, 0 °C, 94%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t., 73% (3 steps); (n) AgNO3, 2,6-lutidine, THF, H2O, 65 °C, 85%.
Scheme 10.
Synthesis of aurilide analogue 48. Reagents and conditions: (a) H2, Pd/C, EtOH, r.t.; (b) ε-Boc-α-Fmoc-l-Lys, PyBOP, DIPEA, CH2Cl2, r.t., 100% (2 steps); (c) Et2NH, CH3CN, r.t.; (d) sodium salt of allo-d-isoleucic acid, EDCI∙HCl, HOBt, DMF, r.t., 98% (2 steps); (e) TMSOTf, 2,6-lutidine, 0 °C; (f) (Boc)2O, 1 M aq NaOH, r.t.; (g) l-Val-OCH2CCl3, HOBt, Et3N, DMF, CH2Cl2, r.t., 76% (3 steps); (h) 55, EDCI∙HCl, DMAP, CH2Cl2, r.t., 74%; (i) HF∙pyridine, pyridine, 40 °C, 78%, (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, 0 °C, 94%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t., 73% (3 steps); (n) AgNO3, 2,6-lutidine, THF, H2O, 65 °C, 85%.
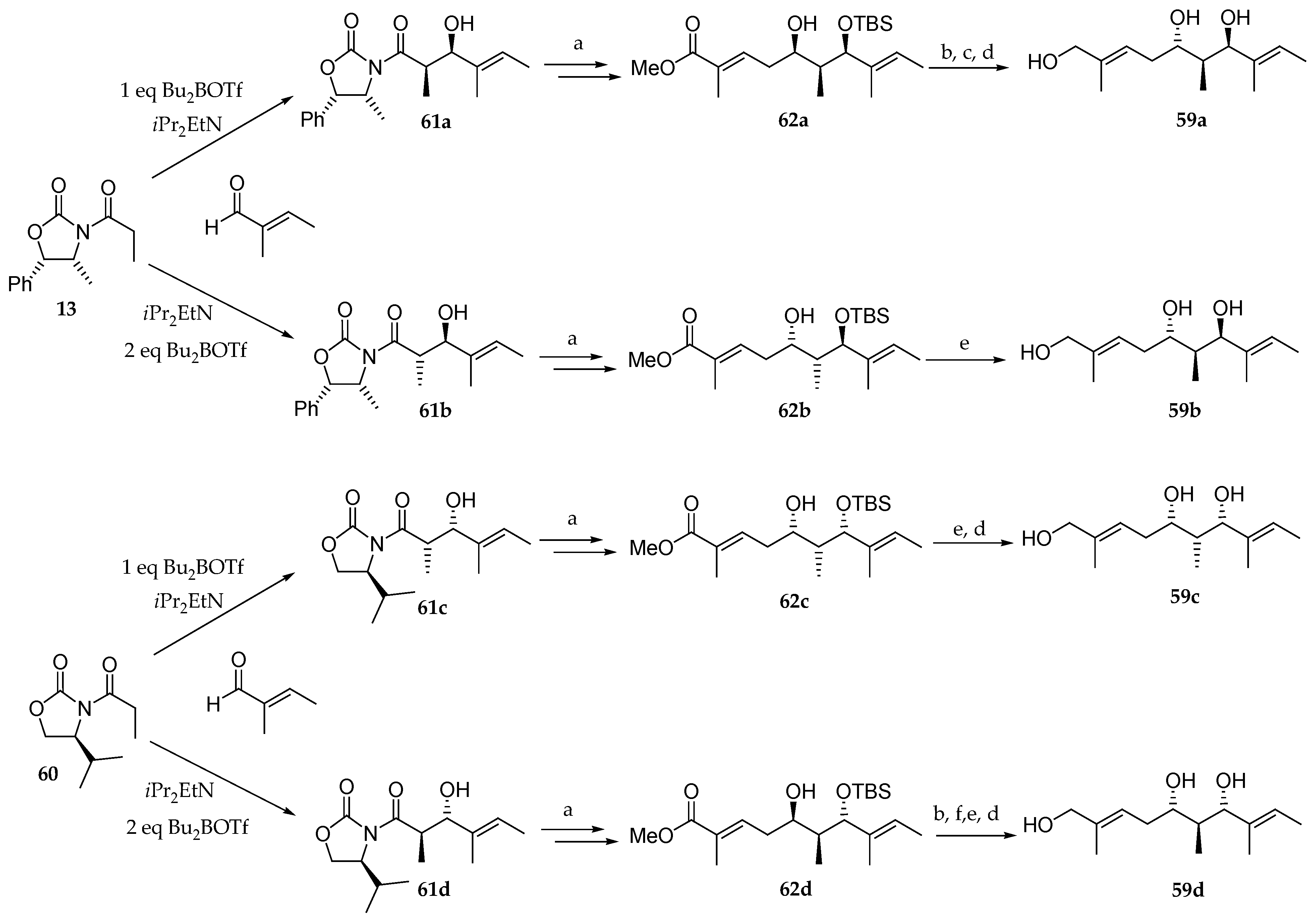
Scheme 11.
Synthesis of triols
59a–
d. Reagents and conditions: (
a) [
11]; (
b) DMSO, DCC; (
c) LiAlH
4; (
d) PPTS, MeOH; (
e) DiBAL-H; (
f) NaBH
4. DCC =
N,
N’-dicyclohexylcarbodiimide; PPTS = pyridinium
p-toluenesulfonate.
Scheme 11.
Synthesis of triols
59a–
d. Reagents and conditions: (
a) [
11]; (
b) DMSO, DCC; (
c) LiAlH
4; (
d) PPTS, MeOH; (
e) DiBAL-H; (
f) NaBH
4. DCC =
N,
N’-dicyclohexylcarbodiimide; PPTS = pyridinium
p-toluenesulfonate.
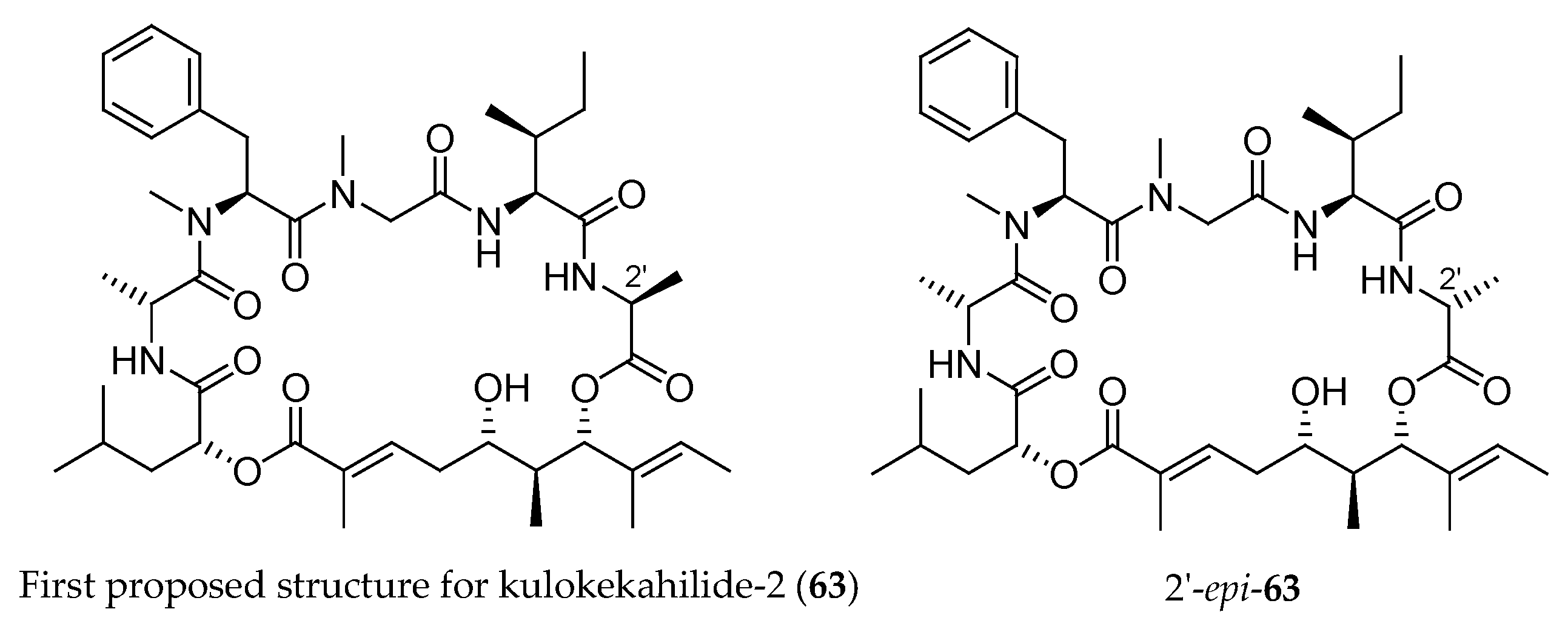
Figure 4.
First structure (63) proposed for kulokekahilide-2 and its epimer at 2′ (2′-epi-63).
Figure 4.
First structure (63) proposed for kulokekahilide-2 and its epimer at 2′ (2′-epi-63).
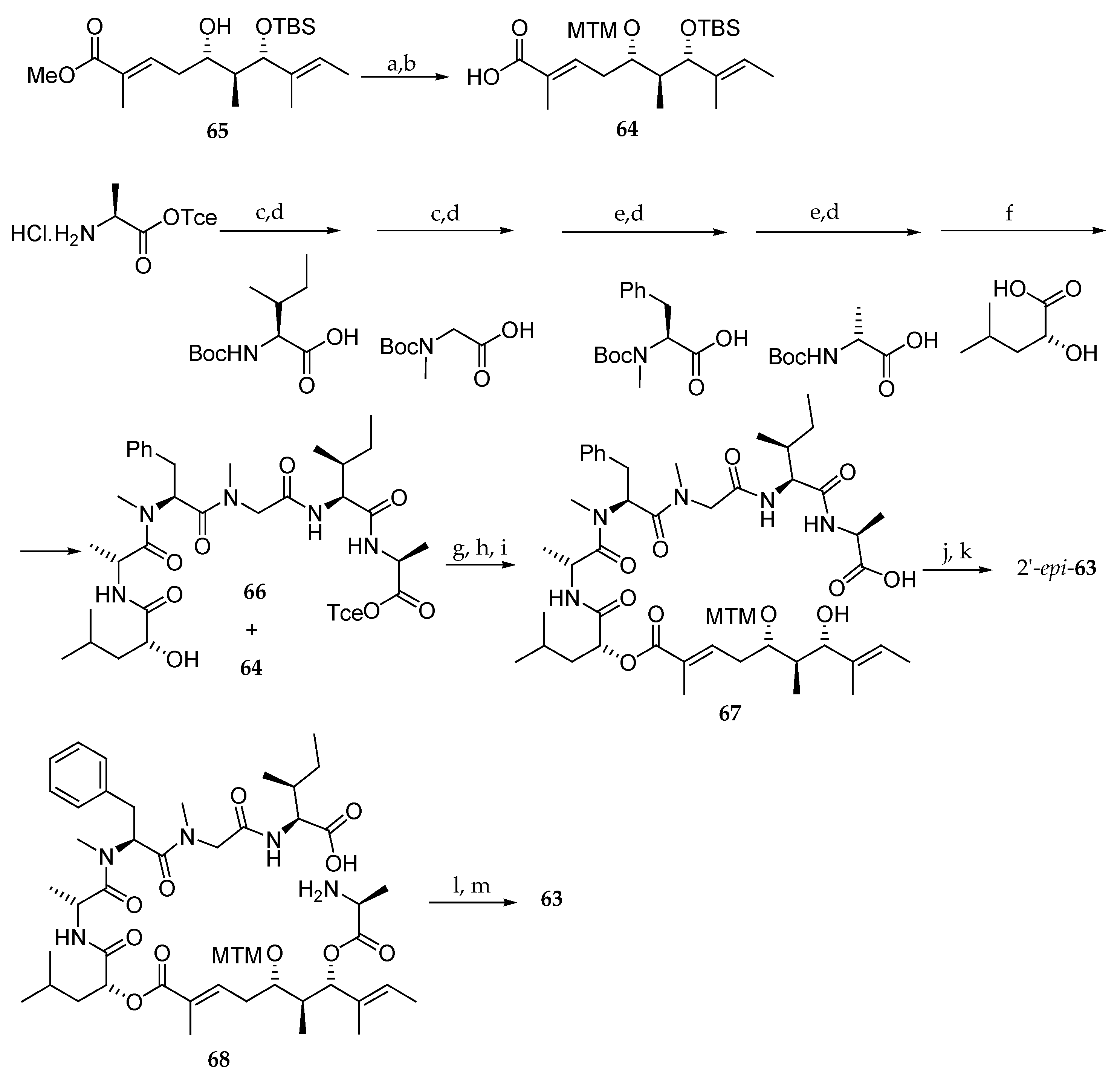
Scheme 12.
Synthesis of 2-epi-63 and 63. Reagents and conditions: (a) Ac2O, AcOH, DMSO, r.t., 84%; (b) LiOH, H2O, MeOH, H2O, r.t., 88%; (c) EDCI∙HCl, HOBt, Et3N, DMF-CH2Cl2 (1:1), r.t.; (d) 4M HCl, dioxane, r.t.; (e) PyBOP, DIPEA, DMF-CH2Cl2 (1:1), r.t.; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t.; (g) EDCI∙HCl, DMAP, CH2Cl2, 45%; (h) HF∙pyridine, pyridine, THF, 97%; (i) Zn powder, AcONH4, 93%; (j) MNBA, DMAP, CH2Cl2, 60%; (k) AgNO3, 2,6-lutidine, THF, H2O, 65%; (l) EDCI∙HCl, HOAt, DMF, CH2Cl2, 70%; (m) AgNO3, 2,6-lutidine, THF, H2O, 81%. MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 12.
Synthesis of 2-epi-63 and 63. Reagents and conditions: (a) Ac2O, AcOH, DMSO, r.t., 84%; (b) LiOH, H2O, MeOH, H2O, r.t., 88%; (c) EDCI∙HCl, HOBt, Et3N, DMF-CH2Cl2 (1:1), r.t.; (d) 4M HCl, dioxane, r.t.; (e) PyBOP, DIPEA, DMF-CH2Cl2 (1:1), r.t.; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t.; (g) EDCI∙HCl, DMAP, CH2Cl2, 45%; (h) HF∙pyridine, pyridine, THF, 97%; (i) Zn powder, AcONH4, 93%; (j) MNBA, DMAP, CH2Cl2, 60%; (k) AgNO3, 2,6-lutidine, THF, H2O, 65%; (l) EDCI∙HCl, HOAt, DMF, CH2Cl2, 70%; (m) AgNO3, 2,6-lutidine, THF, H2O, 81%. MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 13.
Synthesis of the
11C-labeled C1-C10 subunit
87. Reagents and conditions: (
a) [
4]; (
b) PMBTCA, Et
2O, −78 °C added to CF
3CO
3H, then r.t., 2 h, 49%; (
c) HF∙pyridine, pyridine, THF, r.t., 40 °C, overnight, 78%; (
d) Grubbs II catalyst (1.3 eq), toluene (0.01 M), 80 °C, 24 h, 14%; (
e) [
11C]CH
3I, Pd
2(dba)
3, P(
o-tolyl)
3, K
2CO
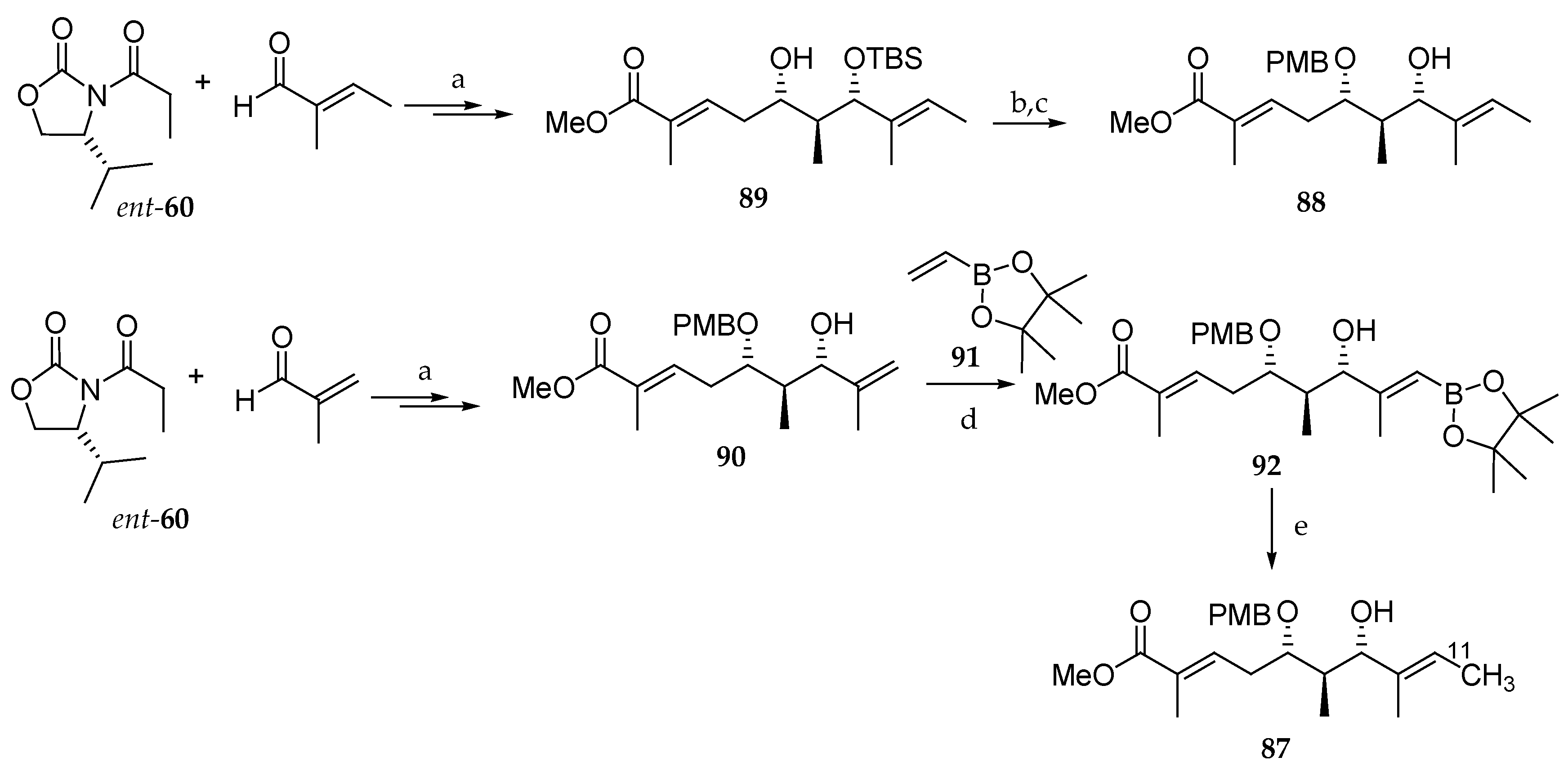
3, DMF, 70 °C, 5 min, 78%. PMBTCA = 4-methoxybenzyl-2,2,2-trichloroacetimidate.
Scheme 13.
Synthesis of the
11C-labeled C1-C10 subunit
87. Reagents and conditions: (
a) [
4]; (
b) PMBTCA, Et
2O, −78 °C added to CF
3CO
3H, then r.t., 2 h, 49%; (
c) HF∙pyridine, pyridine, THF, r.t., 40 °C, overnight, 78%; (
d) Grubbs II catalyst (1.3 eq), toluene (0.01 M), 80 °C, 24 h, 14%; (
e) [
11C]CH
3I, Pd
2(dba)
3, P(
o-tolyl)
3, K
2CO
3, DMF, 70 °C, 5 min, 78%. PMBTCA = 4-methoxybenzyl-2,2,2-trichloroacetimidate.
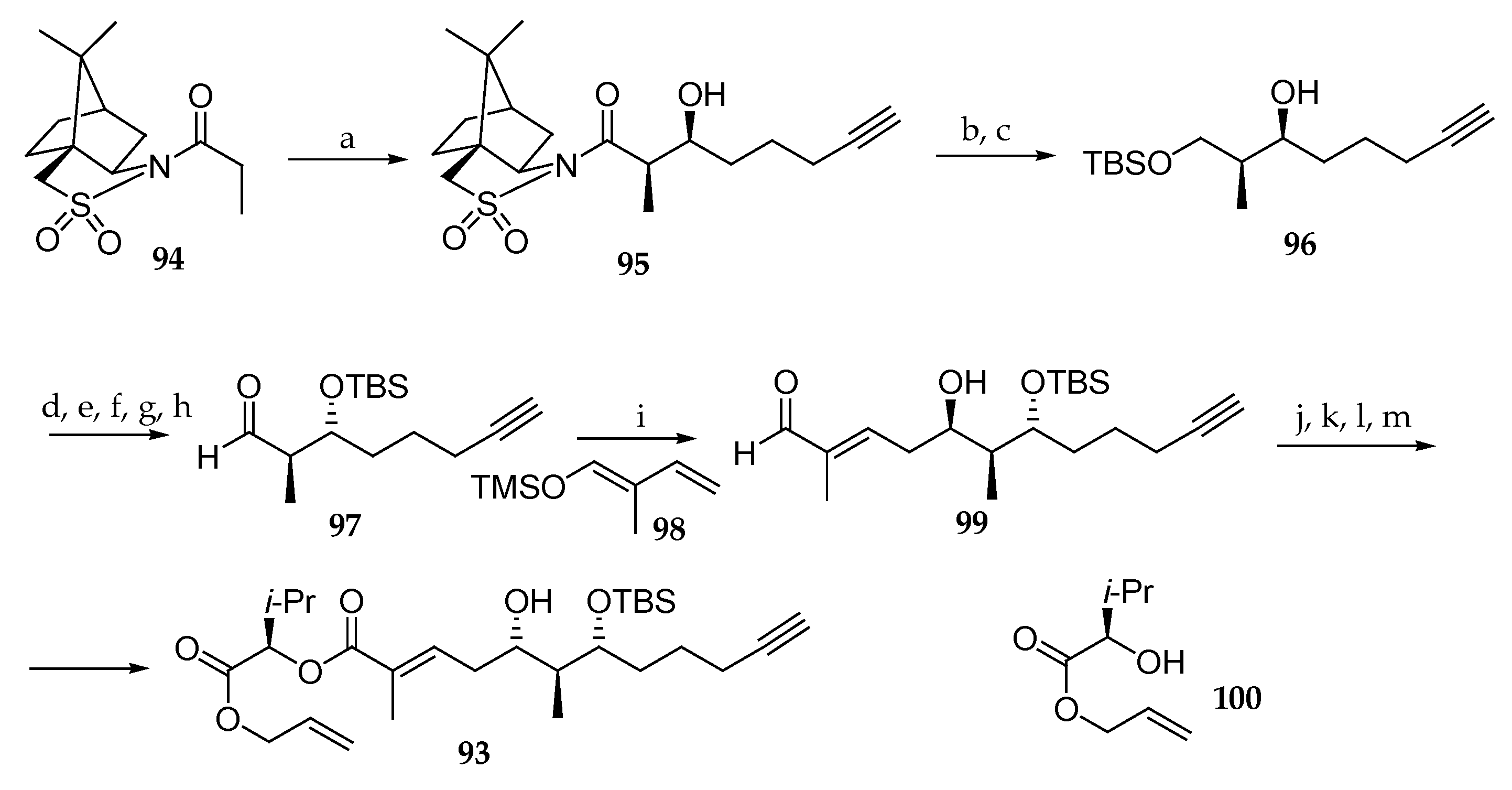
Scheme 14.
