2.1. Structure Elucidation
Compound
1 was isolated as a colorless oil, and its molecular formula was determined to be C
31H
48O
5 using HRESIMS (
m/
z [M + Na]
+ 523.3382, calcd 523.3394), corresponding to eight degrees of unsaturation (DOU). The
1H NMR spectrum of
1 exhibited three singlet methyl groups at
δH 0.77, 0.82, and 1.04; three doublet methyl groups at
δH 1.07, 1.23, and 1.37; and three oxymethines at
δH 4.03, 4.19, and 5.41. Furthermore, unique upfield signals at
δH 0.57 and −0.49 indicated the presence of a cyclopropane. Analysis of the
13C NMR and HSQC spectra revealed the presence of two ester carbons (
δC 174.8 and 171.7), three oxymethine carbons (
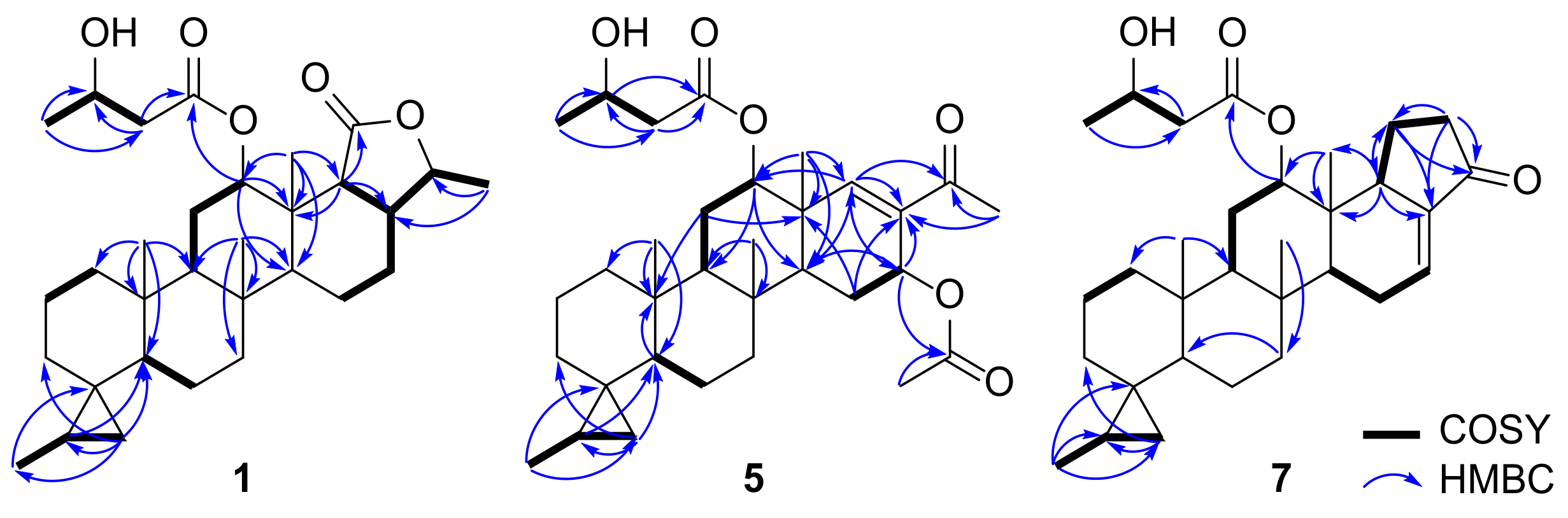
δC 80.4, 74.8, 64.5), 10 methylene carbons, six methine carbons, and six methyl groups. The HMBC data showed notable correlations between the singlet methyl groups and methines from
δH 0.77 to
δC 51.4/50.3,
δH 0.82 to
δC 54.3/51.4, and
δH 1.04 to
δC 54.3, which are known as characteristic correlations occurring from the ring junctions of scalarane-type 6/6/6/6 fused-cyclic systems (
Figure 4). Additional HMBC correlations from the doublet methyl group at
δH 1.37 to
δC 80.4/44.9 and from the methine at
δH 2.34 to
δC 174.8/44.9 suggested the existence of a
γ-valerolactone moiety. Therefore, our preliminary findings led to the hypothesis that compound
1 possessed a honulactone A-like scaffold (B+D type shown in
Figure 2) [
12].
