
Complete Genome Sequence of Two Deep-Sea Streptomyces Isolates from Madeira Archipelago and Evaluation of Their Biosynthetic Potential

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Isolation, Phenotypic Characterization and Sequencing
2.2. Genome Assembly and Annotation
2.3. Phylogenetic Analysis of the Deep-Sea Isolated Strains
2.4. Marine Adaptation Genes
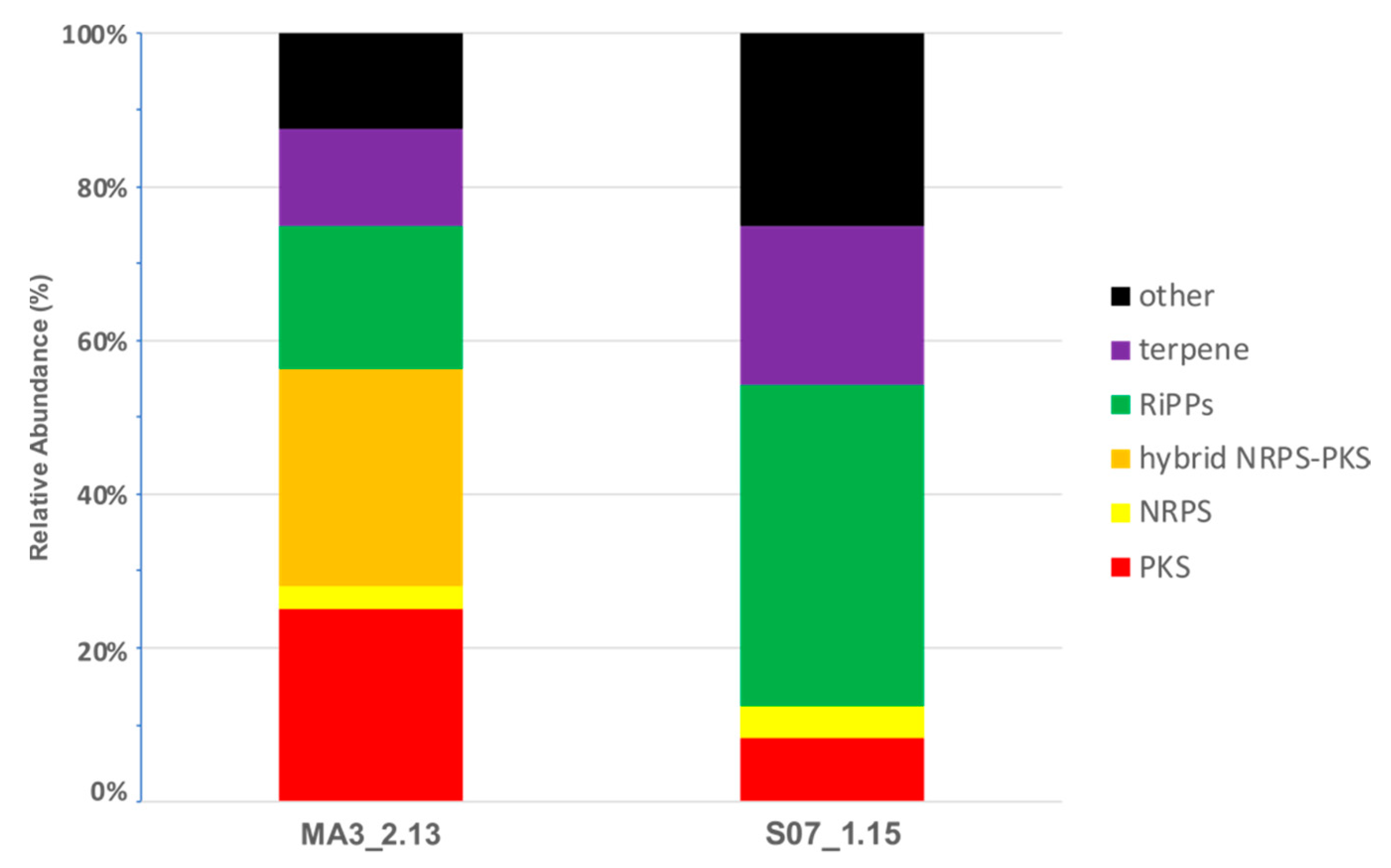
2.5. Secondary Metabolism in Silico profiling
3. Discussion
4. Materials and Methods
4.1. Sampling, Isolation and Microbial Growth
4.2. Genomic DNA Isolation and PCR Amplification
4.3. Short-Read (Illumina) and Long-Read (PacBio) Sequencing
4.4. De Novo Genome Assembly and Annotation
4.5. Identification of Putative Marine Adaptation Genes
4.6. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef]
- Goh, K.M.; Shahar, S.; Chan, K.G.; Chong, C.S.; Amran, S.I.; Sani, M.H.; Zakaria, I.I.; Kahar, U.M. Current status and potential applications of underexplored prokaryotes. Microorganisms 2019, 7, 468. [Google Scholar] [CrossRef]
- Doroghazi, J.R.; Metcalf, W.W. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genom. 2013, 14, 611. [Google Scholar] [CrossRef]
- Belknap, K.C.; Park, C.J.; Barth, B.M.; Andam, C.P. Genome mining of biosynthetic and chemotherapeutic gene clusters in Streptomyces bacteria. Sci. Rep. 2020, 10, 2003. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H. Gifted microbes for genome mining and natural product discovery. J. Ind. Microbiol. Biotechnol. 2017, 44, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Gross, H. Strategies to unravel the function of orphan biosynthesis pathways: Recent examples and future prospects. Appl. Microbiol. Biotechnol. 2007, 75, 267–277. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, N.; Jeong, Y.; Lee, Y.; Kim, W.; Cho, S.; Palsson, B.O.; Cho, B.K. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 2019, 47, 6114–6129. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef]
- Jayakumar, V.; Sakakibara, Y. Comprehensive evaluation of non-hybrid genome assembly tools for third-generation PacBio long-read sequence data. Brief. Bioinform. 2019, 20, 866–876. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Bellec, A.; Courtial, A.; Cauet, S.; Rodde, N.; Vautrin, S.; Beydon, G.; Arnal, N.; Gautier, N.; Fourment, J.; Prat, E.; et al. Long read sequencing technology to solve complex genomic regions assembly in plants. Next Generat. Sequenc. Applic. 2016, 3, 128. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Lee, N.; Kim, W.; Hwang, S.; Lee, Y.; Cho, S.; Palsson, B.; Cho, B.K. Thirty complete Streptomyces genome sequences for mining novel secondary metabolite biosynthetic gene clusters. Sci. Data 2020, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.V.; Manemann, E.M.; Rowe, S.E.; Callender, M.C.; Soto, W. Marine actinomycetes, new sources of biotechnological products. Mar. Drugs 2021, 19, 365. [Google Scholar] [CrossRef]