Synthesis of palau’amide subunit 93. Reagents and conditions: (a) Et2BOTf, DIPEA, CH2Cl2, −15 °C, then 5-hexynal, −78 °C, 90%; (b) LiAlH4, THF; (c) TBSCl, imidazole, 88% (2 steps); (d) Ph3P, DEAD, p-nitrobenzoic acid; (e) KOH, 86% (2 steps); (f) TBSCl, imidazole; (g) HF∙pyridine, pyridine; (h) Swern oxidation, 80% (3 steps); (i) 98, BF3∙OEt2, CH2Cl2-Et2O 9:1, −78 °C, 65%; (j) NaClO2, NaH2PO4, t-BuOH, 2-methyl-2-butene; (k) 100, EDC; (l) Dess–Martin periodinane, CH2Cl2, r.t.; (m) NaBH4, MeOH, −50 °C, 91%. DEAD = diethyl azodicarboxylate.
Scheme 14.
Synthesis of palau’amide subunit 93. Reagents and conditions: (a) Et2BOTf, DIPEA, CH2Cl2, −15 °C, then 5-hexynal, −78 °C, 90%; (b) LiAlH4, THF; (c) TBSCl, imidazole, 88% (2 steps); (d) Ph3P, DEAD, p-nitrobenzoic acid; (e) KOH, 86% (2 steps); (f) TBSCl, imidazole; (g) HF∙pyridine, pyridine; (h) Swern oxidation, 80% (3 steps); (i) 98, BF3∙OEt2, CH2Cl2-Et2O 9:1, −78 °C, 65%; (j) NaClO2, NaH2PO4, t-BuOH, 2-methyl-2-butene; (k) 100, EDC; (l) Dess–Martin periodinane, CH2Cl2, r.t.; (m) NaBH4, MeOH, −50 °C, 91%. DEAD = diethyl azodicarboxylate.
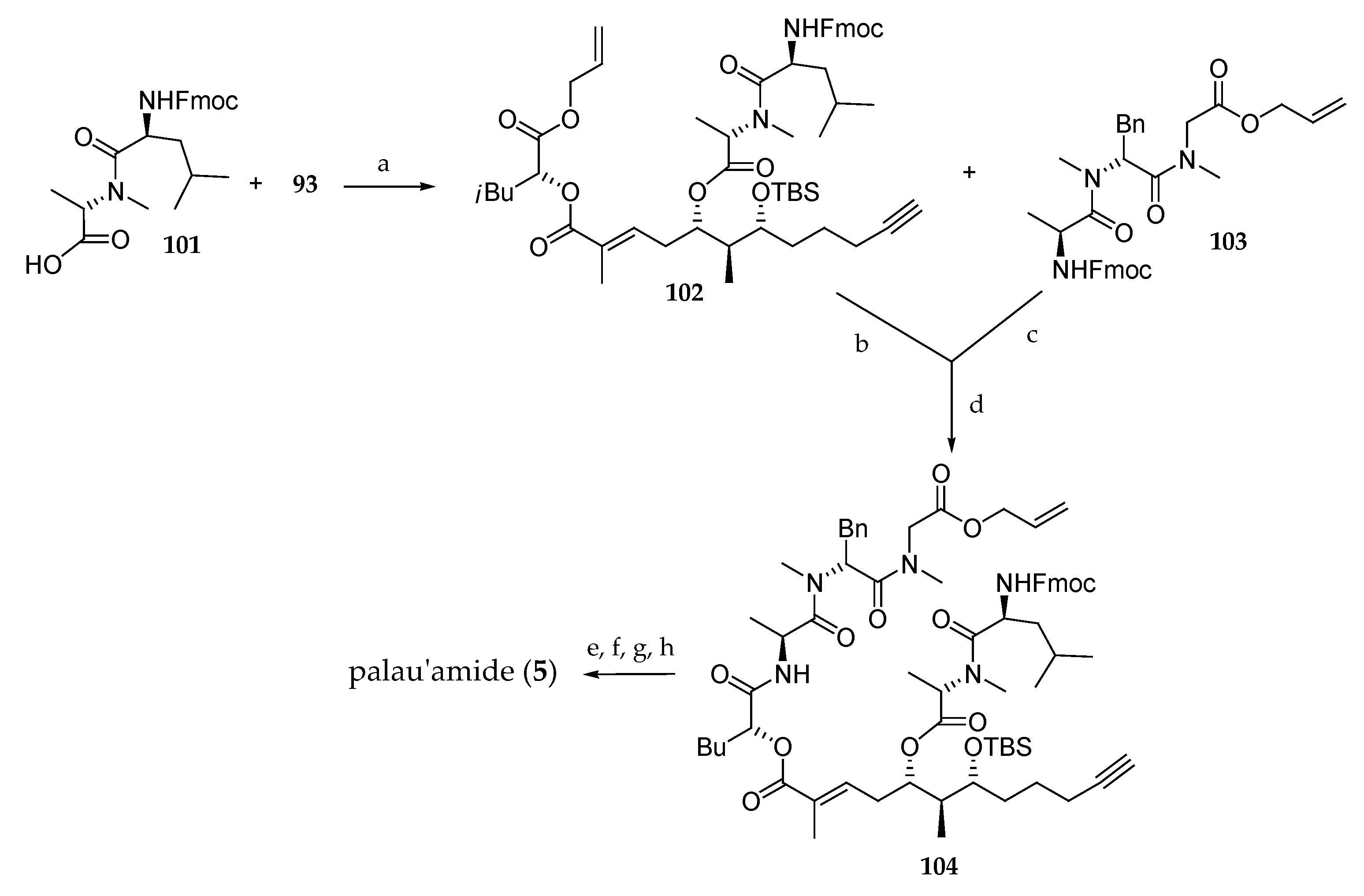
Scheme 15.
Total synthesis of palau’amide (5). Reagents and conditions: (a) 2,4,6-trichlorobenzoyl chloride, DIPEA then 93, DMAP, 85%; (b) Pd(PPh3)4, PhNHMe, THF; (c) Et2NH, CH3CN; (d) HATU, DIPEA, 66% (3 steps); (e) Pd(PPh3)4, PhNHMe; (f) Et2NH, CH3CN; (g) HATU, DIPEA, CH3CN; (h) 5% HF/CH3CN 25% (four steps). HATU = O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate.
Scheme 15.
Total synthesis of palau’amide (5). Reagents and conditions: (a) 2,4,6-trichlorobenzoyl chloride, DIPEA then 93, DMAP, 85%; (b) Pd(PPh3)4, PhNHMe, THF; (c) Et2NH, CH3CN; (d) HATU, DIPEA, 66% (3 steps); (e) Pd(PPh3)4, PhNHMe; (f) Et2NH, CH3CN; (g) HATU, DIPEA, CH3CN; (h) 5% HF/CH3CN 25% (four steps). HATU = O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate.
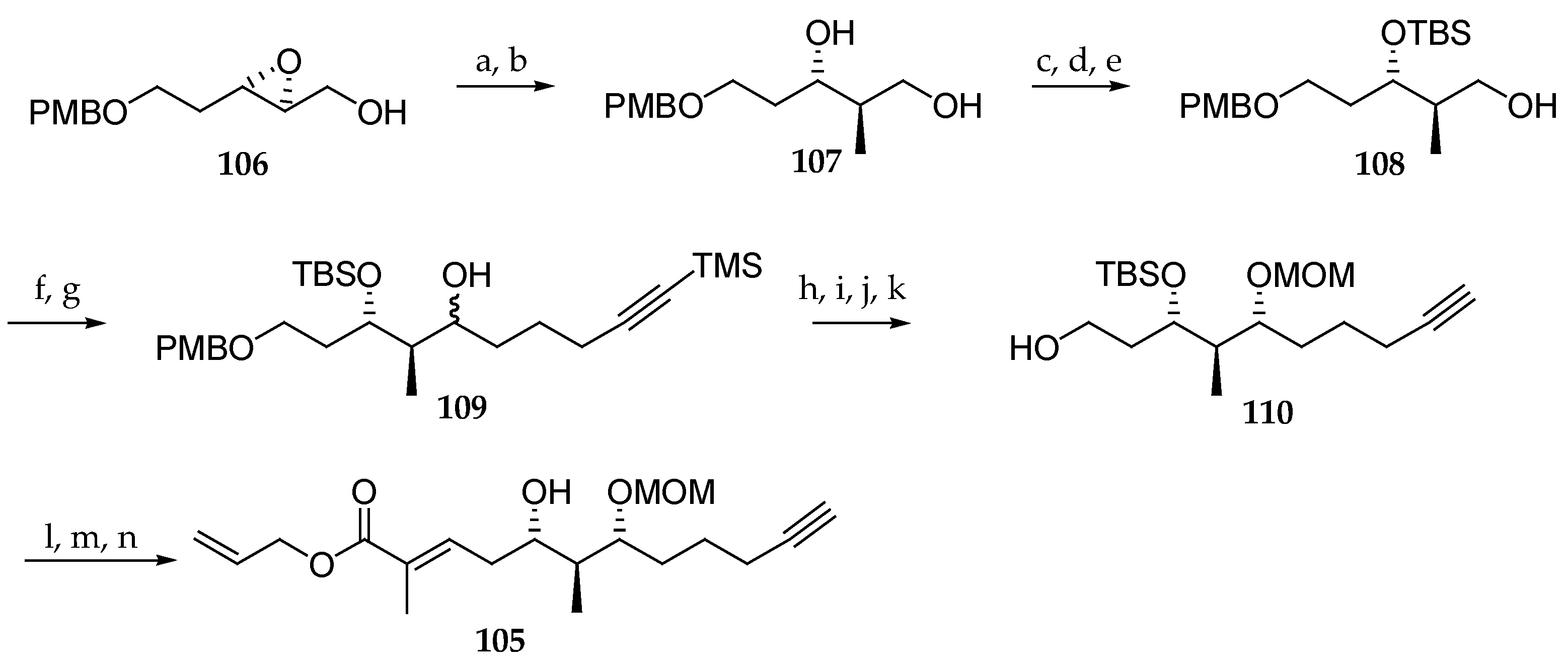
Scheme 16.
Stereoselective synthesis of the C33–C44 fragment 105 of palau’amide (5). Reagents and conditions: (a) Me2CuCNLi2, THF, −20 °C, 6 h; (b) NaIO4, 30 min, 70% (2 steps); (c) BzCl, Et3N, CH2Cl2, 0 °C, 1 h, 97%; (d) TBSOTf, CH2Cl2, 2,6-lutidine, 0 °C, 1 h, 98%; (e) K2CO3, MeOH, r.t., 4 h, 98%; (f) (COCl)2, DMSO, CH2Cl2, Et3N, −78 °C, 3 h; (g) TMS-C8H15MgBr, THF, 0 °C to r.t., 2 h, 84% (2 steps); (h) K2CO3, MeOH, r.t., 2 h, 92%; (i) NaBH4, CeCl3, MeOH, −100 °C, 1 h, 90%; (j) MOMCl, DIPEA, cat. AgNO3, CH2Cl2, r.t., 4 h, 82%; (k) DDQ, CH2Cl2, H2O, r.t., 2 h, 91%; (l) IBX, DMSO, THF, 2 h, r.t.; (m) Ph3P=C(CH3)CO2Allyl, THF, reflux, 2 h, 80% (2 steps); (n) TBAF, THF, cat. AcOH, r.t., 3 h, 86%. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
Scheme 16.
Stereoselective synthesis of the C33–C44 fragment 105 of palau’amide (5). Reagents and conditions: (a) Me2CuCNLi2, THF, −20 °C, 6 h; (b) NaIO4, 30 min, 70% (2 steps); (c) BzCl, Et3N, CH2Cl2, 0 °C, 1 h, 97%; (d) TBSOTf, CH2Cl2, 2,6-lutidine, 0 °C, 1 h, 98%; (e) K2CO3, MeOH, r.t., 4 h, 98%; (f) (COCl)2, DMSO, CH2Cl2, Et3N, −78 °C, 3 h; (g) TMS-C8H15MgBr, THF, 0 °C to r.t., 2 h, 84% (2 steps); (h) K2CO3, MeOH, r.t., 2 h, 92%; (i) NaBH4, CeCl3, MeOH, −100 °C, 1 h, 90%; (j) MOMCl, DIPEA, cat. AgNO3, CH2Cl2, r.t., 4 h, 82%; (k) DDQ, CH2Cl2, H2O, r.t., 2 h, 91%; (l) IBX, DMSO, THF, 2 h, r.t.; (m) Ph3P=C(CH3)CO2Allyl, THF, reflux, 2 h, 80% (2 steps); (n) TBAF, THF, cat. AcOH, r.t., 3 h, 86%. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
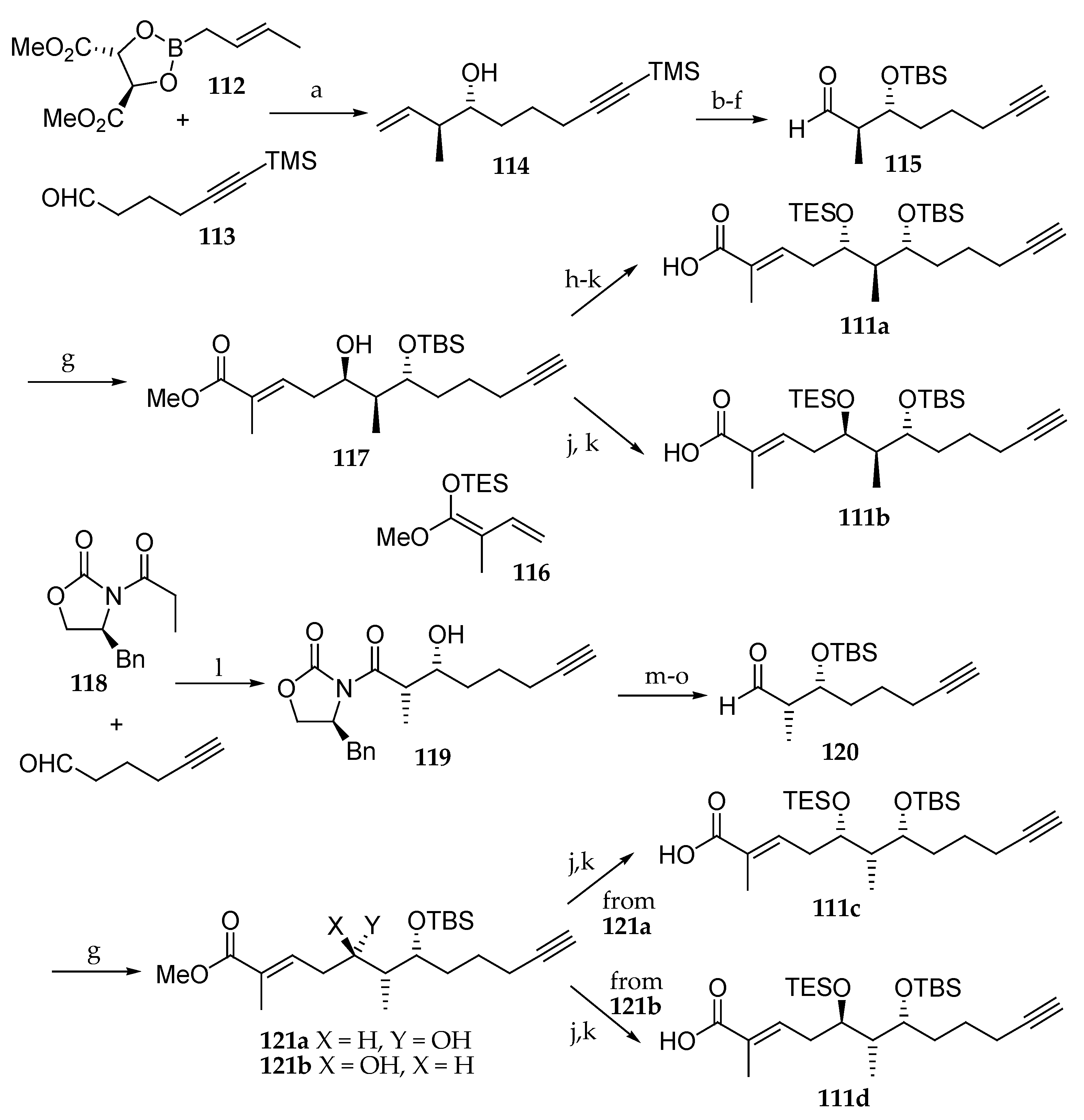
Scheme 17.
Synthesis of the polyketides units 111a, 111b, 111c, and 111d of palau’amide (5). Reagents and conditions: (a) MS 4 Å, toluene, −78 °C, 3 h, quant.; (b) TBSCl, imidazole, DMF, r.t., 3 h, 94%; (c) Bu4NF, THF, r.t., 1 h; (d) TBSCl, imidazole, DMF, r.t., 2.5 h, 100% (two steps); (e) OsO4, NMO, acetone, H2O, r.t., 75 min; (f) NaIO4, acetone, H2O, r.t., 1.5 h, 56% in two steps (recovered diol 28%); (g) 116, BF3∙Et2O, CH2Cl2, Et2O, −40 °C, 4.5 h; then, aq HCl, (117) 80%, (121a) 81% based on recovered starting material (br sm), (121b) 10% br sm; (h) Dess–Martin periodinane, CH2Cl2, r.t., 1 h, quant; (i) NaBH4, MeOH, −78 °C, 1.5 h, 83%; (j) LiOH, H2O, MeOH, 40 °C, 17 h; (k) TESCl, imidazole, DMF, r.t., 4.5 h; (111a) 90% (2 steps), (111b), 72% (2 steps), (111c) 83% (2 steps), (111d) 41% (2 steps); (l) Bu2BOTf, Et3N, CH2Cl2, −78 °C, 2 h, r.t., 1 h, quant; (m) Me2AlN(Me)OMe, THF, toluene, −10 °C, 4 h, 79%; (n) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 4 h, 99%; (o) DiBAL-H, THF, hexane, −78 °C, 3 h, 91%.
Scheme 17.
Synthesis of the polyketides units 111a, 111b, 111c, and 111d of palau’amide (5). Reagents and conditions: (a) MS 4 Å, toluene, −78 °C, 3 h, quant.; (b) TBSCl, imidazole, DMF, r.t., 3 h, 94%; (c) Bu4NF, THF, r.t., 1 h; (d) TBSCl, imidazole, DMF, r.t., 2.5 h, 100% (two steps); (e) OsO4, NMO, acetone, H2O, r.t., 75 min; (f) NaIO4, acetone, H2O, r.t., 1.5 h, 56% in two steps (recovered diol 28%); (g) 116, BF3∙Et2O, CH2Cl2, Et2O, −40 °C, 4.5 h; then, aq HCl, (117) 80%, (121a) 81% based on recovered starting material (br sm), (121b) 10% br sm; (h) Dess–Martin periodinane, CH2Cl2, r.t., 1 h, quant; (i) NaBH4, MeOH, −78 °C, 1.5 h, 83%; (j) LiOH, H2O, MeOH, 40 °C, 17 h; (k) TESCl, imidazole, DMF, r.t., 4.5 h; (111a) 90% (2 steps), (111b), 72% (2 steps), (111c) 83% (2 steps), (111d) 41% (2 steps); (l) Bu2BOTf, Et3N, CH2Cl2, −78 °C, 2 h, r.t., 1 h, quant; (m) Me2AlN(Me)OMe, THF, toluene, −10 °C, 4 h, 79%; (n) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 4 h, 99%; (o) DiBAL-H, THF, hexane, −78 °C, 3 h, 91%.

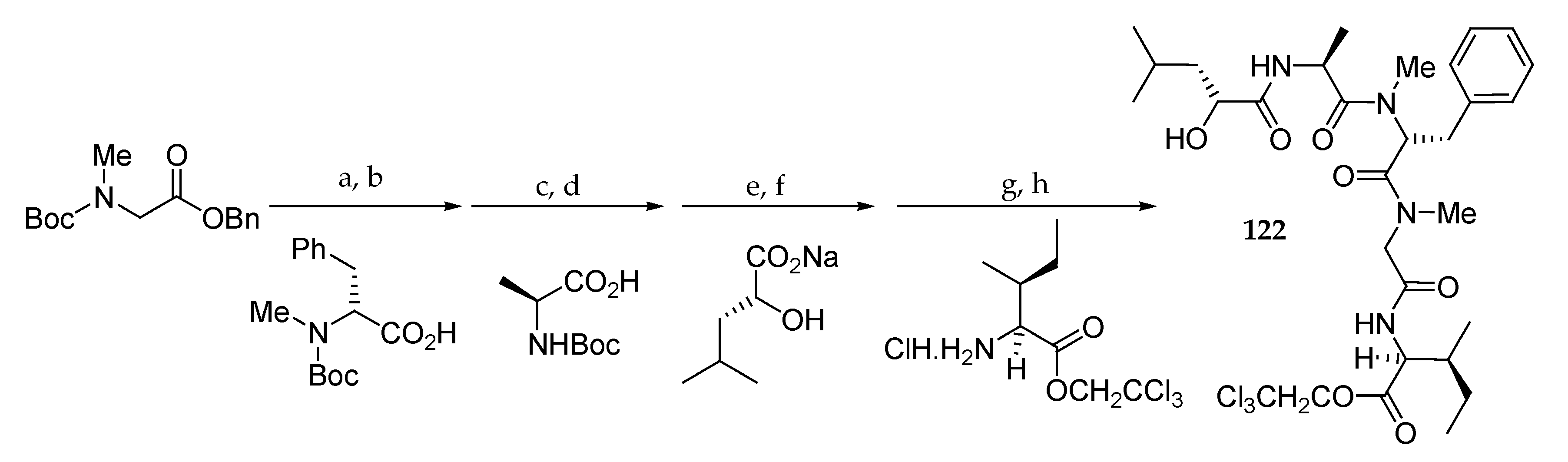
Scheme 18.