While the △17,18-olefin in honulactones is considered one of the structural features that forms the unsaturated lactone E-ring, the initially identified γ-valerolactone and DOU suggest the possibility of a saturated terminal lactone in compound 1. This speculation was confirmed by the 1H-1H COSY cross peak observed for H2-15–H2-16–H-17–H-18, as well as HMBC correlations from CH3-23 (δH 1.04) to C-18 (δC 52.5) and from H-18 (δH 2.34) to C-13 (δC 38.7). In addition, the cyclopropane moiety inferred from the 1H NMR data was positioned at C-4 based on the HMBC correlations from H2-19 (δH 0.57, and –0.49) to C-3 (δC 33.2)/C-5 (δC 50.3) and from CH3-27 (δH 1.07) to C-4 (δC 22.7), and the spin system for CH3-27–H-20 (δH 0.72)–H2-19 (δH 0.57, −0.49) in the 1H-1H COSY spectrum. Interpretation of the remaining HMBC correlations from CH3-4′ (δH 1.23) to C-2′ (δC 43.3)/C-3′ (δC 64.5), H2-2′ (δH 2.49/2.42) to C-1′ (δC 171.7)/C-3′, and H-12 (δH 5.41) to C-1′ elucidated the 3-hydroxyl butanoate group at C-12.
The trans-fused cyclic scaffold in
1 was determined from the NOESY cross peaks observed between H-11
β (
δH 1.71) and CH
3-21 (
δH 0.82)/CH
3-22 (
δH 0.77), and CH
3-23 and CH
3-21/H-17 (
δH 1.86) (
Figure 5). The NOESY correlations between H-12 and CH
3-23, and H-18 and H-14 (
δH 1.23)/H-24 (
δH 4.03) suggested the
β-orientations of H-12 and CH
3-26, respectively. Moreover, the 20
S* configuration of CH
3-27 was determined based on the NOESY signals observed between H-19
cis (δ
H –0.49) and H-3
β (
δH 1.24)/CH
3-27, and H-19
trans (
δH 0.57) and H
2-6.
Compound
2 was isolated as a colorless oil, and its molecular formula was determined to be C
31H
46O
6 by HRESIMS (
m/
z [M + Na]
+ 537.3167, calcd 537.3187), corresponding to nine degrees of unsaturation. Analysis of the 1D and 2D NMR spectra obtained for
2 indicated a similar carbon framework to
1, but the higher oxidation state of the lactone in E ring appeared as a major difference. HMBC correlations from CH
3-23 (
δH 1.22) to C-18 (
δC 133.7) and from CH
3-26 (
δH 1.56) to C-17 (
δC 162.9) revealed an
α,
β-unsaturated lactone in the E ring, which was responsible for the one degree higher DOU than that of
1. In addition, the
13C chemical shift of C-24 (
δC 104.4) was characteristic of a ketal carbon atom, of which the position was confirmed by HMBC correlations from CH
3-26 to C-24. The
β-configuration of OH-24 was determined by the NOESY correlation observed between H-16
α (
δH 2.28) and CH
3-26 (
Figure S3,
Supplementary Materials).