- Yang, Z.; He, J.; Wei, X.; Ju, J.; Ma, J. Exploration and genome mining of natural products from marine Streptomyces. Appl. Microbiol. Biotechnol. 2020, 104, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Kamjam, M.; Sivalingam, P.; Deng, Z.; Hong, K. Deep sea actinomycetes and their secondary metabolites. Front. Microbiol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Wang, Y.N.; Meng, L.H.; Wang, B.G. Progress in research on bioactive secondary metabolites from deep-sea derived microorganisms. Mar. Drugs 2020, 18, 614. [Google Scholar] [CrossRef]
- Zhou, T.; Komaki, H.; Ichikawa, N.; Hosoyama, A.; Sato, S.; Igarashi, Y. Biosynthesis of akaeolide and lorneic acids and annotation of type I polyketide synthase gene clusters in the genome of Streptomyces sp. NPS554. Mar. Drugs 2015, 13, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Li, W.J.; Jiao, W.C.; Li, Y.; Yuan, W.J.; Zhang, Y.Q.; Klenk, H.P.; Suh, J.W.; Bai, F.W. Streptomyces xinghaiensis sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 2009, 59, 2870–2874. [Google Scholar] [CrossRef][Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Davó, A.; Fenical, W.; Jensen, P.R. Comparative actinomycete diversity in marine sediments. Aquatic. Microbial. Ecol. 2008, 52, 1–11. [Google Scholar] [CrossRef]
- Rodriguez, R.L.; Gunturu, S.; Harvey, W.T.; Rossello-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, W.; Liu, Z.; Huang, Y.; Zhang, Q.; Tian, X.; Zhang, C.; Zhu, Y. Complete genome sequence of Streptomyces sp. SCSIO 03032 isolated from Indian Ocean sediment, producing diverse bioactive natural products. Mar. Genom. 2021, 55, 100803. [Google Scholar] [CrossRef]
- Chen, L.Y.; Wang, X.Q.; Wang, Y.M.; Geng, X.; Xu, X.N.; Su, C.; Yang, Y.L.; Tang, Y.J.; Bai, F.W.; Zhao, X.Q. Genome mining of Streptomyces xinghaiensis NRRL B-24674(T) for the discovery of the gene cluster involved in anticomplement activities and detection of novel xiamycin analogs. Appl. Microbiol. Biotechnol. 2018, 102, 9549–9562. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D. GToTree: A user-friendly workflow for phylogenomics. Bioinformatics 2019, 35, 4162–4164. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; de Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Penn, K.; Jensen, P.R. Comparative genomics reveals evidence of marine adaptation in Salinispora species. BMC Genom. 2012, 13, 86. [Google Scholar] [CrossRef]