Synthesis of subunit 122. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C, 1.5 h; (b) DEPC, Et3N, DMF, r.t., 16 h, 94% (2 steps); (c) TFA, CH2Cl2, 0 °C, 1.5 h; (d) DEPC, Et3N, DMF, r.t., 17 h, 94% (2 steps); (e) TFA, CH2Cl2, 0 °C, 1.5 h; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 17 h, 75% (2 steps); (g) H2, 5% Pd/C, EtOH, r.t., 5.5 h; (h) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 12.5 h, 96% (2 steps).
Scheme 18.
Synthesis of subunit 122. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C, 1.5 h; (b) DEPC, Et3N, DMF, r.t., 16 h, 94% (2 steps); (c) TFA, CH2Cl2, 0 °C, 1.5 h; (d) DEPC, Et3N, DMF, r.t., 17 h, 94% (2 steps); (e) TFA, CH2Cl2, 0 °C, 1.5 h; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 17 h, 75% (2 steps); (g) H2, 5% Pd/C, EtOH, r.t., 5.5 h; (h) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 12.5 h, 96% (2 steps).
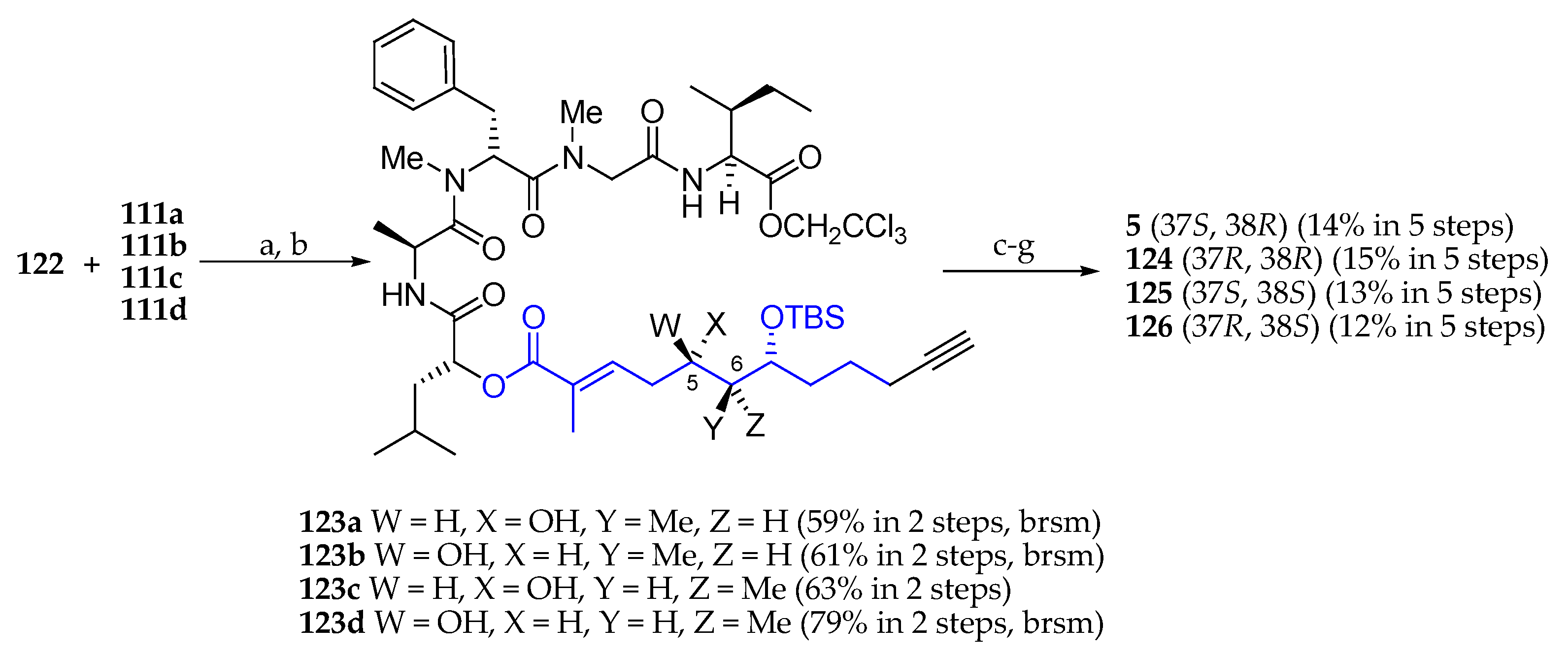
Scheme 19.
Synthesis of palau’amide (5) and stereoisomers 124, 125, and 126. Reagents and conditions: (a) EDCI∙HCl, DMAP, CH2Cl2, r.t., 16 h; (b) AcOH, H2O, THF, r.t., 6 h; (c) Fmoc-N-Me-l-Ala, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, toluene, r.t., 16 h; (d) Zn, NH4OAc, THF, r.t., 3 h; (e) Et2NH, MeCN, r.t. 3 h; (f) EDCI∙HCl, HOAt, Et3N, DMF-CH2Cl2 (1:10, 1 mM), r.t., 45 h; (g) HF∙pyridine, pyridine, r.t., 2 h.
Scheme 19.
Synthesis of palau’amide (5) and stereoisomers 124, 125, and 126. Reagents and conditions: (a) EDCI∙HCl, DMAP, CH2Cl2, r.t., 16 h; (b) AcOH, H2O, THF, r.t., 6 h; (c) Fmoc-N-Me-l-Ala, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, toluene, r.t., 16 h; (d) Zn, NH4OAc, THF, r.t., 3 h; (e) Et2NH, MeCN, r.t. 3 h; (f) EDCI∙HCl, HOAt, Et3N, DMF-CH2Cl2 (1:10, 1 mM), r.t., 45 h; (g) HF∙pyridine, pyridine, r.t., 2 h.
Scheme 20.
Synthesis of polyketide subunits 127a, 127b, 127c, and 127d. Reagents and conditions: (a) ent-118, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 80% (2 steps); (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to r.t., 89% (131) and 87% (141); (c) LiBH4, MeOH, THF, 0 °C to r.t., 69% (132) and 80% (142); (d) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (e) ethyltriphenylphosphonium bromide, n-BuLi, THF, r.t., 69% (2 steps, Z/E = 7:1); (f) Pd/C, H2, EtOH, r.t., 92% (134a) and 85% (134b); (g) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (h) 135, BF3·OEt2, CH2Cl2, Et2O, −78 °C, 54% (127c) and 69% (127d) (two steps); (i) Dess–Martin periodinane, CH2Cl2, r.t.; (j) NaBH4, MeOH, −78 °C, 83% (two steps, dr > 97:3); (k) TBAF, THF, r.t., 81%; (l) 2,2-dimethoxypropane, PPTS, CH2Cl2, r.t., 90%; (m) DiBAL-H, toluene, THF, −78 °C, 90%; (n) HCl, MeOH, H2O, r.t., 51%; (o) 129, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 72% (2 steps); (p) TsCl, Et3N, Me3N·HCl, CH2Cl2, r.t.; (q) LiAlH4, THF, 0 °C to r.t., 70% (2 steps).
Scheme 20.
Synthesis of polyketide subunits 127a, 127b, 127c, and 127d. Reagents and conditions: (a) ent-118, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 80% (2 steps); (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to r.t., 89% (131) and 87% (141); (c) LiBH4, MeOH, THF, 0 °C to r.t., 69% (132) and 80% (142); (d) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (e) ethyltriphenylphosphonium bromide, n-BuLi, THF, r.t., 69% (2 steps, Z/E = 7:1); (f) Pd/C, H2, EtOH, r.t., 92% (134a) and 85% (134b); (g) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (h) 135, BF3·OEt2, CH2Cl2, Et2O, −78 °C, 54% (127c) and 69% (127d) (two steps); (i) Dess–Martin periodinane, CH2Cl2, r.t.; (j) NaBH4, MeOH, −78 °C, 83% (two steps, dr > 97:3); (k) TBAF, THF, r.t., 81%; (l) 2,2-dimethoxypropane, PPTS, CH2Cl2, r.t., 90%; (m) DiBAL-H, toluene, THF, −78 °C, 90%; (n) HCl, MeOH, H2O, r.t., 51%; (o) 129, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 72% (2 steps); (p) TsCl, Et3N, Me3N·HCl, CH2Cl2, r.t.; (q) LiAlH4, THF, 0 °C to r.t., 70% (2 steps).

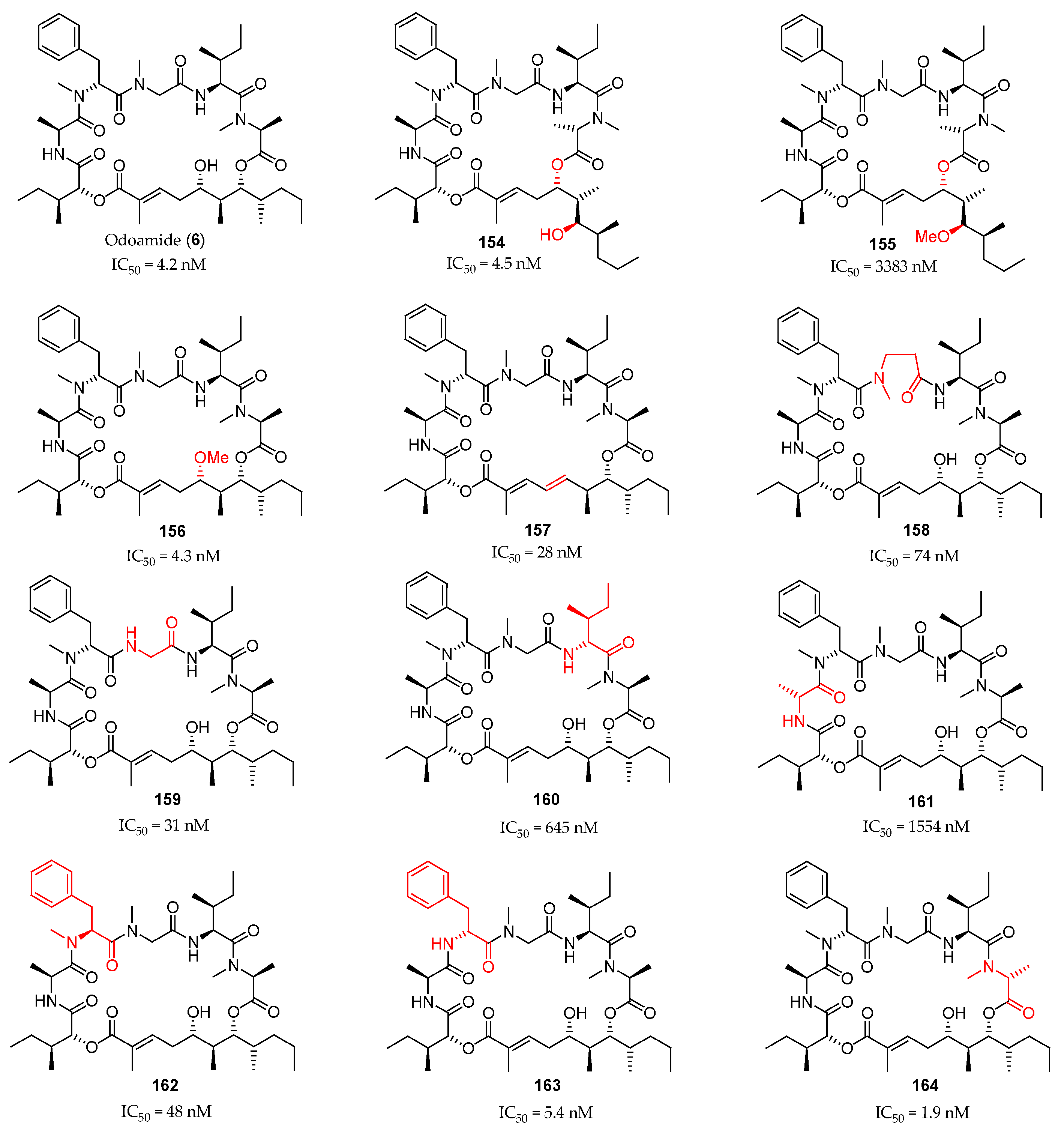
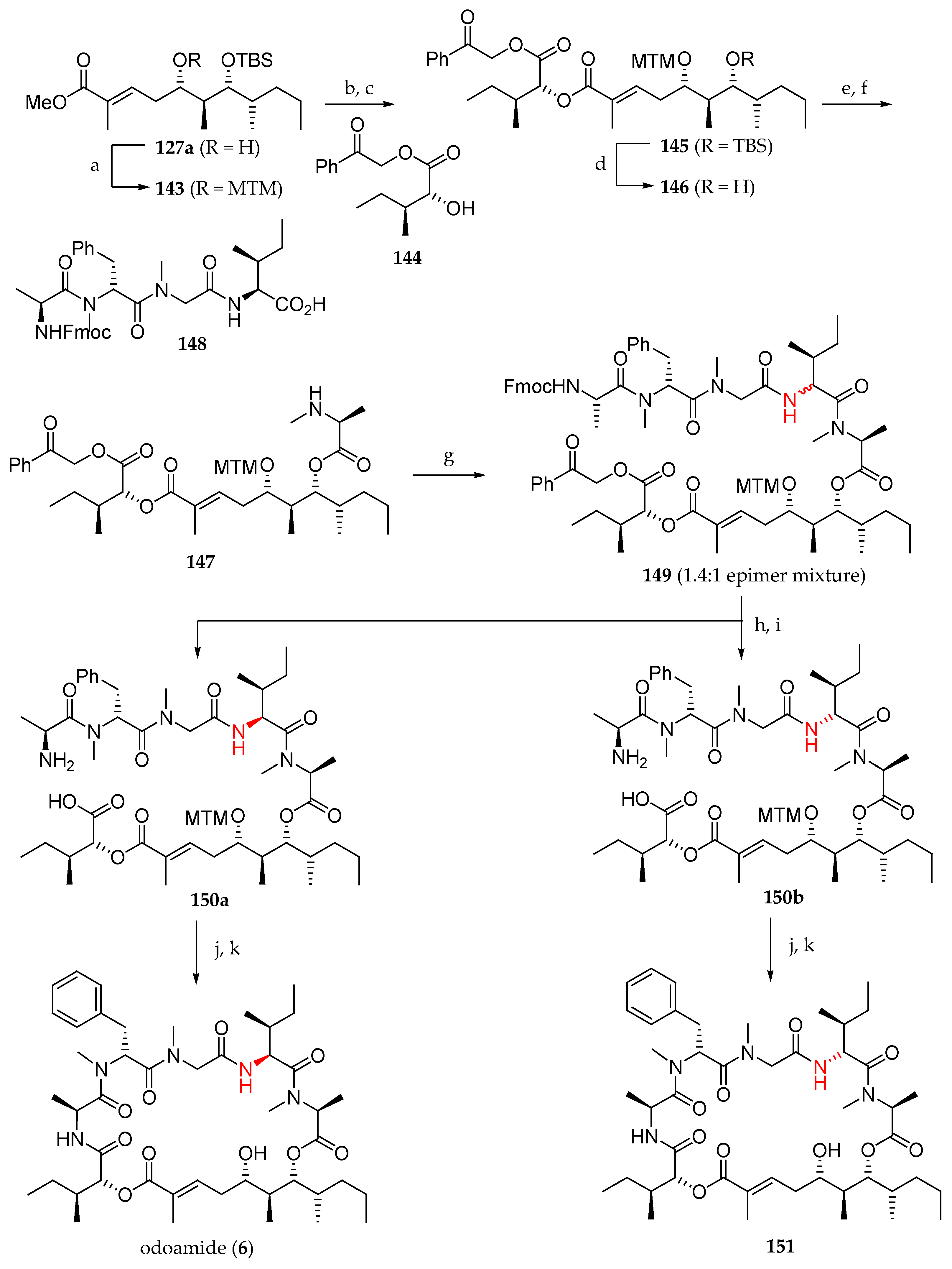
Scheme 21.
Total synthesis of odoamide (6) and its synthetic analogue 151. Reagents and conditions: (a) Ac2O, DMSO, AcOH, r.t., 59%; (b) LiOH, THF, MeOH, H2O, 0 to 30 °C; (c) 144, MNBA, DMAP, CH2Cl2, r.t., 87% (2 steps); (d) HF·pyridine, THF, pyridine, 0 °C to r.t., 74%; (e) Fmoc-N-Me-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (f) Et2NH, MeCN, 0 °C to r.t., 54% (2 steps); (g) 148, EDCI∙HCl, HOAt, CH2Cl2, 0 °C to r.t.; (h) Zn, CH3COOH, H2O, EtOAc, r.t.; (i) Et2NH, MeCN, 0 °C to r.t., 30% (150a) and 24% (150b) (3 steps); (j) HATU, HOAt, collidine, DMF, r.t.; (k) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C, 85% (6), and 62% (151) (2 steps).
Scheme 21.
Total synthesis of odoamide (6) and its synthetic analogue 151. Reagents and conditions: (a) Ac2O, DMSO, AcOH, r.t., 59%; (b) LiOH, THF, MeOH, H2O, 0 to 30 °C; (c) 144, MNBA, DMAP, CH2Cl2, r.t., 87% (2 steps); (d) HF·pyridine, THF, pyridine, 0 °C to r.t., 74%; (e) Fmoc-N-Me-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (f) Et2NH, MeCN, 0 °C to r.t., 54% (2 steps); (g) 148, EDCI∙HCl, HOAt, CH2Cl2, 0 °C to r.t.; (h) Zn, CH3COOH, H2O, EtOAc, r.t.; (i) Et2NH, MeCN, 0 °C to r.t., 30% (150a) and 24% (150b) (3 steps); (j) HATU, HOAt, collidine, DMF, r.t.; (k) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C, 85% (6), and 62% (151) (2 steps).
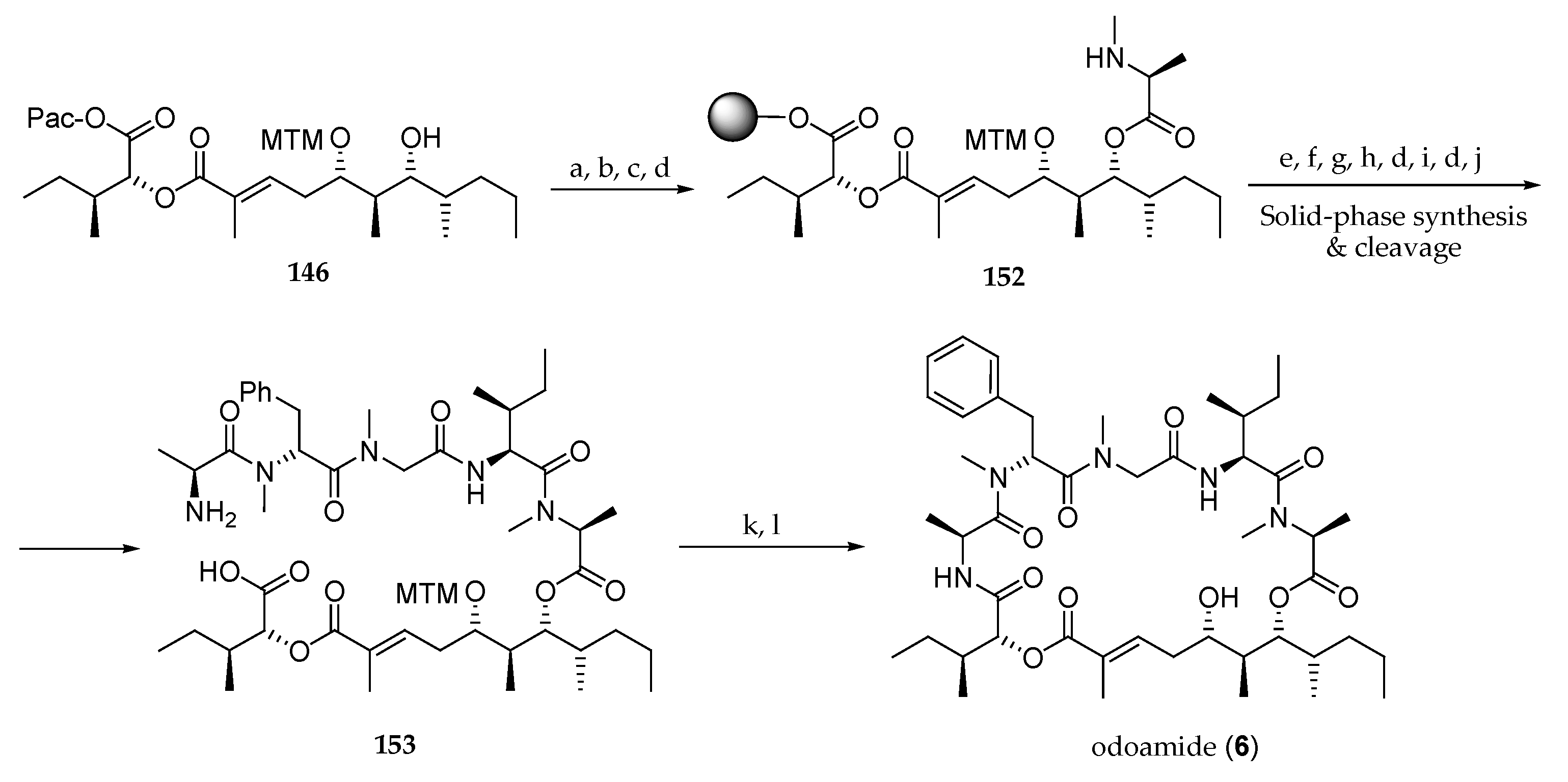
Scheme 22.
Odoamide (6) synthesis using solid-phase peptide synthesis. Reagents and conditions: (a) Fmoc-N-Me-l-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (b) Zn, AcOH, r.t.; (c) Cl-(2-Cl)Trt resin, DIPEA, CH2Cl2, r.t.; (d) 20% piperidine, DMF, r.t.; (e) Alloc-l-Ile-OH, DIC, HOAt, DMF, r.t.; (f) Pd(PPh3)4, PhSiH3, CH2Cl2, r.t.; (g) Fmoc-Sar-OH, HOBt∙H2O, DIC, DMF, r.t.; (h) Fmoc-N-Me-d-Phe-OH, HATU, DIPEA, DMF, r.t.; (i) Fmoc-l-Ala-OH, HATU, DIPEA, DMF, r.t.; (j) 30% HFIP, CH2Cl2, r.t.; (k) HATU, HOAt, collidine, DFM, r.t.; (l) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C.
Scheme 22.
Odoamide (6) synthesis using solid-phase peptide synthesis. Reagents and conditions: (a) Fmoc-N-Me-l-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (b) Zn, AcOH, r.t.; (c) Cl-(2-Cl)Trt resin, DIPEA, CH2Cl2, r.t.; (d) 20% piperidine, DMF, r.t.; (e) Alloc-l-Ile-OH, DIC, HOAt, DMF, r.t.; (f) Pd(PPh3)4, PhSiH3, CH2Cl2, r.t.; (g) Fmoc-Sar-OH, HOBt∙H2O, DIC, DMF, r.t.; (h) Fmoc-N-Me-d-Phe-OH, HATU, DIPEA, DMF, r.t.; (i) Fmoc-l-Ala-OH, HATU, DIPEA, DMF, r.t.; (j) 30% HFIP, CH2Cl2, r.t.; (k) HATU, HOAt, collidine, DFM, r.t.; (l) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C.