Compound
3 was isolated as a mixture of two inseparable epimers. The molecular formula of
3 was deduced to be C
31H
44O
6 by HRESIMS (
m/
z [M + Na]
+ 535.3011, calcd 535.3030), corresponding to 10 degrees of unsaturation. An initial inspection of the
13C NMR spectrum revealed that most of the peaks were split into a doublet-like shape, indicating a 1:1 mixture of diastereomers. The 1D and 2D NMR spectra obtained for compound
3 exhibited most of the structural features of
2, except for one more disubstituted olefin observed at
δH (6.38/6.37)/
δC (138.84/138.80) and
δH (6.29/6.25)/
δC (118.6/118.4). The location of the double bond was determined to be △
15,16 using the consecutive
1H-
1H COSY correlations observed for H-14 (
δH 2.69/2.62)–H-15 (
δH 6.38/6.37)–H-16 (
δH 6.29/6.25). The splittings observed in the
13C NMR spectrum were most prominent at CH
3-26 (Δ
δC 1.13 ppm), informing a mixture of C-24 epimers. This phenomenon has often been observed in the case of 24-homoscalaranes, which possess both an △
15,16-olefin and 24-hydroxy pentenolide E-ring [
25,
26]. Since the △
15,16-olefin increases the planarity of the D-ring and renders the C-24 stereocenter more isolated, the 24
R* and 24
S* diastereomers exhibit almost identical spectroscopic and chromatographic behaviors to give an inseparable mixture.
Compound 4 was isolated as an inseparable mixture and its molecular formula was determined to be C32H46O6 by HRESIMS (m/z [M + Na]+ 549.3163, calcd 549.3187), indicating 10 degrees of unsaturation. The NMR spectra of 4 were only discriminated from those of 3 by the extra methylene group observed at δH 1.50 and δC 29.5/29.4, which was also supported by the mass difference of +14. The extra methylene group was observed in the ester side chain located at C-12, which formed a 3-hydroxypentanoate moiety, as supported by the spin system for H2-2′ (δH 2.35)–H-3′ (δH 3.90/3.86)–H2-4′ (δH 1.50)–CH3-5′ (δH 0.95) in the 1H-1H COSY spectrum.
Compound
5 was isolated as a colorless oil. Its molecular formula was determined to be C
32H
48O
6 by HRESIMS (
m/
z [M + Na]
+ 551.3310, calcd 551.3343), corresponding to nine degrees of unsaturation. Our initial analysis of the
1H NMR spectrum obtained for compound
5 indicated the presence of the scalarane-type scaffold: five singlet methyl groups at
δH 0.80, 0.84, 1.06, 2.02, and 2.22; two doublet methyl groups at
δH 1.08 and 1.25; three oxymethines at
δH 4.19, 5.11, and 5.76; unique cyclopropane signals at
δH 0.59 and –0.49; and one singlet olefin at
δH 6.72. The
13C NMR and HSQC spectra showed one ketone (
δC 197.7), two ester carbons (
δC 172.0, 170.2), one trisubstituted olefin carbon (
δC 153.2, 135.1), three oxymethine carbons (
δC 76.8, 65.3, 64.6), eight methylenes, four methines, and seven methyl groups. In addition, the HMBC correlation from the singlet methyl at
δH 2.22 to
δC 197.7 suggested the presence of a methyl ketone moiety, instead of the lactone E-ring observed in compounds
1–
4, leading to the conclusion that
5 had a B+F type skeleton, as shown in
Figure 2.
Detailed interpretation of the combined spectral data of
5 revealed that the features related to the A-B-C ring system were identical to those of
1–
4. As anticipated, the methyl ketone was positioned at C-17 to form an unsaturated ketone in the D ring on the basis of HMBC correlations between CH
3-26 (
δH 2.22) and C-17 (
δC 135.1), and H-18 (
δH 6.72) and C-17/C-24 (
δC 197.7) (
Figure 4). Moreover, the
1H-
1H COSY cross peak for H-14 (
δH 1.76)–H
2-15 (
δH 1.89, 1.61)–H-16 (
δH 5.76) and HMBC correlations from
δH 2.02 (C
H3CO
2–) to
δC 170.2 (CH
3CO
2–) and from H-16 to
δC 170.2 positioned an acetate substituent at C-16, of which the relative configuration was assigned to be
α-orientation based on the small coupling constants between H
2-15 and H-16 (dd,
JH-15–H-16 = 4.3, 1.6 Hz).