- Ian, E.; Malko, D.B.; Sekurova, O.N.; Bredholt, H.; Ruckert, C.; Borisova, M.E.; Albersmeier, A.; Kalinowski, J.; Gelfand, M.S.; Zotchev, S.B. Genomics of sponge-associated Streptomyces spp. closely related to Streptomyces albus J1074: Insights into marine adaptation and secondary metabolite biosynthesis potential. PLoS ONE 2014, 9, e96719. [Google Scholar] [CrossRef]
- Almeida, E.L.; Carrillo Rincon, A.F.; Jackson, S.A.; Dobson, A.D.W. Comparative genomics of marine sponge-derived Streptomyces spp. isolates SM17 and SM18 with their closest terrestrial relatives provides novel insights into environmental niche adaptations and secondary metabolite biosynthesis potential. Front. Microbiol. 2019, 10, 1713. [Google Scholar] [CrossRef]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Munoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Arulprakasam, K.R.; Dharumadurai, D. Genome mining of biosynthetic gene clusters intended for secondary metabolites conservation in actinobacteria. Microb. Pathog. 2021, 161, 105252. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yang, Z.; Zhang, C.; Liu, Z.; He, J.; Liu, Q.; Zhang, T.; Ju, J.; Ma, J. Genome mining of Streptomyces atratus SCSIO ZH16: Discovery of atratumycin and identification of its biosynthetic gene cluster. Org. Lett. 2019, 21, 1453–1457. [Google Scholar] [CrossRef]
- Yang, Z.; Wei, X.; He, J.; Sun, C.; Ju, J.; Ma, J. Characterization of the noncanonical regulatory and transporter genes in atratumycin biosynthesis and production in a heterologous host. Mar. Drugs 2019, 17, 560. [Google Scholar] [CrossRef]
- Twigg, F.F.; Cai, W.; Huang, W.; Liu, J.; Sato, M.; Perez, T.J.; Geng, J.; Dror, M.J.; Montanez, I.; Tong, T.L.; et al. Identifying the biosynthetic gene cluster for triacsins with an N-hydroxytriazene moiety. ChemBioChem 2019, 20, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.J.; Pechersky, Y.; Wang, P.; Wang, J.X.; Balskus, E.P. The cremeomycin biosynthetic gene cluster encodes a pathway for diazo formation. ChemBioChem 2015, 16, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Tomita, T.; Shin-Ya, K.; Wakimoto, T.; Kuzuyama, T.; Nishiyama, M. Discovery of unprecedented hydrazine-forming machinery in bacteria. J. Am. Chem. Soc. 2018, 140, 9083–9086. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Morales, P.; Kopp, J.F.; Martinez-Guerrero, C.; Yanez-Guerra, L.A.; Selem-Mojica, N.; Ramos-Aboites, H.; Feldmann, J.; Barona-Gomez, F. Phylogenomic analysis of natural products biosynthetic gene clusters allows discovery of arseno-organic metabolites in model Streptomycetes. Genome Biol. Evol. 2016, 8, 1906–1916. [Google Scholar] [CrossRef]
- Wang, B.; Guo, F.; Huang, C.; Zhao, H. Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W.; Zhang, Q.; Shi, T.; Ma, L.; Zhu, Y.; Li, S.; Zhang, H.; Zhao, Y.L.; Shi, R.; et al. Mechanistic insights into polycycle formation by reductive cyclization in ikarugamycin biosynthesis. Angew. Chem. Int. Ed. Engl. 2014, 53, 4840–4844. [Google Scholar] [CrossRef]
- Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G.L.; Aigle, B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. USA 2011, 108, 6258–6263. [Google Scholar] [CrossRef]
- Huang, S.; Ma, L.; Tong, M.H.; Yu, Y.; O’Hagan, D.; Deng, H. Fluoroacetate biosynthesis from the marine-derived bacterium Streptomyces xinghaiensis NRRL B-24674. Org. Biomol. Chem. 2014, 12, 4828–4831. [Google Scholar] [CrossRef]
- Deng, H.; O’Hagan, D.; Schaffrath, C. Fluorometabolite biosynthesis and the fluorinase from Streptomyces cattleya. Nat. Prod. Rep. 2004, 21, 773–784. [Google Scholar] [CrossRef]
- Wang, P.; Wang, D.; Zhang, R.; Wang, Y.; Kong, F.; Fu, P.; Zhu, W. Novel macrolactams from a deep-sea-derived Streptomyces species. Mar. Drugs 2020, 19, 13. [Google Scholar] [CrossRef]
- Schorn, M.A.; Alanjary, M.M.; Aguinaldo, K.; Korobeynikov, A.; Podell, S.; Patin, N.; Lincecum, T.; Jensen, P.R.; Ziemert, N.; Moore, B.S. Sequencing rare marine actinomycete genomes reveals high density of unique natural product biosynthetic gene clusters. Microbiology 2016, 162, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Hwang, S.; Kim, J.; Cho, S.; Palsson, B.; Cho, B.K. Mini review: Genome mining approaches for the identification of secondary metabolite biosynthetic gene clusters in Streptomyces. Comput. Struct. Biotechnol. J. 2020, 18, 1548–1556. [Google Scholar] [CrossRef]
- Thrash, A.; Hoffmann, F.; Perkins, A. Toward a more holistic method of genome assembly assessment. BMC Bioinform. 2020, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Cohan, F.M.; Lawrence, J.G.; Spratt, B.G.; Coenye, T.; Feil, E.J.; Stackebrandt, E.; van de Peer, Y.; Vandamme, P.; Thompson, F.L.; et al. Opinion: Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 2005, 3, 733–739. [Google Scholar] [CrossRef]
- Nindita, Y.; Cao, Z.; Fauzi, A.A.; Teshima, A.; Misaki, Y.; Muslimin, R.; Yang, Y.; Shiwa, Y.; Yoshikawa, H.; Tagami, M.; et al. The genome sequence of Streptomyces rochei 7434AN4, which carries a linear chromosome and three characteristic linear plasmids. Sci. Rep. 2019, 9, 10973. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.L.; Li, Y.; Zhang, Y.J.; Zhou, Z.M.; Zhang, W.X.; Chen, X.L.; Zhang, X.Y.; Zhou, B.C.; Wang, L.; Zhang, Y.Z. Comparative genomics reveals a deep-sea sediment-adapted life style of Pseudoalteromonas sp. SM9913. ISME J. 2011, 5, 274–284. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Montalban-Lopez, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef]
- Agrawal, P.; Khater, S.; Gupta, M.; Sain, N.; Mohanty, D. RiPPMiner: A bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. [Google Scholar] [CrossRef]