Figure 5.
Odoamide (6) and synthetic analogues prepared by Kaneda and co-workers and their cytotoxicity against A549 cells.
Figure 5.
Odoamide (6) and synthetic analogues prepared by Kaneda and co-workers and their cytotoxicity against A549 cells.
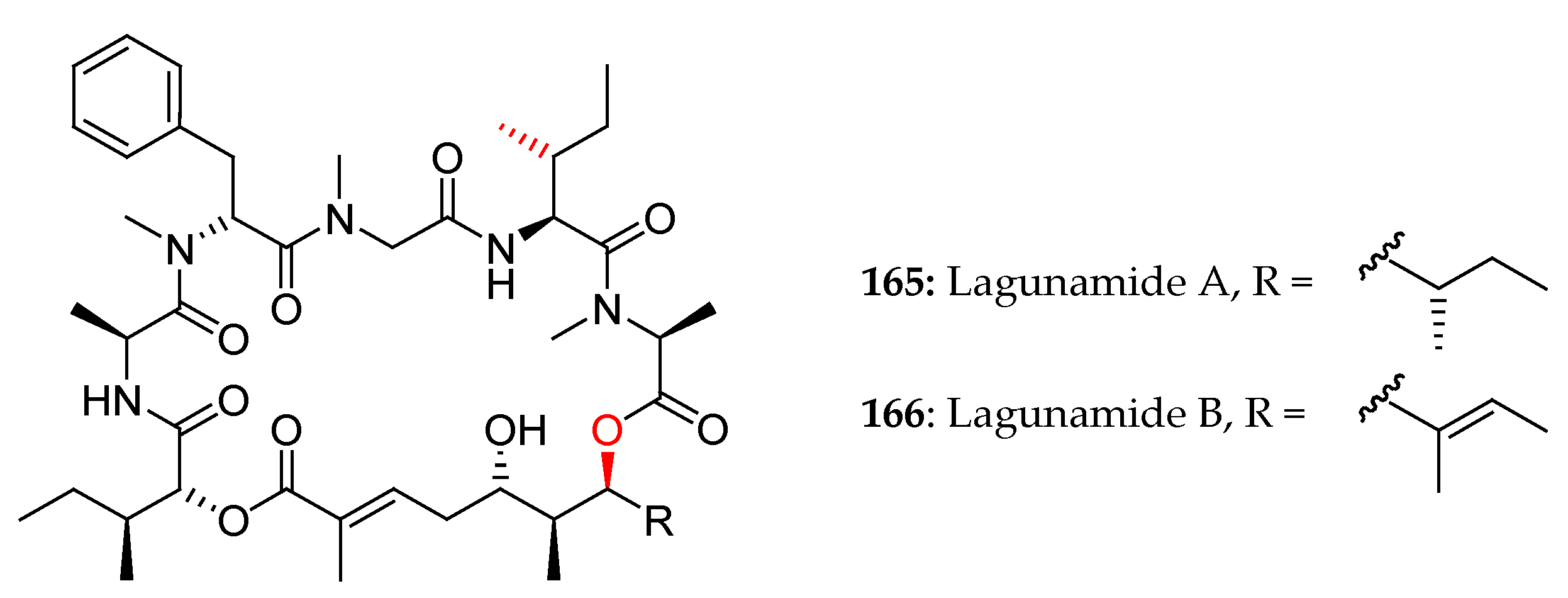
Figure 6.
First proposed structures for lagunamides A and B.
Figure 6.
First proposed structures for lagunamides A and B.
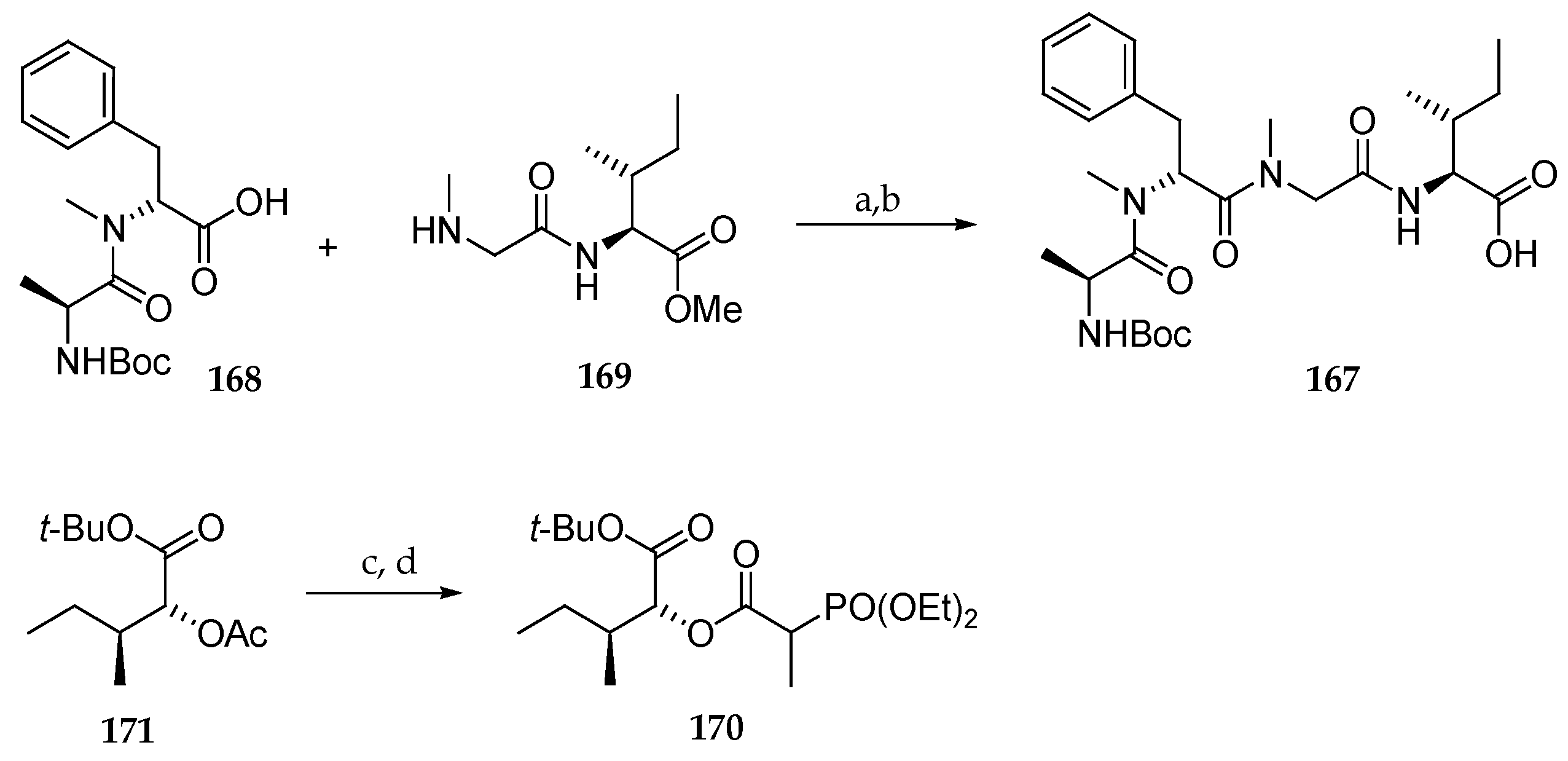
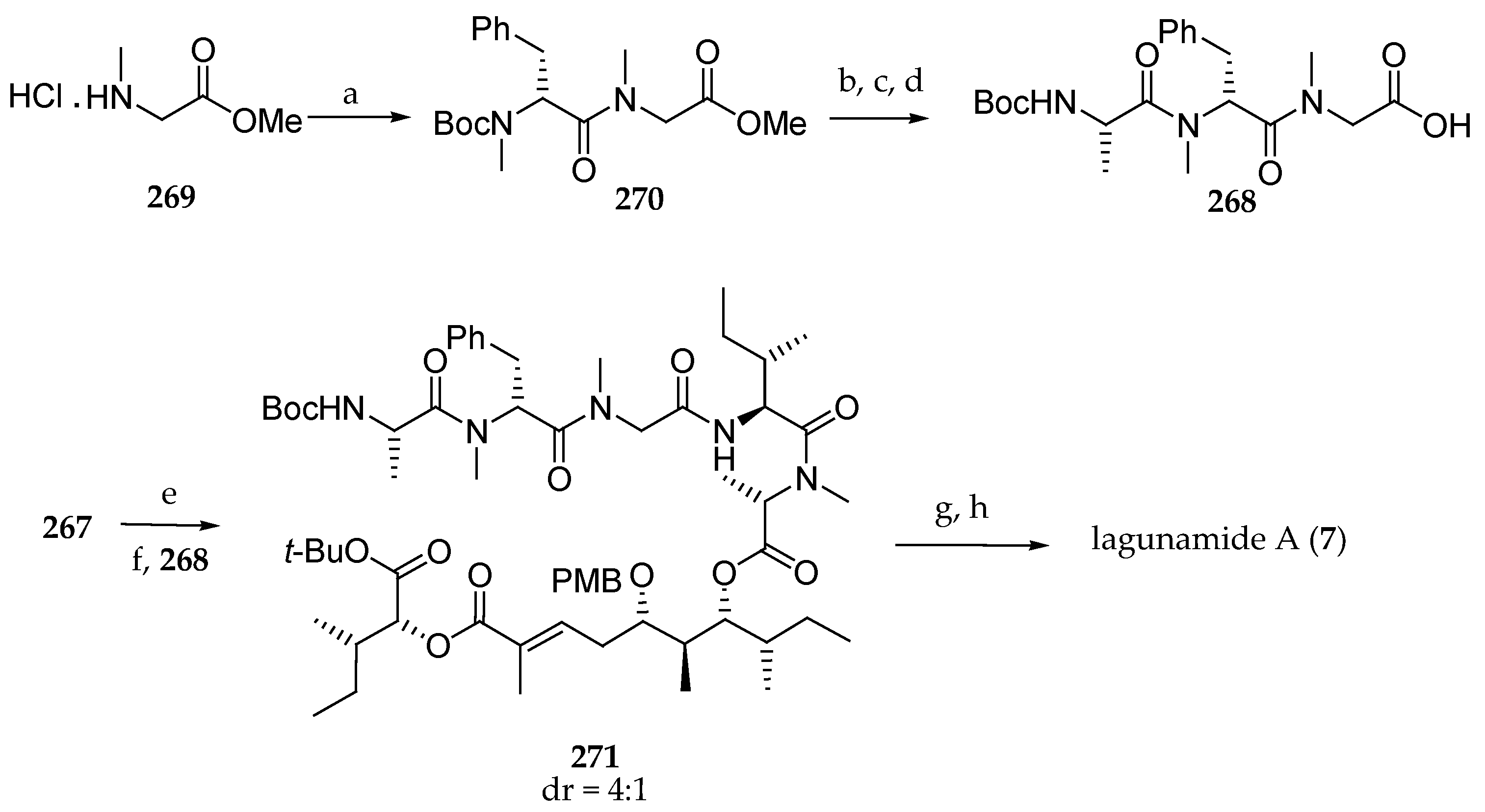
Scheme 23.
Synthesis of subunits 167 and 170 of lagunamide A (7). Reagents and conditions: (a) HATU, HOAt, DIPEA, CH2Cl2; (b) LiOH, THF, H2O, MeOH, then H+; (c) K2CO3, MeOH, H2O; (d) 2-(diethoxyphosphoryl)propanoic acid, DIPC, collidine, CH2Cl2.
Scheme 23.
Synthesis of subunits 167 and 170 of lagunamide A (7). Reagents and conditions: (a) HATU, HOAt, DIPEA, CH2Cl2; (b) LiOH, THF, H2O, MeOH, then H+; (c) K2CO3, MeOH, H2O; (d) 2-(diethoxyphosphoryl)propanoic acid, DIPC, collidine, CH2Cl2.
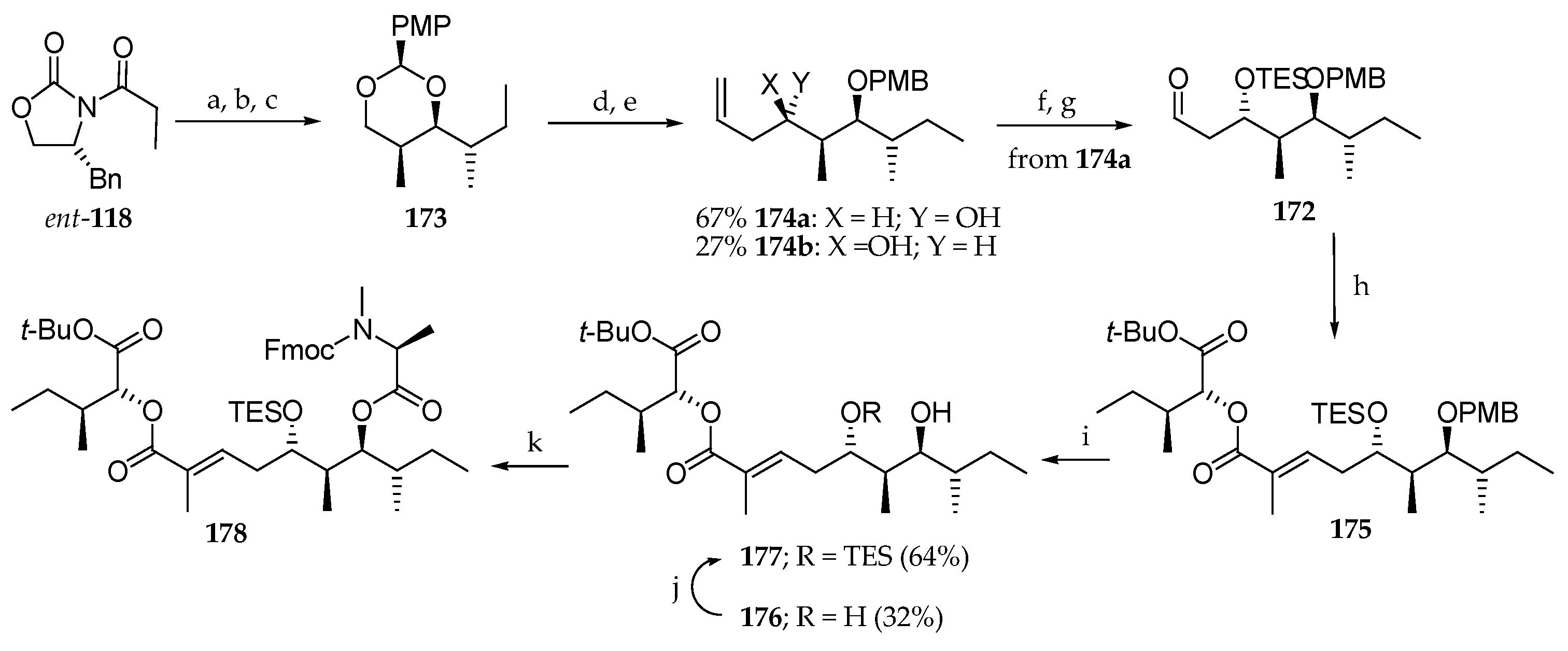
Scheme 24.
Synthesis of subunit 178 of lagunamide A (7). Reagents and conditions: (a) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, then S-2-methylbutanal, −78 °C; (b) NaBH4, Et2O, MeOH; (c) anisaldehyde dimethyl acetal, PPTS, CH2Cl2 (61% from ent-118); (d) DiBAL-H, CH2Cl2, −78 to −10 °C; (e) Dess–Martin periodinane, NaHCO3, CH2Cl2 then allyltributylstannane, BF3∙OEt2, CH2Cl2, −78 °C (67% from 173); (f) TESOTf, 2,6-lutidine, CH2Cl2; (g) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (h) 170, DIPEA, LiCl, MeCN; (i) DDQ, phosphate buffer; (j) TESOTf, 2,6-lutidine, CH2Cl2; (k) Fmoc-N-Me-Ala-Cl, DMAP, toluene, 60 °C.
Scheme 24.
Synthesis of subunit 178 of lagunamide A (7). Reagents and conditions: (a) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, then S-2-methylbutanal, −78 °C; (b) NaBH4, Et2O, MeOH; (c) anisaldehyde dimethyl acetal, PPTS, CH2Cl2 (61% from ent-118); (d) DiBAL-H, CH2Cl2, −78 to −10 °C; (e) Dess–Martin periodinane, NaHCO3, CH2Cl2 then allyltributylstannane, BF3∙OEt2, CH2Cl2, −78 °C (67% from 173); (f) TESOTf, 2,6-lutidine, CH2Cl2; (g) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (h) 170, DIPEA, LiCl, MeCN; (i) DDQ, phosphate buffer; (j) TESOTf, 2,6-lutidine, CH2Cl2; (k) Fmoc-N-Me-Ala-Cl, DMAP, toluene, 60 °C.
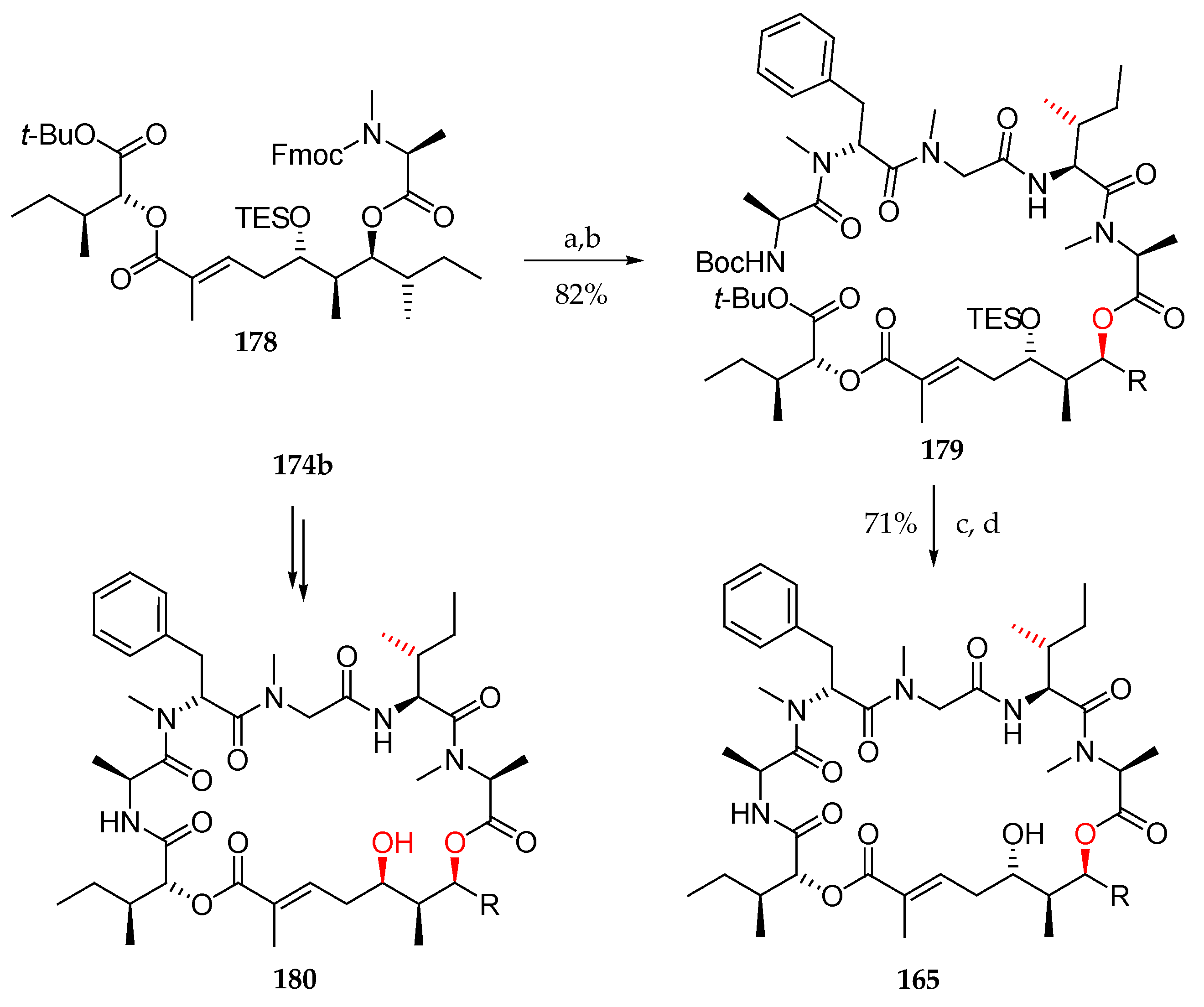
Scheme 25.
Synthesis of 165, the first supposed structure for lagunamide A, and its epimer 180. Reagents and conditions: (a) Et2NH, MeCN; (b) 167, HATU, HOAt, collidine, DMF; (c) TFA, DCM; (d) HATU, HOAt, collidine, DMF.
Scheme 25.
Synthesis of 165, the first supposed structure for lagunamide A, and its epimer 180. Reagents and conditions: (a) Et2NH, MeCN; (b) 167, HATU, HOAt, collidine, DMF; (c) TFA, DCM; (d) HATU, HOAt, collidine, DMF.