Compound 6 was isolated as an amorphous solid. Its molecular formula was determined to be C30H46O5 by HRESIMS (m/z [M + Na]+ 509.3215, calcd 509.3237), corresponding to eight degrees of unsaturation. The 1H and 13C NMR spectra obtained for compound 6 were almost identical to those of 5. However, the absence of one ester carbon and the singlet methyl group at δH 2.02 suggested deacetylation from 5, which was further supported by an upfield shift of H-16 (δH 4.62). The relative configuration of OH-16 was assigned as β-orientation based on the large coupling constant observed between H-16 and H-15β (dd, JH-15–H-16 = 9.6, 5.1 Hz).
Compound
7 was isolated as a yellow oil. Its molecular formula was determined to be C
31H
44O
4 by HRESIMS (
m/
z [M + Na]
+ 503.3113, calcd 503.3132), corresponding to 10 degrees of unsaturation. Preliminary analysis of the
1H and
13C NMR data revealed that the scalarane-type scaffold had a cyclopropane substituent on the A ring. Interpretation of the
13C NMR and HSQC spectra exhibited the sp
2 carbons in the enone systems: three sp
2 methines at
δH 7.38/
δC 157.5,
δH 6.34/
δC 137.4, and
δH 6.64/
δC 130.4; and one trisubstituted sp
2 carbon atom at
δC 136.4. Therefore, HMBC correlations observed from H-25 (
δH 7.38) to C-17 (
δC 136.4)/C-18 (
δC 49.3)/C-24 (
δC 195.9), H-26 (
δH 6.34) to C-17/C-24/C-25 (
δC 157.5), and H-18 (
δH 3.35) to C-17/C-23 (
δC 14.5), as well as the