- Rottig, M.; Medema, M.H.; Blin, K.; Weber, T.; Rausch, C.; Kohlbacher, O. NRPSpredictor2--a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39, W362–W367. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing, G.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, M.A.; Beyvers, S.; Nkouamedjo-Fankep, R.C.; Hanel, P.H.G.; Jelonek, L.; Blom, J.; Goesmann, A. EDGAR3.0: Comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 2021, 49, W185–W192. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010, 11, 595. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ichikawa, N.; Oguchi, A.; Ikeda, H.; Ishikawa, J.; Kitani, S.; Watanabe, Y.; Nakamura, S.; Katano, Y.; Kishi, E.; Sasagawa, M.; et al. Genome sequence of Kitasatospora setae NBRC 14216T: An evolutionary snapshot of the family Streptomycetaceae. DNA Res. 2010, 17, 393–406. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Ponten, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Isolate | Genome Size (bp) | Fold Coverage (x) | G+C Content (%) | No. of CDS 1 | No. of rRNA Operons | No. of tRNA Genes | No. of BGCs 2 | GenBank Accession Number |
|---|---|---|---|---|---|---|---|---|
| MA3_2.13 | 7,653,710 | 139 | 72.1 | 6412 | 5 | 55 | 32 | CP082362 |
| S07_1.15 | 7,094,148 | 159 | 73.2 | 6492 | 6 | 62 | 24 | JAJBZK000000000 |
| 160,397 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albuquerque, P.; Ribeiro, I.; Correia, S.; Mucha, A.P.; Tamagnini, P.; Braga-Henriques, A.; Carvalho, M.d.F.; Mendes, M.V. Complete Genome Sequence of Two Deep-Sea Streptomyces Isolates from Madeira Archipelago and Evaluation of Their Biosynthetic Potential. Mar. Drugs 2021, 19, 621. https://doi.org/10.3390/md19110621
Albuquerque P, Ribeiro I, Correia S, Mucha AP, Tamagnini P, Braga-Henriques A, Carvalho MdF, Mendes MV. Complete Genome Sequence of Two Deep-Sea Streptomyces Isolates from Madeira Archipelago and Evaluation of Their Biosynthetic Potential. Marine Drugs. 2021; 19(11):621. https://doi.org/10.3390/md19110621
Chicago/Turabian StyleAlbuquerque, Pedro, Inês Ribeiro, Sofia Correia, Ana Paula Mucha, Paula Tamagnini, Andreia Braga-Henriques, Maria de Fátima Carvalho, and Marta V. Mendes. 2021. "Complete Genome Sequence of Two Deep-Sea Streptomyces Isolates from Madeira Archipelago and Evaluation of Their Biosynthetic Potential" Marine Drugs 19, no. 11: 621. https://doi.org/10.3390/md19110621
APA StyleAlbuquerque, P., Ribeiro, I., Correia, S., Mucha, A. P., Tamagnini, P., Braga-Henriques, A., Carvalho, M. d. F., & Mendes, M. V. (2021). Complete Genome Sequence of Two Deep-Sea Streptomyces Isolates from Madeira Archipelago and Evaluation of Their Biosynthetic Potential. Marine Drugs, 19(11), 621. https://doi.org/10.3390/md19110621