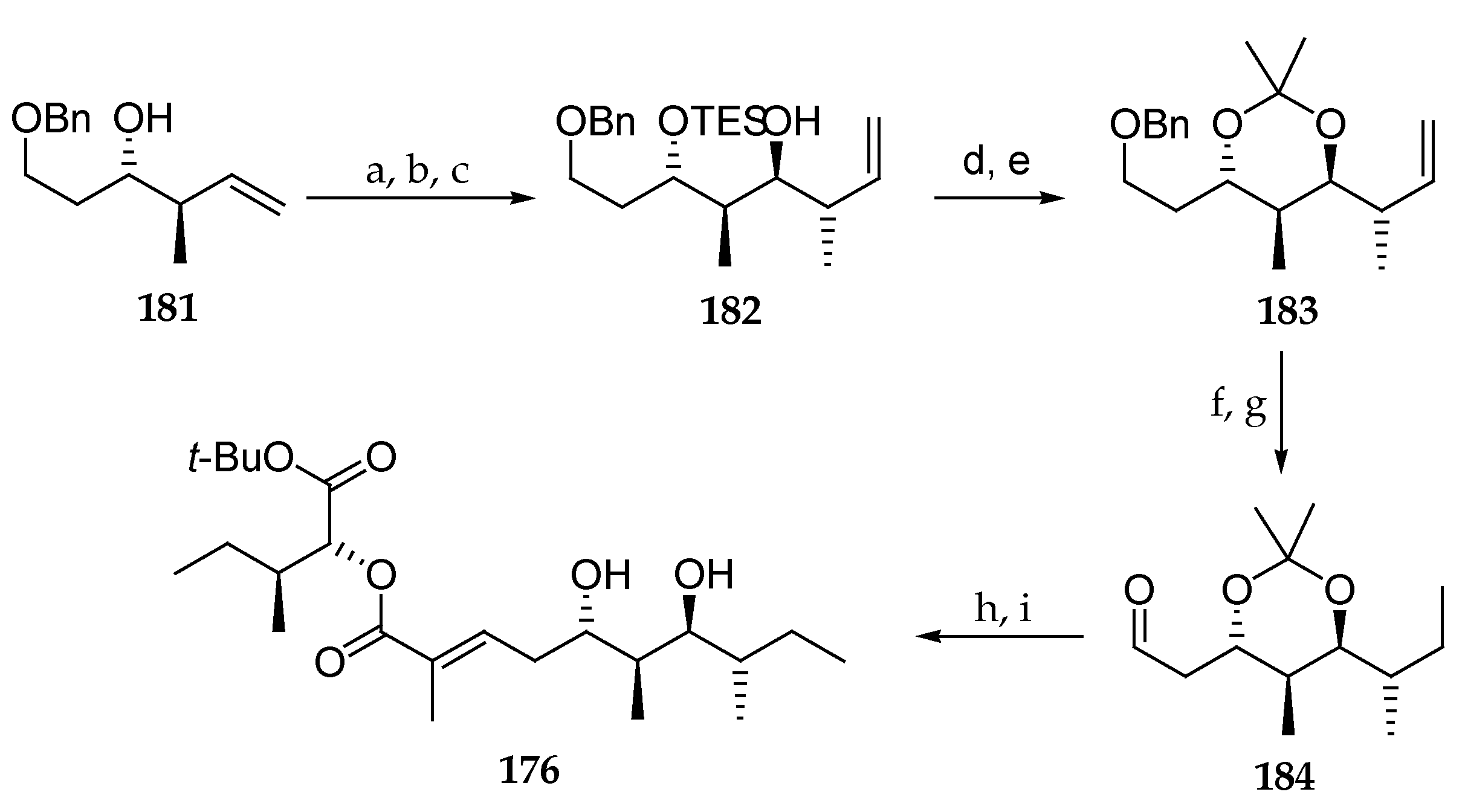
Scheme 26.
Synthesis of polyketide subunit 176. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) E-2-butene, KOBut, n-BuLi, −78 °C, (-)-Ipc2-BOMe, BF3∙OEt2, then Et3N, H2O2, 62% (3 steps); (d) HCl, MeOH; (e) DMP, PPTS, 60 °C, 89% (2 steps); (f) H2, Pd/C, MeOH; (g) Dess–Martin periodinane, NaHCO3, CH2Cl2, 92% (2 steps); (h) 170, DIPEA, LiCl MeCN; (i) PTSA, MeOH, 81% (2 steps). DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid.
Scheme 26.
Synthesis of polyketide subunit 176. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) E-2-butene, KOBut, n-BuLi, −78 °C, (-)-Ipc2-BOMe, BF3∙OEt2, then Et3N, H2O2, 62% (3 steps); (d) HCl, MeOH; (e) DMP, PPTS, 60 °C, 89% (2 steps); (f) H2, Pd/C, MeOH; (g) Dess–Martin periodinane, NaHCO3, CH2Cl2, 92% (2 steps); (h) 170, DIPEA, LiCl MeCN; (i) PTSA, MeOH, 81% (2 steps). DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid.
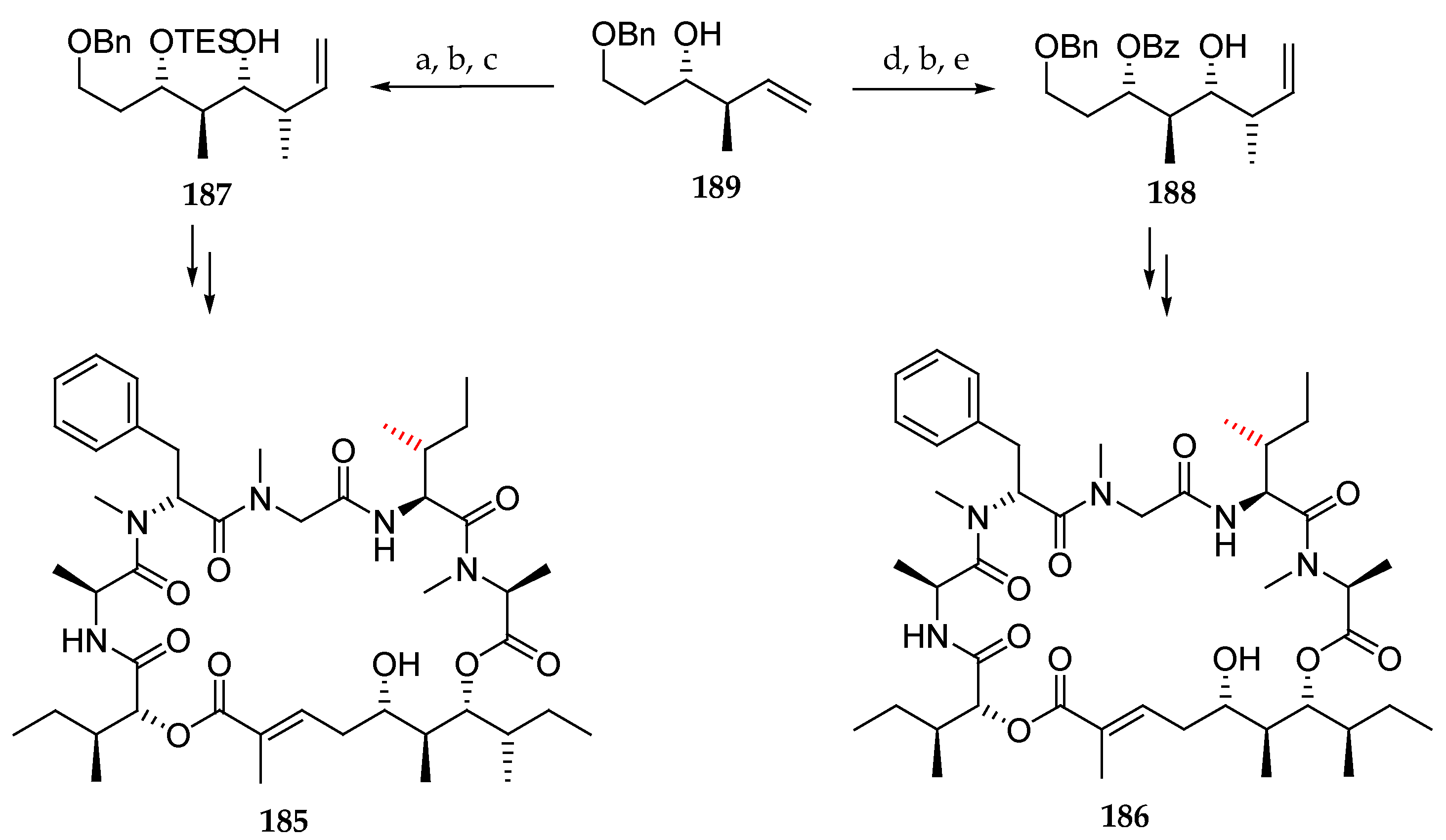
Scheme 27.
Synthesis of epimers 185 and 186. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) Z-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2, 86% (3 steps); (d) BzCl, Et3N, DMAP, toluene, 80 °C, 86%; (e) E-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2.
Scheme 27.
Synthesis of epimers 185 and 186. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) Z-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2, 86% (3 steps); (d) BzCl, Et3N, DMAP, toluene, 80 °C, 86%; (e) E-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2.
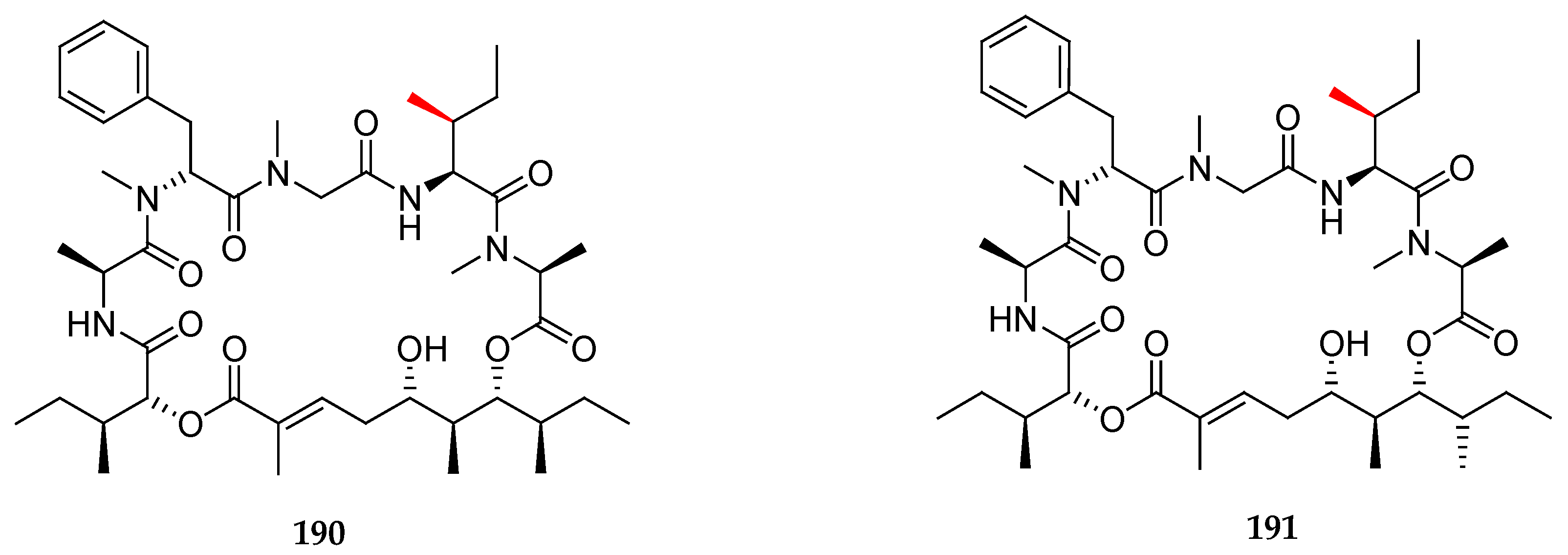
Figure 7.
Two diasteromers of lagunamide A. The stereochemistry of 191 turned out to be the same as that of the natural lagunamide A (7).
Figure 7.
Two diasteromers of lagunamide A. The stereochemistry of 191 turned out to be the same as that of the natural lagunamide A (7).
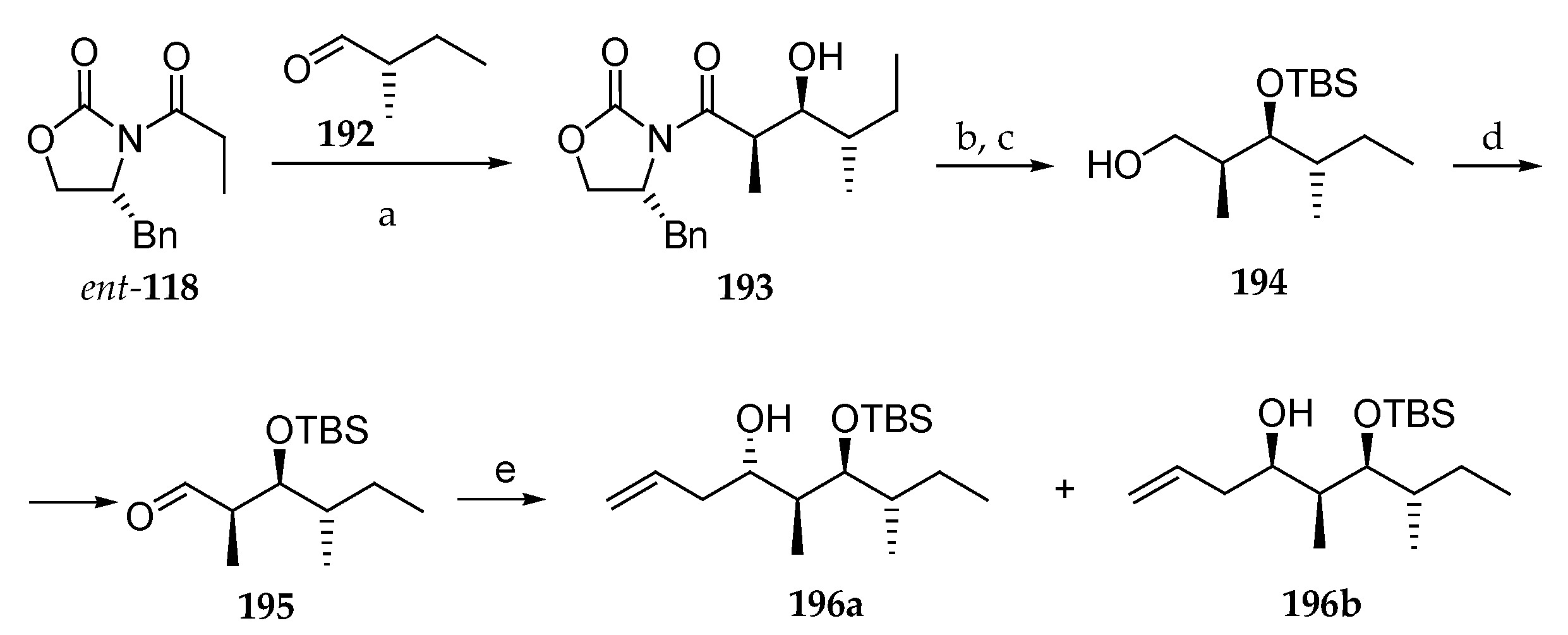
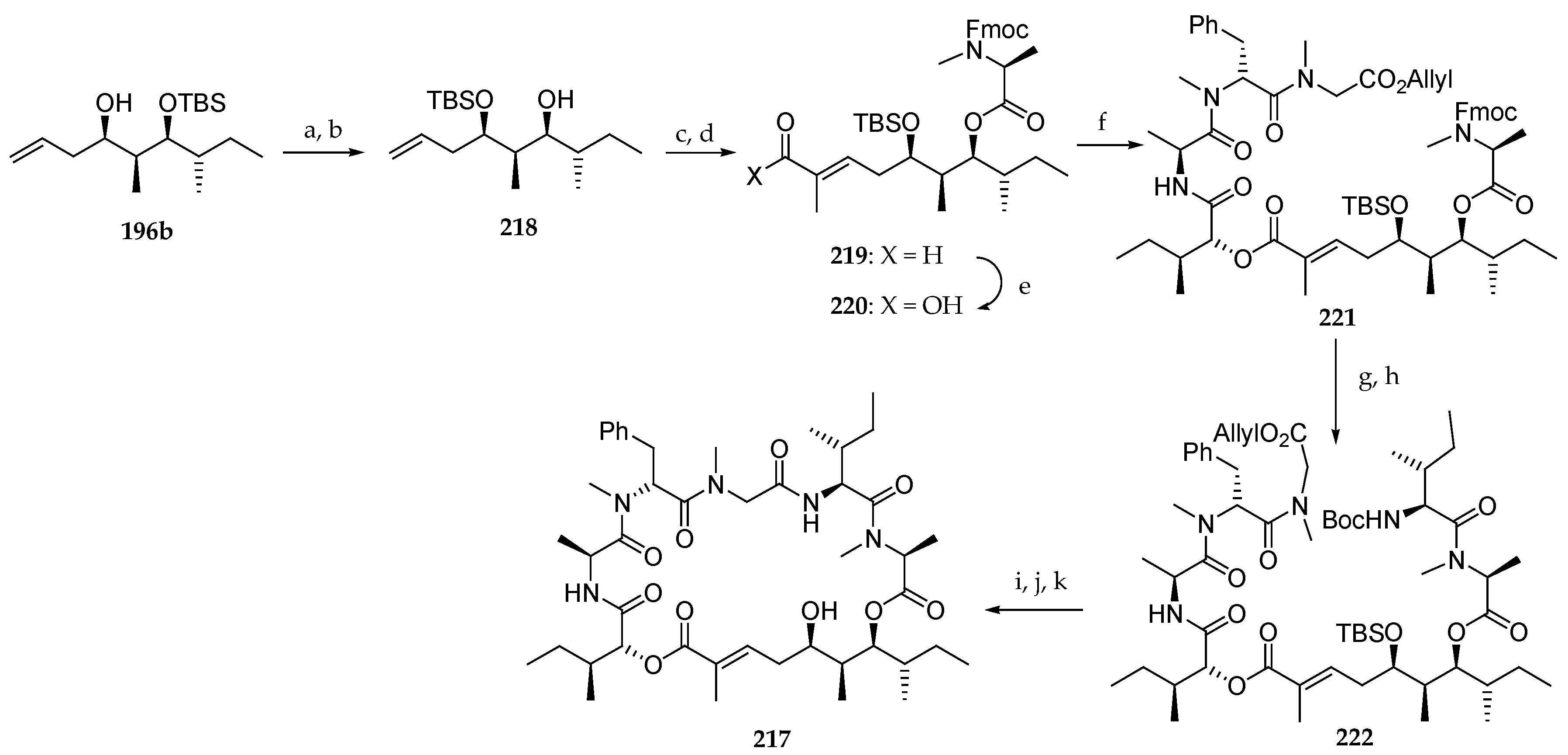
Scheme 28.
Synthesis of homoallylic alcohol 196a. Reagents and conditions: (a) Bu2BOTf, TEA, CH2Cl2, −78 °C, 93%; (b) TBSOTf, 2,6-lutidine, 0 °C, 95%; (c) LiBH4, MeOH, THF, 0 °C; (d) (COCl)2, DMSO, TEA, −78 °C, 92% (two steps); (e) AllylMgCl (1.5 eq), THF, 78%C, 11% (196a), 81% (196b) or ZnCl2 (2.5 eq), allylMgCl, THF, −78 °C, 90% of 196a and 196b (90:10).
Scheme 28.
Synthesis of homoallylic alcohol 196a. Reagents and conditions: (a) Bu2BOTf, TEA, CH2Cl2, −78 °C, 93%; (b) TBSOTf, 2,6-lutidine, 0 °C, 95%; (c) LiBH4, MeOH, THF, 0 °C; (d) (COCl)2, DMSO, TEA, −78 °C, 92% (two steps); (e) AllylMgCl (1.5 eq), THF, 78%C, 11% (196a), 81% (196b) or ZnCl2 (2.5 eq), allylMgCl, THF, −78 °C, 90% of 196a and 196b (90:10).
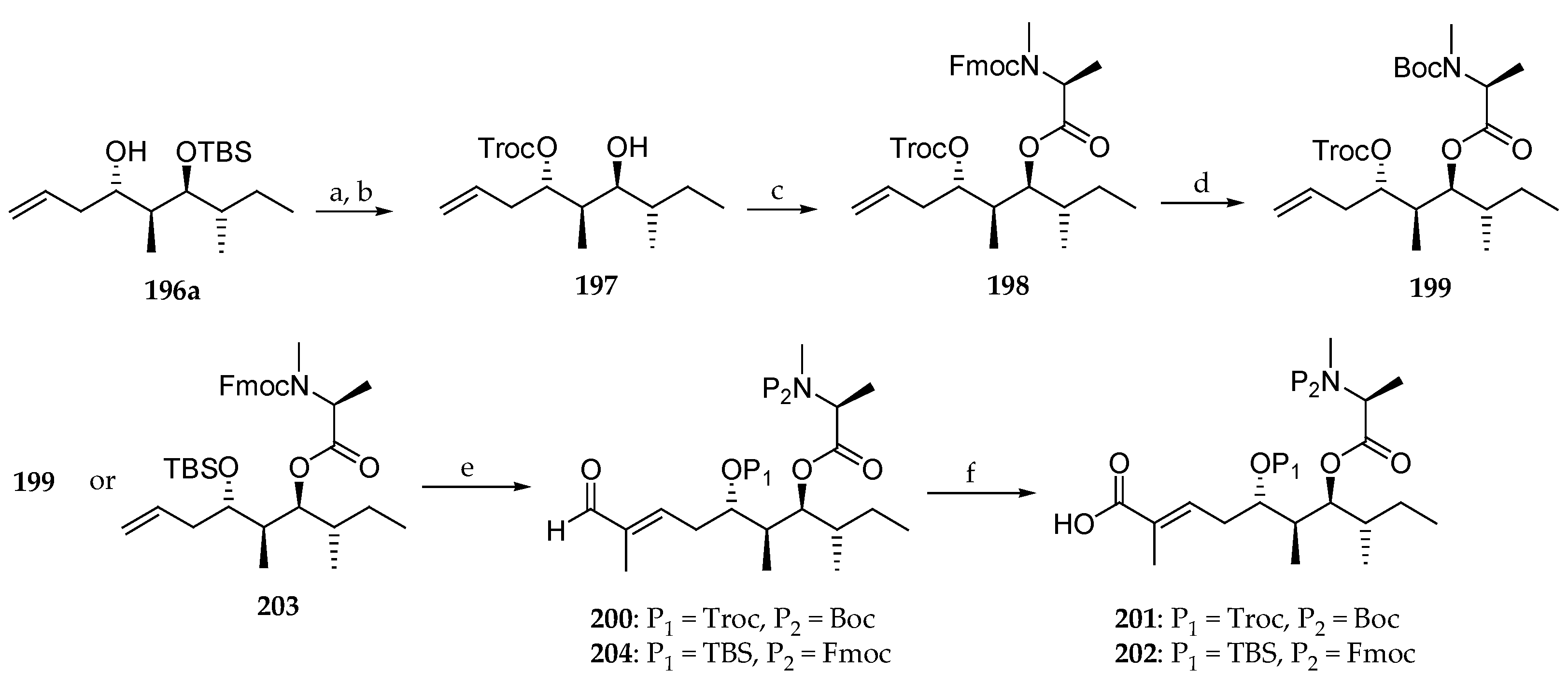
Scheme 29.
Synthesis of polyketides subunits 201 and 202. Reagents and conditions: (a) TrocCl, DMAP (cat.), pyridine, CH2Cl2, 96%; (b) 40% aq HF, CH3CN, 95%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 61%; (d) (i) Et2NH, CH3CN, r.t.; (ii) Boc2O, TEA, CH2Cl2, r.t., 75% (two steps); (e) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, E:Z > 99:1, 81% for 200, 80% for 204; (f) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 93% for 201, 87% for 202.
Scheme 29.
Synthesis of polyketides subunits 201 and 202. Reagents and conditions: (a) TrocCl, DMAP (cat.), pyridine, CH2Cl2, 96%; (b) 40% aq HF, CH3CN, 95%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 61%; (d) (i) Et2NH, CH3CN, r.t.; (ii) Boc2O, TEA, CH2Cl2, r.t., 75% (two steps); (e) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, E:Z > 99:1, 81% for 200, 80% for 204; (f) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 93% for 201, 87% for 202.