1H-
1H COSY cross peak for H-18–H-25–H-26, confirmed the presence of a △
25,26-cyclopenten-24-one subunit for the E-ring and the trisubstituted double bond at △
16,17 (
Figure 4).
Compound
8 was isolated as a yellow oil, and its molecular formula was determined to be C
34H
52O
8 by HRESIMS (
m/
z [M + Na]
+ 611.3541, calcd 611.3554), corresponding to nine degrees of unsaturation. The
1H NMR spectrum obtained for compound
8 showed similar patterns to that of
5. However, the upfield peaks observed for the cyclopropane moiety in
5 were substituted by an oxymethine at
δH 5.35, a methyl singlet at
δH 1.09, and an acetate at
δH 2.03, suggesting the C+F type scaffold shown in
Figure 2. Therefore, the connectivity of C-27–C-20–C-4–C-19 was determined using the HMBC correlations observed from CH
3-19 (
δH 0.99) to C-20 (
δC 73.2) and from CH
3-27 (
δH 1.09) to C-4 (
δC 39.4)/C-20 (
Figure 6). In addition, the acetate at
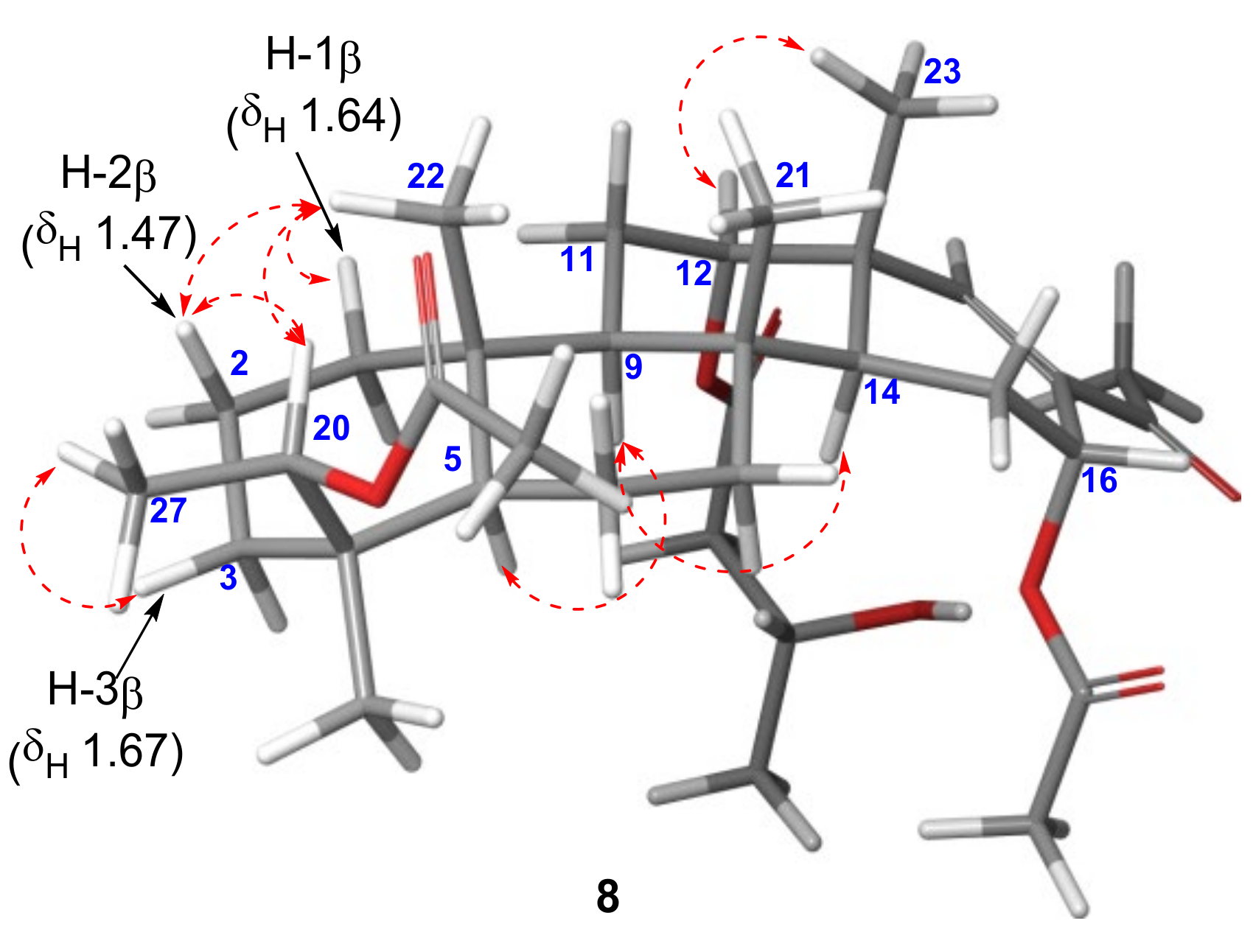
δH 2.03 exhibited a HMBC correlation with C-20 to be located at C-20. The relative configuration at C-20 was assigned as 20
R* from the NOESY correlations observed between H-20 (
δH 5.35) and H-2
β (
δH 1.47)/CH
3-22 (
δH 0.87), and H-3
β (
δH 1.67) and CH
3-27 (
Figure 7). Similarly, the configuration of the acetate group at C-16 was assigned as
α-orientation based on the small coupling constant observed for H-16 (dd,
JH-15–H-16 = 4.3, 1.6 Hz).
Compound 9 was isolated as a colorless oil, and its molecular formula was determined to be C30H48O6 by HRESIMS (m/z [M + NH4]+ 522.3810, calcd 522.3789) corresponding to seven degrees of unsaturation. Analysis of the 1D and 2D NMR data provided almost identical features to those of 8 to determine the carbon skeleton of compound 9. In this case, only one ester carbon atom (δC 172.2) was observed in the 13C NMR spectrum, and the acetate groups shown in the 1H NMR spectrum of 8 disappeared. This information indicated that compound 9 was the deacetylation product of 8. Accordingly, the upfield shifts of H-20 (δH 4.32) and H-16 (δH 4.55) were the major differences, compared to compound 8.
Compound
10 was isolated as a yellow oil, and its molecular formula was determined to be C
33H
50O
8 by HRESIMS (
m/
z [M + Na]
+ 597.3404, calcd 597.3398), corresponding to nine degrees of unsaturation. Preliminary inspection of the
13C NMR and HSQC data of
10 identified four singlet methyl groups (
δH 0.87/
δC 16.6,
δH 0.87/
δC 16.8,
δH 0.96/
δC 23.3,
δH 1.13/
δC 19.8), three doublet methyl groups (
δH 1.07/
δC 16.0
, δH 1.18/
δC 22.5,
δH 1.39/
δC 18.2), and one acetate group (
δH 2.03/
δC 22.0), indicating a honulactone C-like scaffold (C+D type shown in
Figure 2) [
12]. A detailed analysis of the
1H NMR spectrum identified an oxymethine group at
δH 4.44 as a major difference from honulactone C. The location of the oxymethine was determined to be C-16, as indicated by the HMBC correlations from H-16 (
δH 4.44) to C-17 (
δC 162.1)/C-18 (
δC 135.6) and
1H-
1H COSY cross peak for H
2-15 (
δH 1.91, 1.84)–H-16 (
Figure 6). The configuration of the OH-16 group was assigned as
α-orientation based on the small coupling constant observed for H-16 (dd,
JH-15–H-16 = 4.7, 1.4 Hz), and compound
10 was named as 16
α-hydroxyhonulactone C [
12].
Compound
11 was isolated as a yellow oil. Its molecular formula was determined as C
33H
50O
8 by HRESIMS (
m/
z [M + Na]
+ 597.3396, calcd 597.3398), corresponding to nine degrees of unsaturation. The
1H and
13C NMR data of
11 were almost identical to those of
10, but a ketal moiety (
δC 104.4) was observed instead of one doublet methyl group and two oxymethines in compound
10. As shown in compounds
2–
4, the hemiketal functionality in the scalarane-type scaffold usually occurs at C-24 in the E-ring, which was also applicable in this case, as indicated by the HMBC correlations from CH
3-26 (
δH 1.56) to C-17 (
δC 162.9)/C-24 (
δC 104.4). The
α-orientation of the hydroxyl group at C-24 was determined by the NOESY correlation between H-16
β (
δH 2.33) and CH
3-26. Thus, compound
11 was named 24
α-hydroxyhonulactone C [
12].
Compound 12 was isolated as an inseparable mixture. Its molecular formula was determined as C33H48O8 by HRESIMS (m/z [M + Na]+ 595.3241, calcd 595.3241), corresponding to 10 degrees of unsaturation. Compared to 11, two more sp2 methines at δC 138.9/138.7 and δH 6.38, and δC 118.44/118.35 and δH 6.28/6.26 were observed in the 13C NMR and HSQC spectra, indicating the presence of a disubstituted double bond. These sp2 protons were involved in a spin system for H-14 (δH 2.66/2.62)–H-15 (δH 6.38)–H-16 (δH 6.28/6.26) in the 1H-1H COSY spectrum and used to confirm the presence of the △15,16-olefin, which was further supported by HMBC correlations from H-15 to C-13 (δC 40.1/40.0)/C-14 (δC 53.96/53.90)/C-17 (δC 157.3) and from H-16 to C-14/C-18 (δC 130.9). As discussed in the cases of 3 and 4, the presence of the olefin at △15,16 and the hemiketal at C-24 rendered compound 12 an inseparable mixture of C-24 epimers.