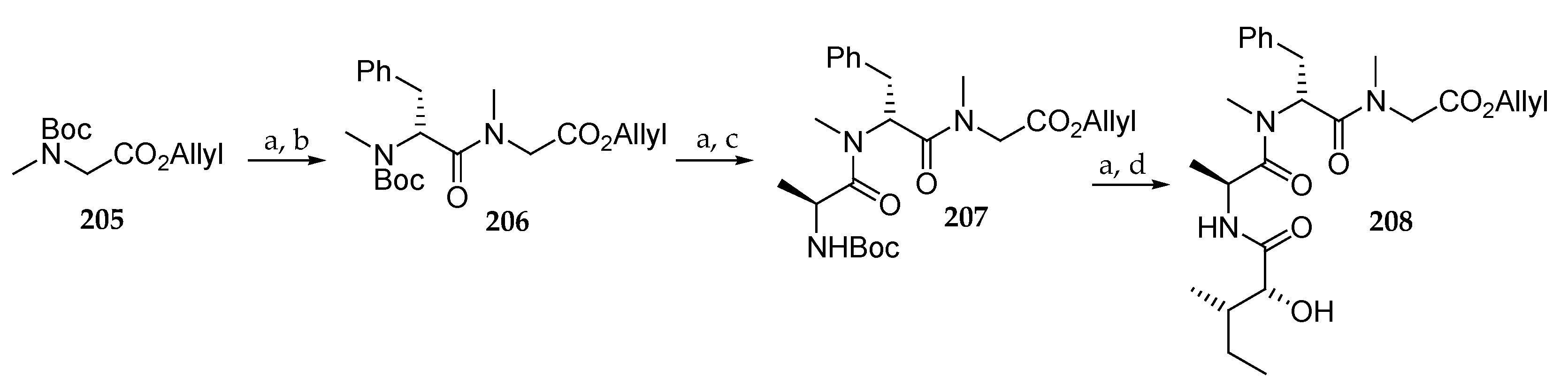
Scheme 30.
Synthesis of tripeptide 208. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C to r.t.; (b) Boc-N-Me-d-Phe-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 89% (2 steps); (c) Boc-L-Ala-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 68% from 206; (d) (2R,3S)-2-hydroxy-3-methylpentanoic acid, HOBt, EDC, DMF, -15 °C to r.t., 56% from 207.
Scheme 30.
Synthesis of tripeptide 208. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C to r.t.; (b) Boc-N-Me-d-Phe-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 89% (2 steps); (c) Boc-L-Ala-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 68% from 206; (d) (2R,3S)-2-hydroxy-3-methylpentanoic acid, HOBt, EDC, DMF, -15 °C to r.t., 56% from 207.
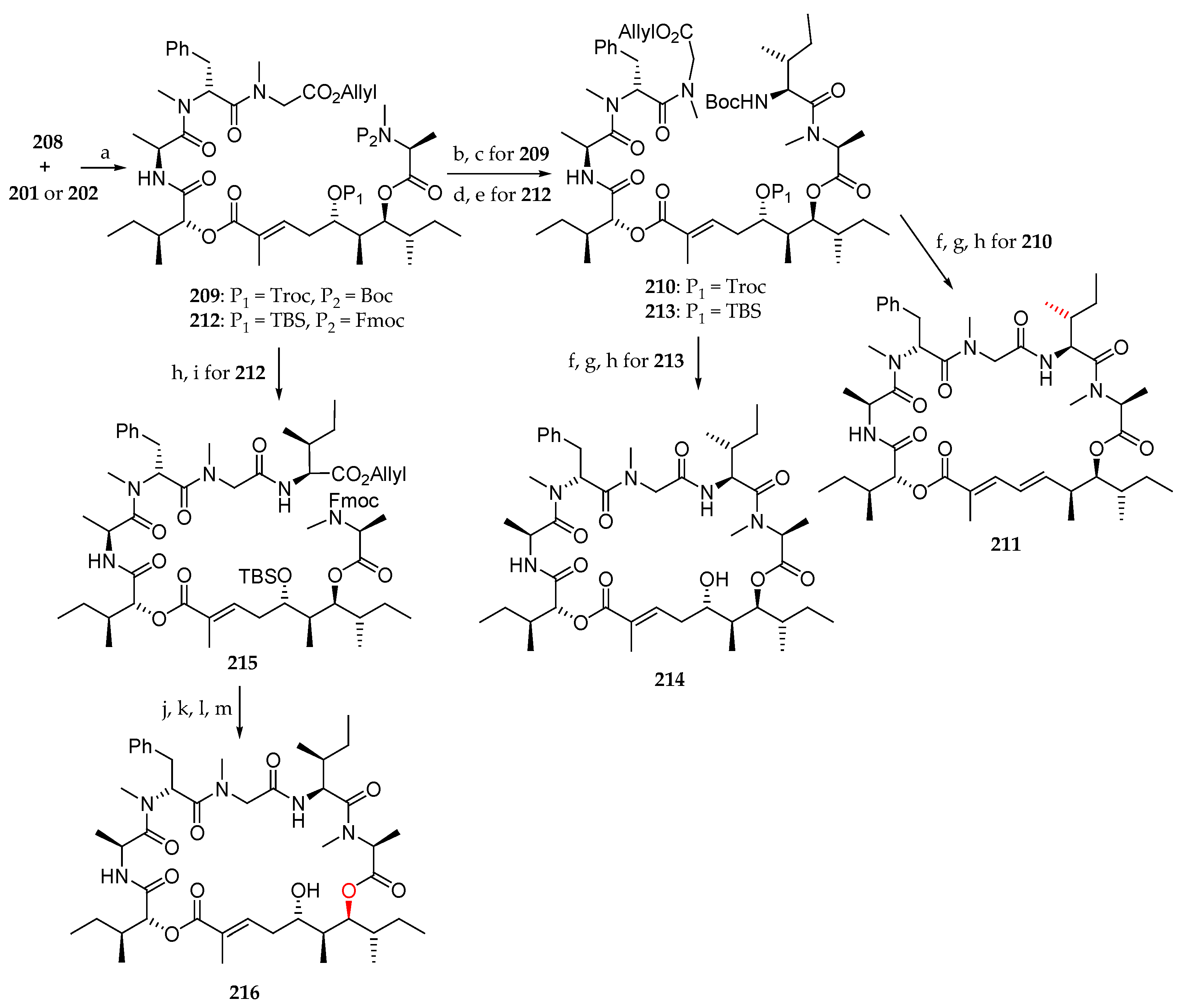
Scheme 31.
Synthesis of lagunamide A analogues 211, 214, and 216. Reagents and conditions: (a) 208, MNBA, DMAP, CH2Cl2, r.t., 66% for 209, 60% for 212; (b) TFA, CH2Cl2, r.t.; (c) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 82% for (2 steps); (d) Et2NH, CH3CN, r.t.; (e) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 76% (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) TFA, CH2Cl2, 0 °C to r.t.; (h) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 22% for 211, 30% for 214 (3 steps). (i) L-lle-O-allyl, HATU, DIPEA, CH2Cl2, r.t., 85% (2 steps). (j) Pd(PPh3)4, PhNHMe, THF, r.t.; (k) Et2NH, CH3CN, r.t.; (l) HATU, DIPEA, CH2Cl2, r.t.; (m) 40% aq HF, CH3CN, 38% (4 steps). MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 31.
Synthesis of lagunamide A analogues 211, 214, and 216. Reagents and conditions: (a) 208, MNBA, DMAP, CH2Cl2, r.t., 66% for 209, 60% for 212; (b) TFA, CH2Cl2, r.t.; (c) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 82% for (2 steps); (d) Et2NH, CH3CN, r.t.; (e) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 76% (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) TFA, CH2Cl2, 0 °C to r.t.; (h) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 22% for 211, 30% for 214 (3 steps). (i) L-lle-O-allyl, HATU, DIPEA, CH2Cl2, r.t., 85% (2 steps). (j) Pd(PPh3)4, PhNHMe, THF, r.t.; (k) Et2NH, CH3CN, r.t.; (l) HATU, DIPEA, CH2Cl2, r.t.; (m) 40% aq HF, CH3CN, 38% (4 steps). MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 32.
Synthesis of lagunamide analogue 217. Reagents and conditions: (a) 40% aq HF, CH3CN, r.t., 92%; (b) TBSOTf, 2,6-lutidine, −78 °C, 75%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 58%; (d) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 83%, E:Z > 99:1; (e) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 78%; (f) 208, MNBA, DMAP, CH2Cl2, r.t., 63%; (g) Et2NH, CH3CN, r.t.; (h) Boc-l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 75% (2 steps); (i) Pd(PPh3)4, PhNHMe, THF, r.t.; (j) TFA, CH2Cl2, 0 °C to r.t.; (k) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 32% (3 steps).
Scheme 32.
Synthesis of lagunamide analogue 217. Reagents and conditions: (a) 40% aq HF, CH3CN, r.t., 92%; (b) TBSOTf, 2,6-lutidine, −78 °C, 75%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 58%; (d) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 83%, E:Z > 99:1; (e) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 78%; (f) 208, MNBA, DMAP, CH2Cl2, r.t., 63%; (g) Et2NH, CH3CN, r.t.; (h) Boc-l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 75% (2 steps); (i) Pd(PPh3)4, PhNHMe, THF, r.t.; (j) TFA, CH2Cl2, 0 °C to r.t.; (k) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 32% (3 steps).
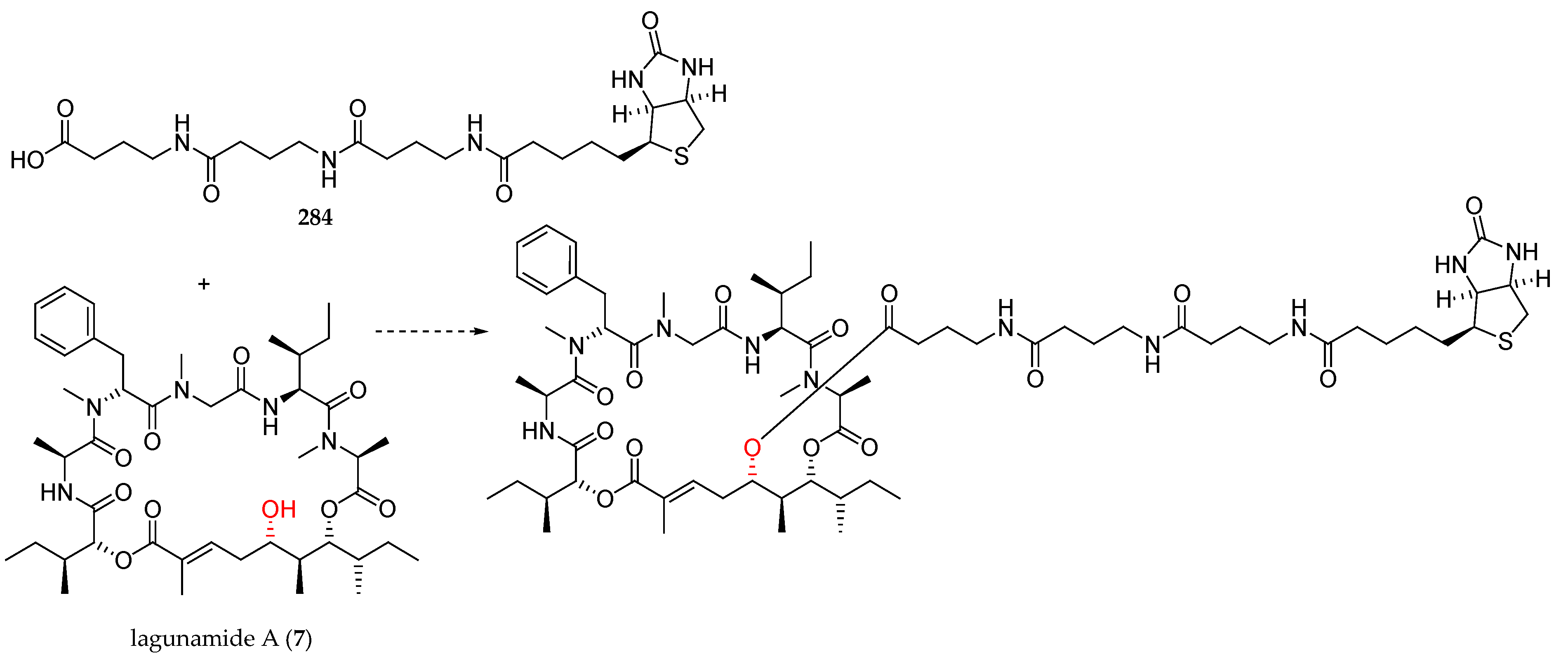
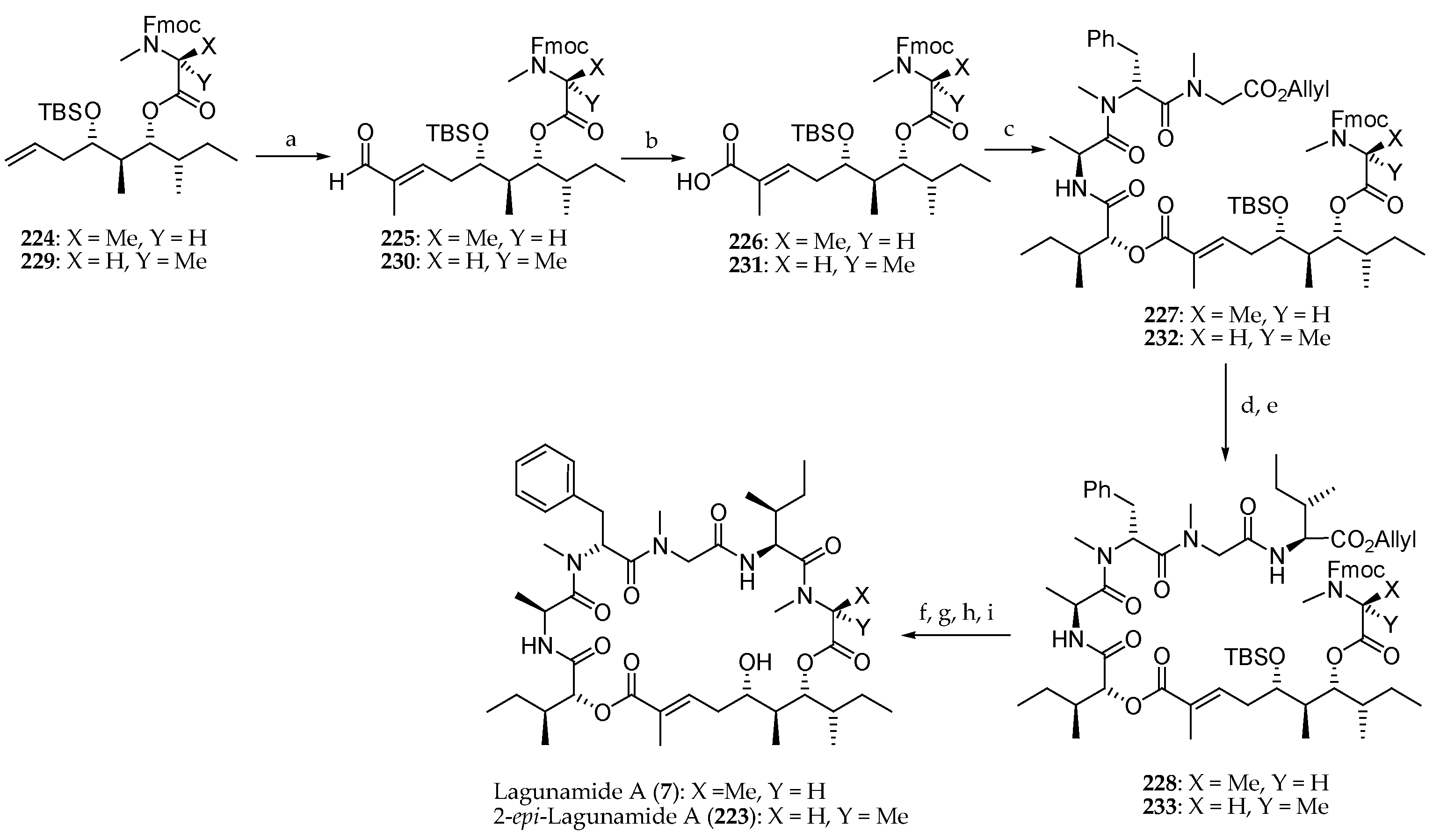
Scheme 33.
Synthesis of lagunamide A (7) and 2-epi-lagunamide A (223). Reagents and conditions: (a) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 87% for 225, 83% for 230, E:Z > 99:1; (b) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 80% for 226, 81% for 231; (c) 208, MNBA, DMAP, CH2Cl2, r.t., 56% for 227, 58% for 232. (d) Pd(PPh3)4; (e) Boc l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 91% for 228, 80% for 233 (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) Et2NH, CH3CN, r.t.; (h) HATU, DIPEA, CH2Cl2, r.t.; (i) 40% aq HF, CH3CN, 38% for 7, 45% for 223 (4 steps).
Scheme 33.
Synthesis of lagunamide A (7) and 2-epi-lagunamide A (223). Reagents and conditions: (a) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 87% for 225, 83% for 230, E:Z > 99:1; (b) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 80% for 226, 81% for 231; (c) 208, MNBA, DMAP, CH2Cl2, r.t., 56% for 227, 58% for 232. (d) Pd(PPh3)4; (e) Boc l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 91% for 228, 80% for 233 (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) Et2NH, CH3CN, r.t.; (h) HATU, DIPEA, CH2Cl2, r.t.; (i) 40% aq HF, CH3CN, 38% for 7, 45% for 223 (4 steps).
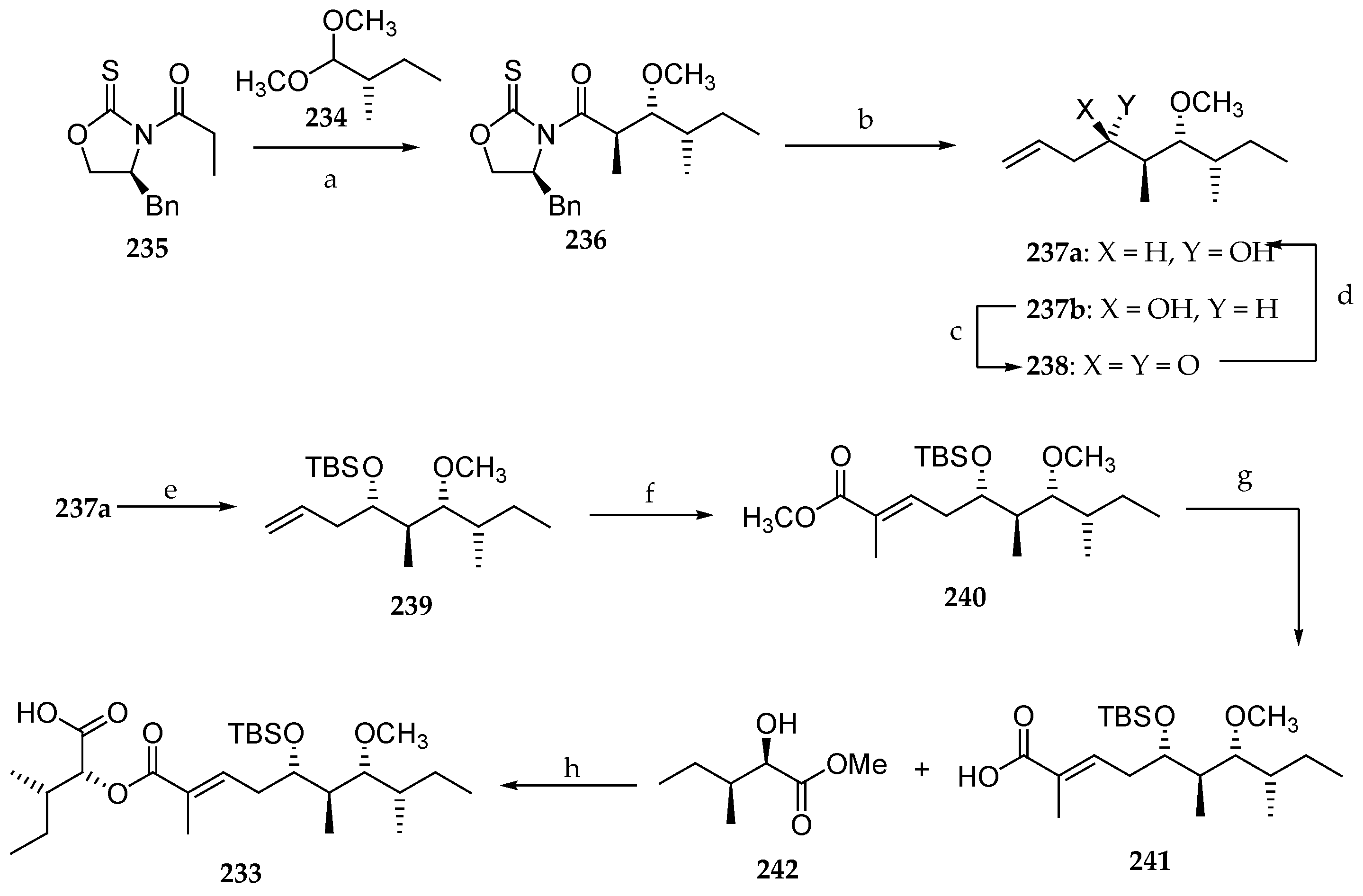
Scheme 34.
Synthesis of C27–C45 fragment of lagunamide A 233. Reagents and conditions: (a) TiCl4, DIPEA, CH2Cl2, 0 °C, 1 h, then SnCl4, 234, −84 °C to 0 °C, 62%; (b) (i) DiBAL-H, CH2Cl2, −84 °C, 10 min, (ii) allylmagnesium chloride, THF, 0 °C, 1 h, 99% (237a/237b = 2:3) (two steps); (c) Dess–Martin periodinane, CH2Cl2, 0 °C, 30 min, 74%; (d) NaBH4, CH3OH, 0 °C, 15 min, 95% (237a/237b = 8:1); (e) TBSOTf, Et3N, CH2Cl2, 0 °C, 1 h, 94%; (f) methyl methacrylate, Grubbs II catalyst, CH2Cl2, reflux, 16 h, 59%; (g) aq KOH, THF, r.t., 3 d, 97%; (h) 242, DCC, DMAP, CH2Cl2, r.t., 22 h, 87%.
Scheme 34.