Compound
13 was isolated as an amorphous solid. Its molecular formula was determined as C
31H
48O
6 by HRESIMS (
m/
z [M + Na]
+ 539.3325, calcd 539.3343), corresponding to eight degrees of unsaturation. Inspection of the
1H NMR spectrum of
13 revealed most of the structural features of the bishomoscalarane-type skeletons. Precise analysis of the
13C NMR and HSQC data revealed the presence of a triplet methyl group (
δH 0.67/
δC 8.80) and ketal carbon (
δC 104.4), suggesting the A+D type skeleton shown in
Figure 2. While most of the spectral data of
13 were identical to phyllofolactone H, the ketal carbon indicated the oxidation of C-24 to give a 24-hydroxy pentenolide E ring. This insight can be confirmed by the HMBC correlation from CH
3-26 (
δH 1.48) to C-17 (
δC 163.0)/C-24 (
δC 104.4). The configuration of OH-24 was determined to be
α-orientation by the NOESY correlation between H-16
β (
δH 2.33) and CH
3-26. Thus, compound
13 was named 24
α-hydroxyphyllofolactone H [
19].
Compound
14 was isolated as an inseparable mixture. Its molecular formula was determined as C
31H
46O
6 by HRESIMS (
m/
z [M + Na]
+ 537.3175, calcd 537.3187), corresponding to nine degrees of unsaturation. Similar to compound
3, the
13C NMR spectrum of
14 showed a 1:1 splitting pattern corresponding to a mixture of two diastereomers. The distinctive spectral features of
14, differentiated from
13, were observed as the two sp
2 methines at
δH 6.40/6.38 and 6.28/6.27, suggesting an unsaturated derivative of
13. The methines belonged in the
1H-
1H COSY correlation for H-14 (
δH 2.68/2.62)–H-15 (
δH 6.40/6.38)–H-16 (
δH 6.28/6.27) to identify the olefin at C-15 (
Figure 6). In addition,
14 was determined to be a mixture of C-24 epimers, considering the largest splitting observed at CH
3-26 (Δ
δC 1.13 ppm).
Compound 15 was isolated as an inseparable mixture. Its molecular formula was determined to be C32H48O6 by HRESIMS (m/z [M + Na]+ 551.3366, calcd 551.3343), corresponding to nine degrees of unsaturation. The MS data indicated an additional methylene relative to 14, which was further supported by the change observed in the coupling pattern of the terminal methyl group of the side chain at C-12 from a doublet to triplet. The 13C NMR and HSQC data identified the methylene group at δH 1.51/1.25 and δC 29.5/29.4, which were involved in the spin system for H2-2′–H-3′–H2-4′–CH3-5′ in the 1H-1H COSY spectrum to confirm the presence of the 3-hydroxypentanoate side chain. The orientation of the ester at C-12 was assigned as α by the NOESY signal between H-12 (δH 5.55/5.49) and CH3-23 (δH 1.06/1.05), as well as the small coupling constant observed for H-12 (dd, J = 2.3, 1.8 Hz), to identify 12-epi-phyllactone D/E.
Interestingly, the identified structure was previously isolated as a mixture of C-24 epimers by Li et al. in 2007 [
11], but our experimental
13C NMR data showed some discrepancies with the previously reported data at C-9 (Δ 4.28 ppm), C-11 (Δ 2.7 ppm), C-12 (Δ 2.08 ppm), C-14 (Δ 4.46 ppm), and C-23 (Δ 4.35 ppm) (
Figure 8a). In addition, another identification of 12-
epi-phyllactone D/E was reported by Andersen et al. in 2009 [
13]. Although they acquired almost identical experimental NMR data with ours rather than those reported by Li, the isolated compound was estimated to be same as Li’s without consideration of the differences in NMR data (
Tables S17 and S18, Supplementary Materials). Therefore, we investigated the variations in
13C chemical shifts depending on the orientation of the substituents at C-12.