Synthesis of C27–C45 fragment of lagunamide A 233. Reagents and conditions: (a) TiCl4, DIPEA, CH2Cl2, 0 °C, 1 h, then SnCl4, 234, −84 °C to 0 °C, 62%; (b) (i) DiBAL-H, CH2Cl2, −84 °C, 10 min, (ii) allylmagnesium chloride, THF, 0 °C, 1 h, 99% (237a/237b = 2:3) (two steps); (c) Dess–Martin periodinane, CH2Cl2, 0 °C, 30 min, 74%; (d) NaBH4, CH3OH, 0 °C, 15 min, 95% (237a/237b = 8:1); (e) TBSOTf, Et3N, CH2Cl2, 0 °C, 1 h, 94%; (f) methyl methacrylate, Grubbs II catalyst, CH2Cl2, reflux, 16 h, 59%; (g) aq KOH, THF, r.t., 3 d, 97%; (h) 242, DCC, DMAP, CH2Cl2, r.t., 22 h, 87%.
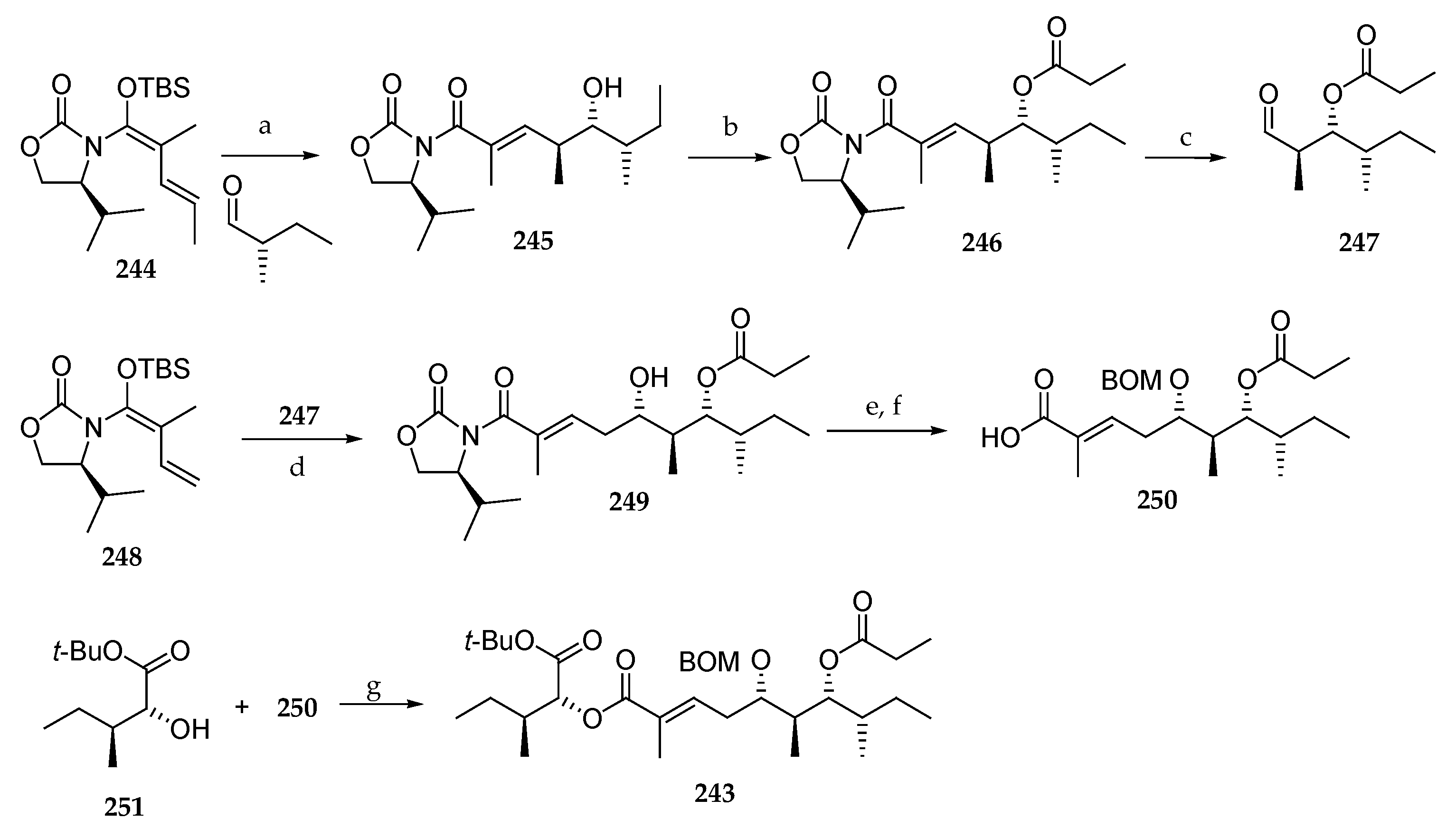
Scheme 35.
Synthesis of fragment C27–C45 243 of lagunamide A (7). Reagents and conditions: (a) TiCl4, CH2Cl2, −78 °C, 26 h, 96%, dr > 98:2; (b) CH3CH2COCl, pyridine, DMAP, CH2Cl2, 25 °C, 13 h, 97%; (c) (i) O3, CH2Cl2, −78 °C, 0.5 h, (ii) Me2S, 95%; (d) TiCl4, PhMe, H2O (10 mol%), −78 to −40 °C, 72 h, 48%, dr = 91:9; (e) BOMCl, DIPEA, TBAI, CH2Cl2, 0 °C, 9 h, 93%; (f) LiOOH, THF, H2O, 0 °C, 1 h; (g) DCC, DMAP, CH2Cl2, 20 °C, 14 h, 88%.
Scheme 35.
Synthesis of fragment C27–C45 243 of lagunamide A (7). Reagents and conditions: (a) TiCl4, CH2Cl2, −78 °C, 26 h, 96%, dr > 98:2; (b) CH3CH2COCl, pyridine, DMAP, CH2Cl2, 25 °C, 13 h, 97%; (c) (i) O3, CH2Cl2, −78 °C, 0.5 h, (ii) Me2S, 95%; (d) TiCl4, PhMe, H2O (10 mol%), −78 to −40 °C, 72 h, 48%, dr = 91:9; (e) BOMCl, DIPEA, TBAI, CH2Cl2, 0 °C, 9 h, 93%; (f) LiOOH, THF, H2O, 0 °C, 1 h; (g) DCC, DMAP, CH2Cl2, 20 °C, 14 h, 88%.
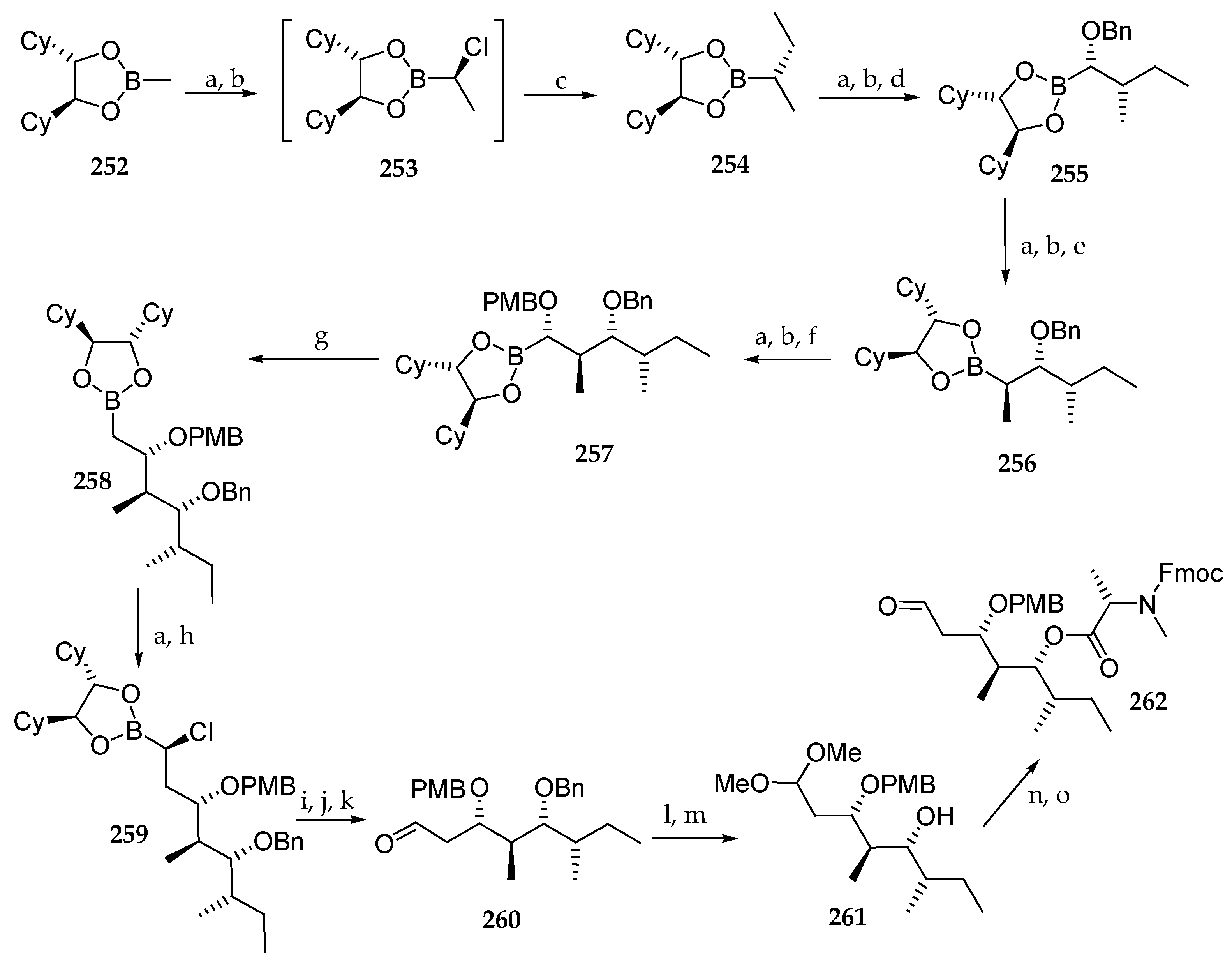
Scheme 36.
Synthesis of aldehyde 262. Reagents and conditions: (a) 3 eq CH2Cl2, 1.25 eq LDA; (b) 2 eq ZnCl2; (c) 2.5 eq EtMgBr, THF, 99% (3 steps); (d) 1.3 eq NaOBn 74% from 254; (e) 2.5 eq MeMgCl, THF, 14 d, 99% from 255; (f) 1.2 eq NaOPMB, THF, DMSO, 66%; (g) 1.25 eq CH2Br2, 1.2 eq n-BuLi, THF, −60 °C to r.t., 98% from 257; (h) 4.0 eq ZnCl2; THF, quant. from 258; (i) 2 eq Na2CO3, 2 eq H2O2; (j) 0.1 eq NaI, 2.0 eq Na2S2O3; (k) MeB(OH)2, MgSO4; (l) 0.1 eq CSA, CH2Cl2, MeOH, 17 h, 96%; (m) Raney-Nickel, H2 (1 bar), EtOH, 2 d, 96%; (n) 2.5 eq Fmoc-N-Me-Ala-Cl, 5 eq DIPEA, CH2Cl2, 20 h, 76%; (o) Amberlist® 15, acetone, 2 h, quant.
Scheme 36.
Synthesis of aldehyde 262. Reagents and conditions: (a) 3 eq CH2Cl2, 1.25 eq LDA; (b) 2 eq ZnCl2; (c) 2.5 eq EtMgBr, THF, 99% (3 steps); (d) 1.3 eq NaOBn 74% from 254; (e) 2.5 eq MeMgCl, THF, 14 d, 99% from 255; (f) 1.2 eq NaOPMB, THF, DMSO, 66%; (g) 1.25 eq CH2Br2, 1.2 eq n-BuLi, THF, −60 °C to r.t., 98% from 257; (h) 4.0 eq ZnCl2; THF, quant. from 258; (i) 2 eq Na2CO3, 2 eq H2O2; (j) 0.1 eq NaI, 2.0 eq Na2S2O3; (k) MeB(OH)2, MgSO4; (l) 0.1 eq CSA, CH2Cl2, MeOH, 17 h, 96%; (m) Raney-Nickel, H2 (1 bar), EtOH, 2 d, 96%; (n) 2.5 eq Fmoc-N-Me-Ala-Cl, 5 eq DIPEA, CH2Cl2, 20 h, 76%; (o) Amberlist® 15, acetone, 2 h, quant.
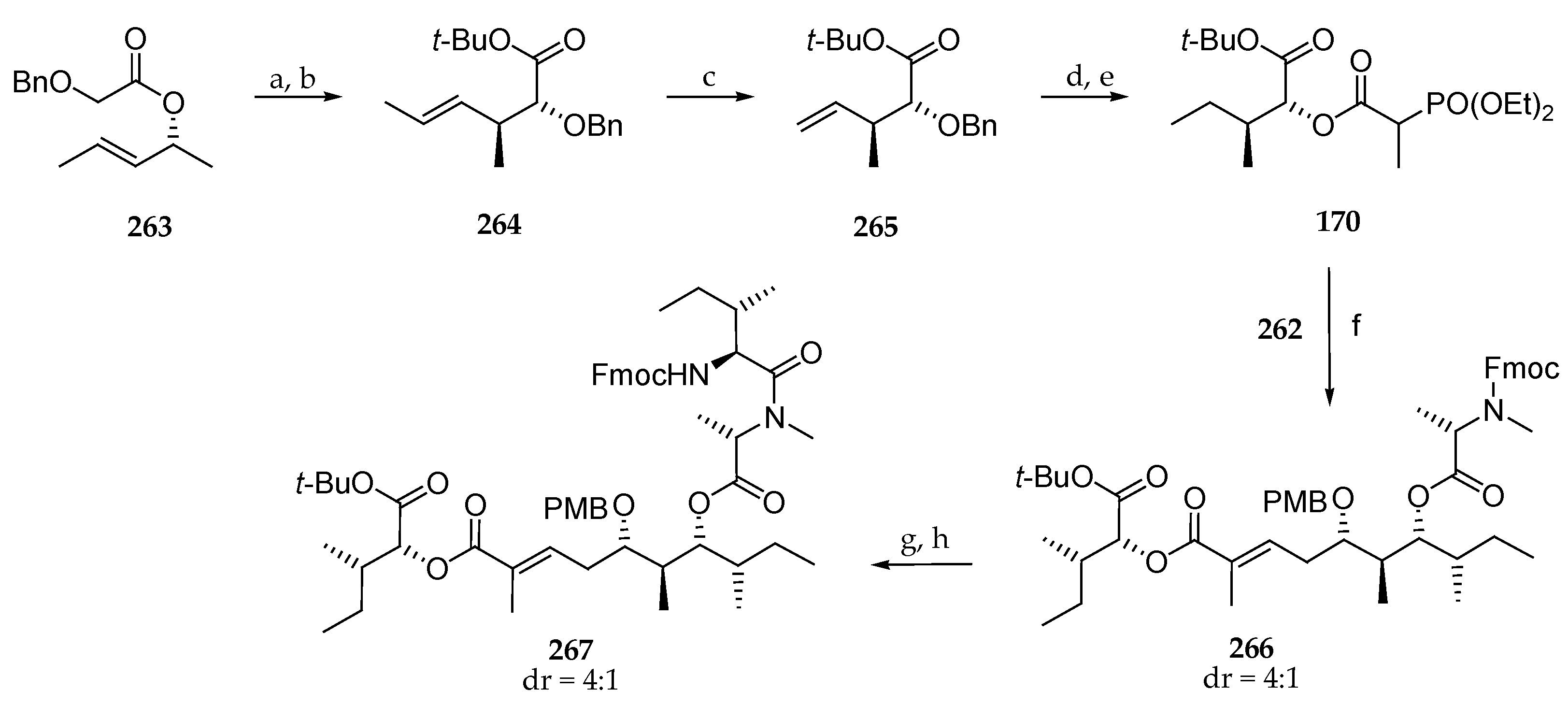
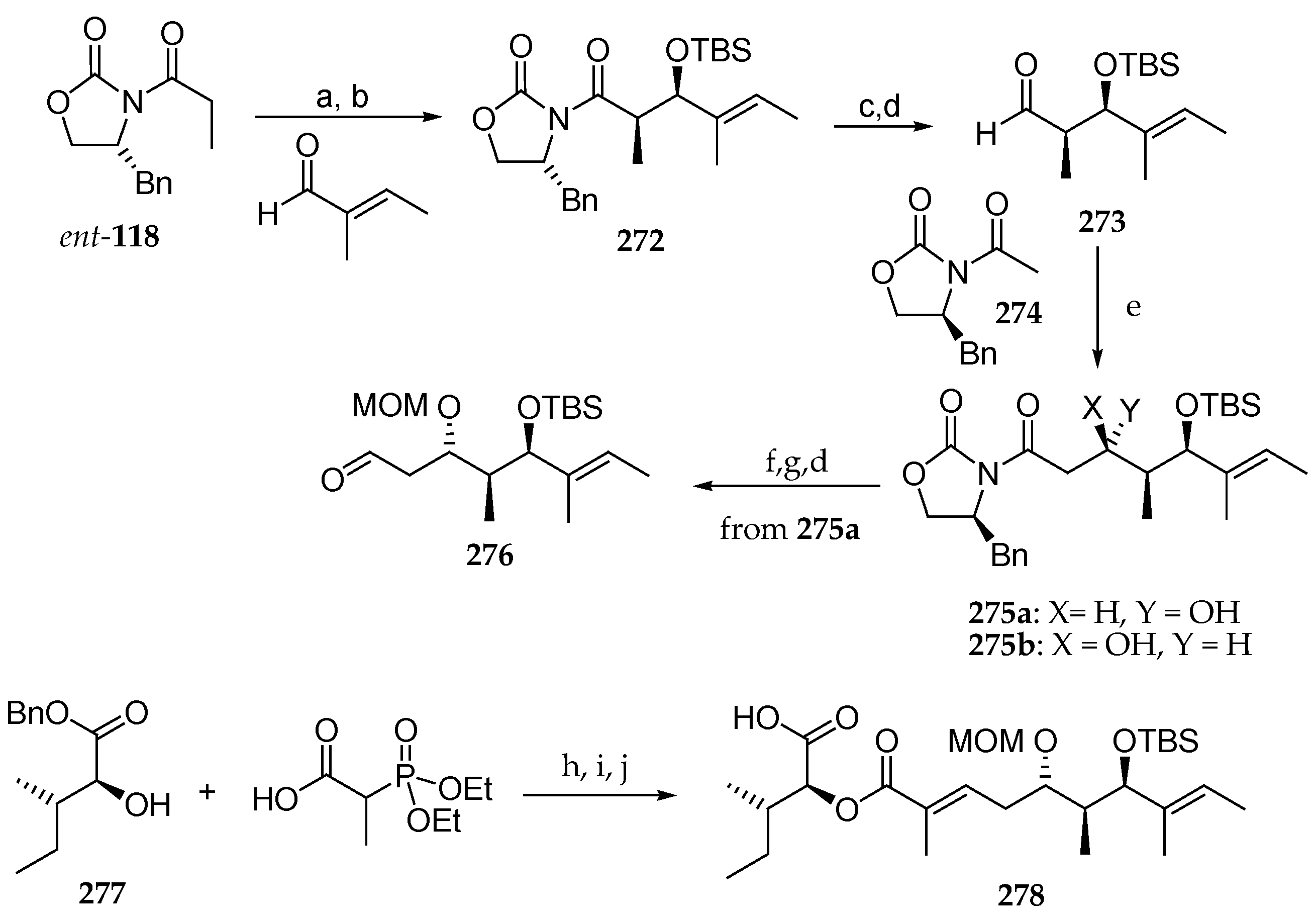
Scheme 37.
Synthesis of phosphonate 170. Reagents and conditions: (a) 1.25 eq LDA, 3 eq TMSCl, 10 min −78 °C, THF, 2 h, r.t.; (b) 4.0 eq Boc2O, 0.5 eq DMAP, t-butanol, r.t., 73%; (c) ethene (2 bar), 5 mol% Grubbs II, toluene, 70 °C, 19 h, 77%; (d) H2, Pd/C, MeOH; (e) (EtO)2P(O)CH(CH3)CO2H, 0.1 eq DMPA, 1.3 eq collidine, 1.3 EDC∙HCl, CH2Cl2, 3 d, 90%; (f) 1.2 equiv 170, 1.24 eq HFIP, 1.18 eq n-BuLi, DME, 4 d, 65%; (g) 80 eq Et2NH, MeCN; (h) 3.0 eq Fmoc-N-Ile-Cl, 6.0 eq DIPE, CH2Cl2, 91%.
Scheme 37.
Synthesis of phosphonate 170. Reagents and conditions: (a) 1.25 eq LDA, 3 eq TMSCl, 10 min −78 °C, THF, 2 h, r.t.; (b) 4.0 eq Boc2O, 0.5 eq DMAP, t-butanol, r.t., 73%; (c) ethene (2 bar), 5 mol% Grubbs II, toluene, 70 °C, 19 h, 77%; (d) H2, Pd/C, MeOH; (e) (EtO)2P(O)CH(CH3)CO2H, 0.1 eq DMPA, 1.3 eq collidine, 1.3 EDC∙HCl, CH2Cl2, 3 d, 90%; (f) 1.2 equiv 170, 1.24 eq HFIP, 1.18 eq n-BuLi, DME, 4 d, 65%; (g) 80 eq Et2NH, MeCN; (h) 3.0 eq Fmoc-N-Ile-Cl, 6.0 eq DIPE, CH2Cl2, 91%.
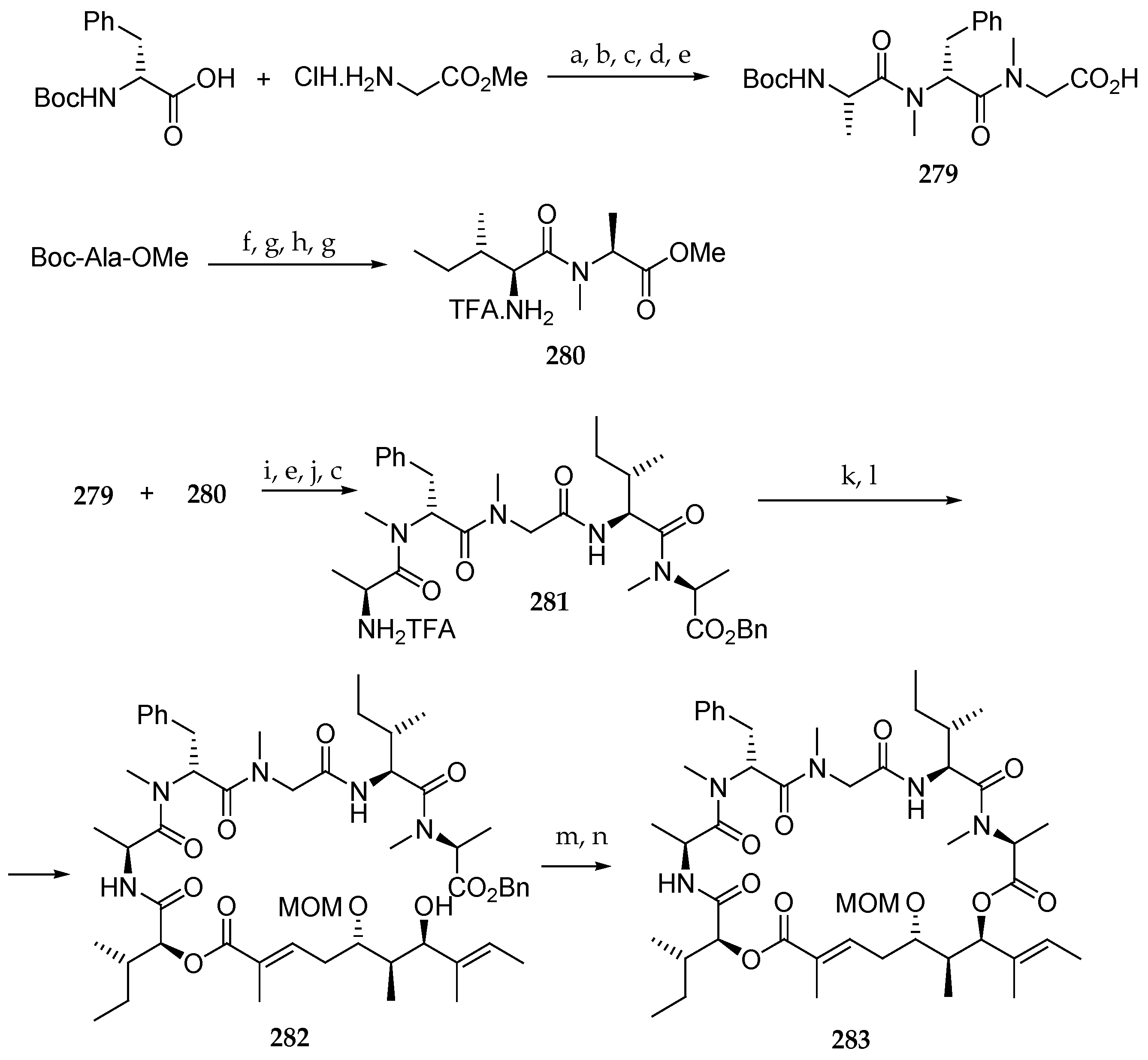
Scheme 38.
Reagents and conditions: (a) 1 eq Boc-N-Me-D-PhOH, 1 eq EDC∙HCl, HOBt, 2 eq DIPEA, CH2Cl2, 82%; (b) HCl, dioxane; (c) Boc-L-Ala-OH, EDC∙HCl, HOAt, DIPEA, DMF; (d) 1.1 eq aq LiOH, 68% (3 steps); (e) Et2NH, MeCN; 1.5 eq 268, 1.5 eq COMU, 3 eq DIPEA, DMF abs 66%; (g) TFA-CH2Cl2 (1:1), 0 °C; (h) 3 eq HATU, 2 eq HOAt, 5 eq collidine, DMF abs, 45%.
Scheme 38.
Reagents and conditions: (a) 1 eq Boc-N-Me-D-PhOH, 1 eq EDC∙HCl, HOBt, 2 eq DIPEA, CH2Cl2, 82%; (b) HCl, dioxane; (c) Boc-L-Ala-OH, EDC∙HCl, HOAt, DIPEA, DMF; (d) 1.1 eq aq LiOH, 68% (3 steps); (e) Et2NH, MeCN; 1.5 eq 268, 1.5 eq COMU, 3 eq DIPEA, DMF abs 66%; (g) TFA-CH2Cl2 (1:1), 0 °C; (h) 3 eq HATU, 2 eq HOAt, 5 eq collidine, DMF abs, 45%.
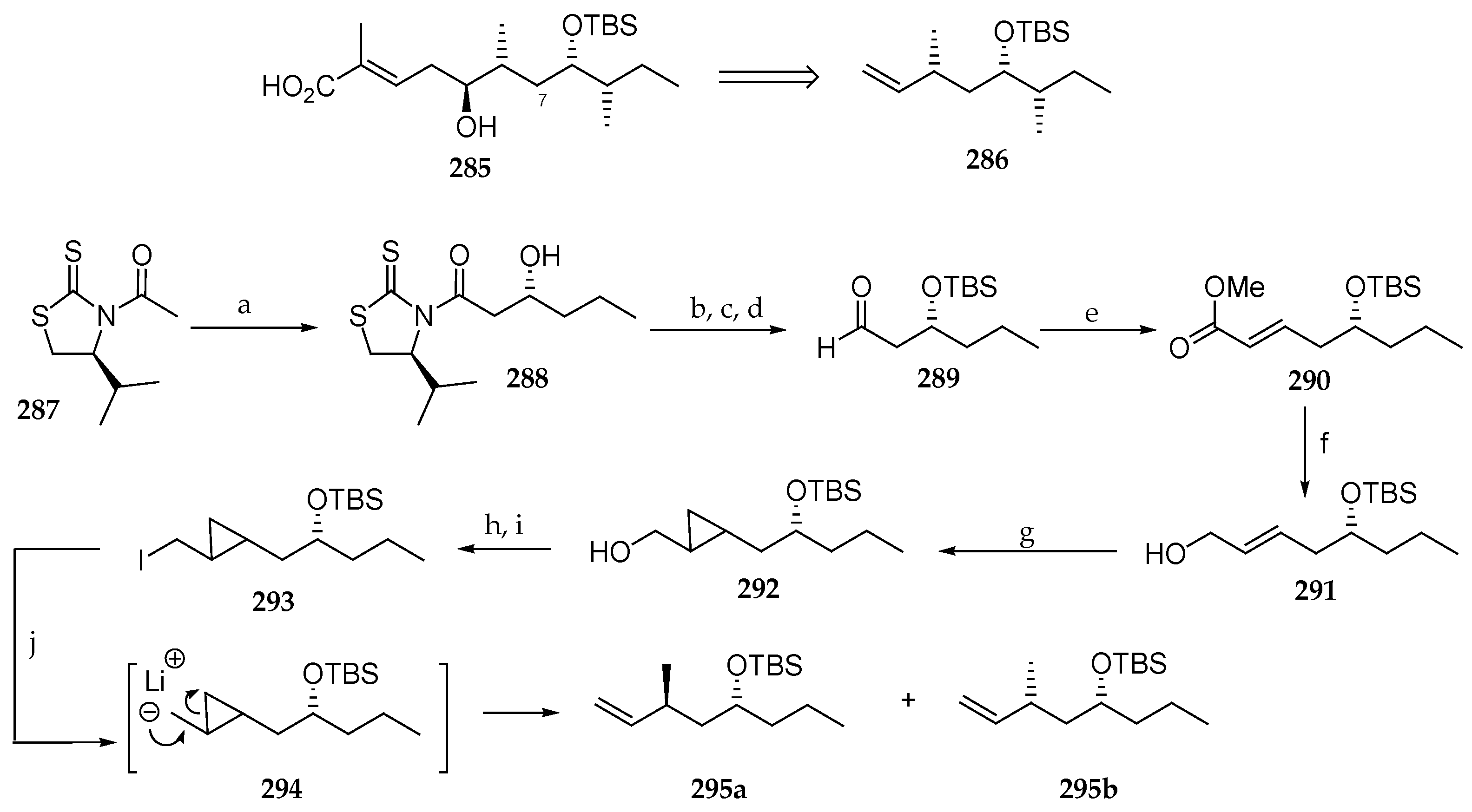
Scheme 39.
Synthesis of the polyketide fragment 278 of lagunamide B (8) by Chakraborty et al. Reagents and conditions: (a) TiCl4, DIPEA, 1-NMP, CH2Cl2, 0 °C, 2 h, 88%, dr 97:3; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 30 min, 93%; (c) LiBH4, THF-MeOH 4:1, 0 °C, 1 h, 81%; (d) py∙SO3, Et3N, DMSO-CH2Cl2 1:0.8, 0 °C, 1 h, 93%; (e) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, 2 h, cooled to −78 °C followed by addition of 273 at −78 °C, 2 h, 63%; (f) MOMCl, DIPEA, CH2Cl2, 0 °C, 1 h, 81%; (g) LiBH4, THF, 0 °C, 1 h, 85%; (h) DIC, DMAP, CH2Cl2, 83%; (i) 276, LiCl, DIEA, ACN, 20 h, 59%; (j) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min.
Scheme 39.
Synthesis of the polyketide fragment 278 of lagunamide B (8) by Chakraborty et al. Reagents and conditions: (a) TiCl4, DIPEA, 1-NMP, CH2Cl2, 0 °C, 2 h, 88%, dr 97:3; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 30 min, 93%; (c) LiBH4, THF-MeOH 4:1, 0 °C, 1 h, 81%; (d) py∙SO3, Et3N, DMSO-CH2Cl2 1:0.8, 0 °C, 1 h, 93%; (e) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, 2 h, cooled to −78 °C followed by addition of 273 at −78 °C, 2 h, 63%; (f) MOMCl, DIPEA, CH2Cl2, 0 °C, 1 h, 81%; (g) LiBH4, THF, 0 °C, 1 h, 85%; (h) DIC, DMAP, CH2Cl2, 83%; (i) 276, LiCl, DIEA, ACN, 20 h, 59%; (j) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min.
Scheme 40.
Reagents and conditions: (a) EDCl, HOBt, DIPEA, CH2Cl2, 8 h, 86%; (b) Ag2O, MeI, DMF, 6 h, 79%; (c) TFA, CH2Cl2, 2 h, 100%; (d) Boc-l-Ala-OH, HATU, DIPEA, CH2Cl2, 8 h, 62%; (e) LiOH, THF, MeOH, H2O, 1 h, 100%; (f) Ag2O, MeI, DMF, 6 h, 74%; (g) TFA, CH2Cl2, 2 h, 100%; (h) Boc-Ile-OH, HATU, DIPEA, CH2Cl2, 8 h, 72%; (i) EDCl, HOBt, DIEA, CH2Cl2, 8 h, 72%; (j) PhCH2OCOCl, Et3N, DMAP, CH2Cl2, 4 h, 78%; (k) 278, EDCl, HOBt, DIEA, CH2Cl2, 8 h, 35%; (l) HF∙pyridine, pyridine, THF, 2 h, 61%; (m) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min; (n) MNBA, DIPEA, CH2Cl2, 48 h, low yield.
Scheme 40.
Reagents and conditions: (a) EDCl, HOBt, DIPEA, CH2Cl2, 8 h, 86%; (b) Ag2O, MeI, DMF, 6 h, 79%; (c) TFA, CH2Cl2, 2 h, 100%; (d) Boc-l-Ala-OH, HATU, DIPEA, CH2Cl2, 8 h, 62%; (e) LiOH, THF, MeOH, H2O, 1 h, 100%; (f) Ag2O, MeI, DMF, 6 h, 74%; (g) TFA, CH2Cl2, 2 h, 100%; (h) Boc-Ile-OH, HATU, DIPEA, CH2Cl2, 8 h, 72%; (i) EDCl, HOBt, DIEA, CH2Cl2, 8 h, 72%; (j) PhCH2OCOCl, Et3N, DMAP, CH2Cl2, 4 h, 78%; (k) 278, EDCl, HOBt, DIEA, CH2Cl2, 8 h, 35%; (l) HF∙pyridine, pyridine, THF, 2 h, 61%; (m) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min; (n) MNBA, DIPEA, CH2Cl2, 48 h, low yield.
Figure 8.
Proposed pre-biotinylated linker for the synthesis of a biosensor for lagunamide A (7).
Figure 8.
Proposed pre-biotinylated linker for the synthesis of a biosensor for lagunamide A (7).
Scheme 41.
Reagents and conditions: (a) TiCl4, LDA (1.2 eq), NMP (1.2 eq), CH2Cl2, then butanal, 70%, dr 20:1; (b) TBSCl, 2,6-lutidine, DMF, 32 °C, 86%; (c) NaBH4, EtOH, 91%; (d) Swern oxidation; (e) MeO2CCH = PPh3, PhMe, 80%; (f) DiBAL-H, CH2Cl2, −78 °C, 92%; (g) Et2Zn, CH2I2, CH2Cl2, 0 °C to r.t., 95%; (h) MsCl, NEt3, CH2Cl2; (i) NaI, acetone, 80% (2 steps); (j) n-BuLi, TMEDA, Et2O, −78 °C, 71%.
Scheme 41.
Reagents and conditions: (a) TiCl4, LDA (1.2 eq), NMP (1.2 eq), CH2Cl2, then butanal, 70%, dr 20:1; (b) TBSCl, 2,6-lutidine, DMF, 32 °C, 86%; (c) NaBH4, EtOH, 91%; (d) Swern oxidation; (e) MeO2CCH = PPh3, PhMe, 80%; (f) DiBAL-H, CH2Cl2, −78 °C, 92%; (g) Et2Zn, CH2I2, CH2Cl2, 0 °C to r.t., 95%; (h) MsCl, NEt3, CH2Cl2; (i) NaI, acetone, 80% (2 steps); (j) n-BuLi, TMEDA, Et2O, −78 °C, 71%.
Table 1.
Marine sources of the aurilide family members.
Table 1.
Marine sources of the aurilide family members.
| Aurilide-Family Member | Year of Isolation | Marine Source | Collection Site |
|---|
| aurilide (1) | 1996 [2] | sea hare Dolabella auricularia | Japanese sea |
| aurilide B (2) | 2006 [3] | cyanobacterium Lyngbya majuscula | Papua New Guinea |
| aurilide C (3) | 2006 [3] | cyanobacterium Lyngbya majuscula | Papua New Guinea |
| kulokekahilide-2 (4) | 2004 [4] | cephalaspidean MolluskPhilinopsis speciosa | |
| palau’amide (5) | 2000 [5] | cyanobacterium Lyngbya (Oscillatoriaceae) | Ulong Channel, Palau |
| odoamide (6) | 2016 [6] | cyanobacterium Okeania sp. | Japanese sea |
| lagunamide A (7) | 2010 [7] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide B (8) | 2010 [7] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide C (9) | 2011 [8] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide D (10) and D’(11) | 2019 [9] | collection of marine cyanobacteria (a mixture of Dichothrix sp. and Lyngbyasp. in a ratio of 1:1 with minor amount of Rivularia sp. present) | Loggerhead Key in the Dry Tortugas in Florida |
Table 2.
Pentapeptide side chains in the different members of the aurilide family.
Table 2.
Pentapeptide side chains in the different members of the aurilide family.
| | R5 | R4 | R2 | C11′ Configuration | X | Y |
|---|
| Aurilide (1) | i-Pr | i-Bu | i-Pr | R | Me | H |
| Aurilide B (2) | i-Pr | (R)-sec-butyl | i-Pr | S | Me | H |
| Aurilide C (3) | i-Pr | (R)-sec-butyl | i-Pr | S | Me | H |
| Kulokekahilide-2 (4) | Me | Bn | (S)-sec-butyl | R | H | Me |
| Palau’amide (5) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Odoamide (6) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide A (7) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide B (8) | Me | Bn | (R)-sec-butyl | R | Me | H |
| Lagunamide C (9) | Me | Bn | (R)-sec-butyl | R | Me | H |
| Lagunamide D (10) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide D’ (11) | Me | Bn | (S)-sec-butyl | R | Me | H |
Table 3.
Cytotoxic activities of the natural aurilide class members (IC50 values in nM, *(LC50) in nM).
Table 3.
Cytotoxic activities of the natural aurilide class members (IC50 values in nM, *(LC50) in nM).
| | HeLaS3 | P388 | BJ | BJ Shp
53 | PC
3 | SK-OV-3 | HCT8 | NCI-H460 | Neuro-2a * | MDA-MB-435 | A-10 | KB | A549 |
|---|
| Aurilide (1) [2] | 11 | | | | | | | | | | | | |
| Aurilide B (2) [3] | | | | | | | | 10 | 40 | | | | |
| Aurilide C (3) [3] | | | | | | | | 50 | 130 | | | | |
| Kulokekalide-2 (4) [4] | | 4.2 | | | | 7.5 | | | | 14.6 | 59.1 | | |
| Palau’amide (5) [5] | | | | | | | | | | | | 13 | |
| Odoamide (6) [6,10] | 26.3 | | | | | | | | | | | | 4.2 |
| Lagunamide A (7) [7,11] | | 6.4 | 20.2 | 58.8 | 2.5 | 3.8 | 1.6 | | | | | | 2.9 |
| Lagunamide B (8) [7,11] | | 20.5 | | | | | 5.2 | | | | | | |
| Lagunamide C (9) [8] | | 24.4 | | | 2.6 | 4.5 | 2.1 | | | | | | 2.4 |
| Lagunamide D (10) [9] | | | | | | | | | | | | | 7.1 |
| Lagunamide D’(11) [9] | | | | | | | | | | | | | 68.2 |
Table 4.
Tetrapeptide sequences used for the library of aurilide derivatives.
Table 4.
Tetrapeptide sequences used for the library of aurilide derivatives.
| | Sequence AA4-AA3-AA2-AA1 | Purity (%) |
|---|
| 1 | d-Val-N-Me-l-Leu-Sar-d-Val | 41 |
| 2 | d-Val-N-Me-l-Leu-Sar-l-Val | 26 |
| 3 | d-Val-N-Me-d-Leu-Sar-d-Val | 42 |
| 4 | d-Val-N-Me-d-Leu-Sar-l-Val | 31 |
| 5 | l-Val-N-Me-l-Leu-Sar-d-Val | 45 |
| 6 | l-Val-N-Me-l-Leu-Sar-l-Val | 42 |
| 7 | l-Val-N-Me-d-Leu-Sar-d-Val | 51 |
| 8 | l-Val-N-Me-d-Leu-Sar-l-Val | 57 |
| 9 | d-Val-l-Leu-Gly-d-Val | 25 |
| 10 | d-Val-l-Leu-Gly-l-Val | 47 |
| 11 | d-Val-d-Leu-Gly-d-Val | 36 |
| 12 | d-Val-D-Leu-Gly-l-Val | 42 |
| 13 | l-Val-l-Leu-Gly-d-Val | 36 |
| 14 | l-Val-l-Leu-Gly-l-Val | 58 |
| 15 | l-Val-d-Leu-Gly-d-Val | 48 |
| 16 | l-Val-d-Leu-Gly-l-Val | 45 |
| 17 | l-Val-Sar-N-Me-d-Leu-l-Val | 29 |
| 18 | N-Me-d-Leu-Sar-l-Val-l-Val | 27 |
| 19 | N-Me-d-Leu-l-Val-Sar-l-Val | 20 |
| 20 | Sar-l-Val-N-Me-d-Leu-l-Val | 22 |
Table 5.
Structures of aurilide (1) and analogues with corresponding cytotoxicity against HeLa S3 cells.
Table 5.
Structures of aurilide (1) and analogues with corresponding cytotoxicity against HeLa S3 cells.
| | R1 | R2 | R3 | X | Y | IC50 (ng/mL) |
|---|
| aurilide (1) | OH | Me | iPr | iPr | H | 2.4–11 |
| 40 | H | Me | iPr | iPr | H | 17 |
| 45 | OH | Me | iPr | H | iPr | >4000 |
| 46 | OH | (CH2)4NHBoc | iPr | iPr | H | 20 |
| 47 | OH | Me | iPr | (CH2)4NHBoc | H | 260 |
| 48 | OH | Me | (CH2)4NHBoc | iPr | H | 32 |
| 49 | OCO(CH2)5NHFmoc | Me | iPr | iPr | H | 420 |
| 50 | OCO(CH2)5NHCO(CH2)2CO2H | Me | iPr | iPr | H | 140 |
| 51 | OCO(CH2)5NHCO(CH2)2CO2H | Me | iPr | H | iPr | >10,000 |
Table 6.
Cytotoxicity of kulokekahilide-2 (4) and its 26-membered ring analogues.
Table 6.
Cytotoxicity of kulokekahilide-2 (4) and its 26-membered ring analogues.
| | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | P388
IC50 (μg/mL) | HeLa
IC50 (μg/mL) | A549
IC50 (nM) | K562
IC50 (nM) | MCF
7IC50 (nM) |
|---|
| Natural | | | | | | | | | 0.004 | - | | | |
| 69 | Me | H | H | Bn | H | Me | H | H | 0.016 | 0.0032 | 0.0021 | 0.0031 | 0.22 |
| 63 | H | Me | Bn | H | Me | H | H | H | 10 | >10 | | | |
| 2′-epi-63 | H | Me | Bn | H | H | Me | H | H | >10 | >10 | | | |
| 70 | Me | H | Bn | H | H | Me | H | H | 0.40 | 0.039 | | | |
| 71 | Me | H | H | Bn | Me | H | H | H | 0.016 | 0.0053 | | | |
| 72 | H | Me | H | Bn | H | Me | H | H | 4.5 | 2 | 1045 | 584 | 1645 |
| 73 | Me | H | H | Bn | Me | H | H | MTM | 0.08 | 0.016 | | | |
| 74 | Me | H | H | Bn | H | Me | H | MTM | 0.016 | 0.0072 | 0.00019 | 0.0063 | 6.3 |
| 75 | Me | H | Bn | H | H | Me | H | MTM | 0.40 | 0.039 | | | |
| 76 | Me | H | H | Bn | H | Me | Me | MTM | 0.04 | 0.04 | | | |
| 77 | H | Me | H | Bn | H | Me | H | MTM | >10 | >10 | 4336 | 1189 | >10,000 |
| 78 | Me | H | H | pClBn | H | Me | H | H | 0.0072 | 0.0014 | 0.000010 | 0.000011 | 0.0030 |
| 79 | Me | H | H | pClBn | H | Me | H | MTM | 0.016 | 0.0072 | 0.73 | 7.1 | 6.5 |
| 80 | Me | H | H | Bn | H | Me | Me | H | 0.0072 | 0.0072 | | | |
| 81 | Me | H | Bn | H | H | Bn | H | MTM | 0.89 | 0.89 | | | |
| 82 | Me | H | H | Bn | H | Me | H | Bz | - | - | 0.00058 | 1.8 | 2.9 |
Table 7.
Cytotoxicity of kulokekahilide-2 (4) 24-membered ring analogues.
Table 7.
Cytotoxicity of kulokekahilide-2 (4) 24-membered ring analogues.
| | R1 | R2 | R3 | R4 | R5 | R6 | P388
(μg/mL) | HeLa
(μg/mL) | A549 * | K562 * | MCF7 * |
|---|
| 83 | Me | H | H | Bn | H | Me | 0.0072 | 0.04 | 0.0014 | 0.012 | 0.29 |
| 84 | H | Me | H | Bn | H | Me | 4.5 | 2 | 1077 | 925 | 3692 |
| 85 | Me | H | H | pClBn | H | Me | 0.016 | 0.0014 | 0.000020 | 0.000216 | 0.025 |
| 86 ** | Me | H | Bn | H | H | Bn | 0.08 | 0.08 | | | |






















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