Phyllactone D (
17) and E (
18), the reported 12
β-epimers of
15, were selected for comparison [
25]. While C-12 in phyllactones D and E was observed at
δC 75.1 and 75.8, respectively, the corresponding chemical shifts of the reported and isolated
15 were observed at
δC 75.3 and 73.2/73.1, respectively. The deviations observed for isolated
15 from phyllactone D/E became more obvious at C-9, C-14, and C-23 (
Figure 8b). However, the reported chemical shifts for
15 were better aligned with those of phyllactone D/E. Furthermore, the differences in the
13C NMR chemical shifts observed between isolated
15 and compounds
3,
4,
12, and
14, which share an identical substructure for the B-E ring system, showed negligible values (< 0.5 ppm) around the C-ring (
Table S19, Supplementary Materials). Accordingly, isolated
15 is more likely to be the 12
α-epimer. Even though Li determined the 12
a-configuration observing the NOESY signal between H-12 and CH
3-23 and
JH-12–H-13 calculation (3.0, 2.5 Hz), the NMR database suggests that the compound previously reported by Li is presumed to be a mixture of phyllactone D (
17) and E (
18).
Compound
16 was isolated as a yellowish oil. Its molecular formula was determined as C
27H
40O
5 by HRESIMS (
m/
z [M + Na]
+ 467.2762, calcd 467.2768), corresponding to eight degrees of unsaturation. The
1H NMR spectrum of
16 revealed five singlet methyl groups at
δH 0.73, 0.74, 0.79, 0.85, and 0.86; one acetate group at
δH 1.95; one oxymethine at
δH 4.80; one olefin at
δH 7.30; and one aldehyde at
δH 9.41. The
13C and HSQC NMR spectra showed characteristic peaks for the aldehyde carbon atom at
δC 196.4, two carbonyl carbons at
δC 169.6 and 169.6, one trisubstituted olefin at
δC 145.8 and 124.2, and one oxymethine at
δC 76.9. The HMBC correlation between the two methyl groups at
δC 33.3 and 21.4 was identified as a characteristic feature of the 4-dimethyl-sesterterpenoid scaffold (
Figure 9). The aldehyde at
δH 9.41 exhibited a HMBC correlation with C-18 (
δC 58.7) to be located at C-25. Additional HMBC correlations from H-18 (
δH 3.07) to C-16 (
δC 145.8)/C-17 (
δC 124.2)/C-24 (
δC 169.6), along with the
1H-
1H COSY cross peak for H-14–H
2-15–H-16, indicated the presence of the acid at C-24 and trisubstituted olefin at C-16. The acetate group (
δC 21.3/
δH 1.95) was positioned at C-12, as indicated by the HMBC correlation from H-12 (
δH 4.80) to C-1′ (
δC 169.6) and
1H-
1H COSY cross peak for H
2-11–H-12. Thus, the planar structure of
16 was found to be the deacetalization product of scalarin (
19) [
3]. The NOESY correlations between CH
3-23 (
δH 0.86) and H-12/H-18 determined the configuration of the C-12 acetate and C-18 formyl groups as
α.
Whereas scalarin (
19) exists only in its hemiacetal form, the formation of 18-
epi-19 or
19 via the acetalization of
16 was not observed. To rationalize the observed difference in reactivity, 18-
epi-
16 was proposed as a plausible precursor of scalarin, and geometrical optimization of
16 and 18-
epi-
16 was performed at the B3LYP/6-31G** level of theory. The atomic distance between O-24 to C-25 was calculated to be 3.37 Å for
16 and 2.68 Å for 18-
epi-
16 (
Figure 10). This result suggests that 18-
epi-
16 can undergo acetalization to form scalarin because the
β-orientation of C-25 increases its proximity to the acid at C-24. However, the acetalization of the 25
α-formyl group in
16 will be restricted due to its remoteness to OH-24 to exist as its aldehyde form.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}