New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica


 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
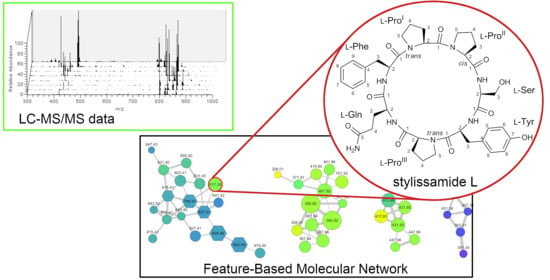
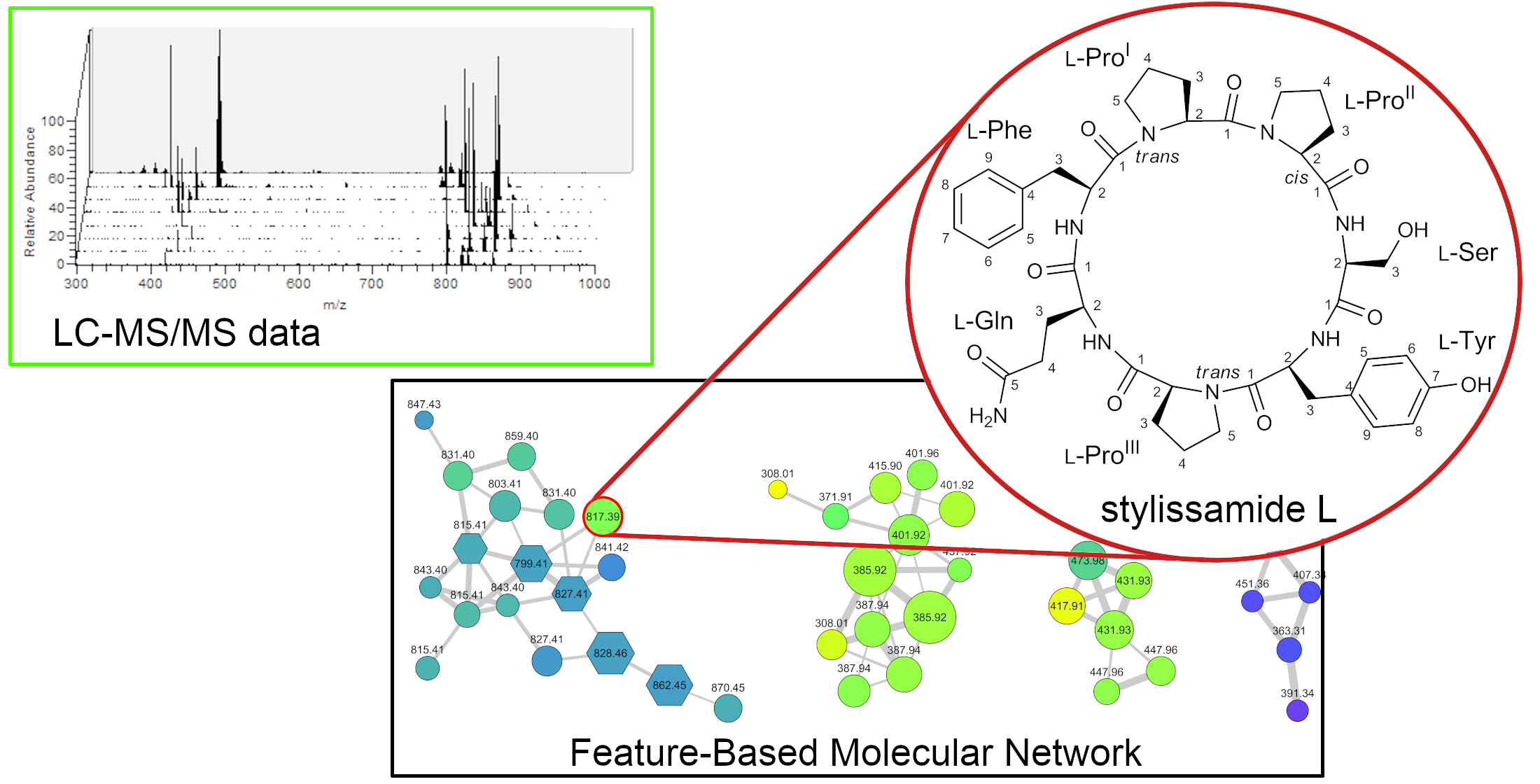
2.1. Collection, Extraction, LC-MS2 Analysis, and Costruction of the Molecular Network
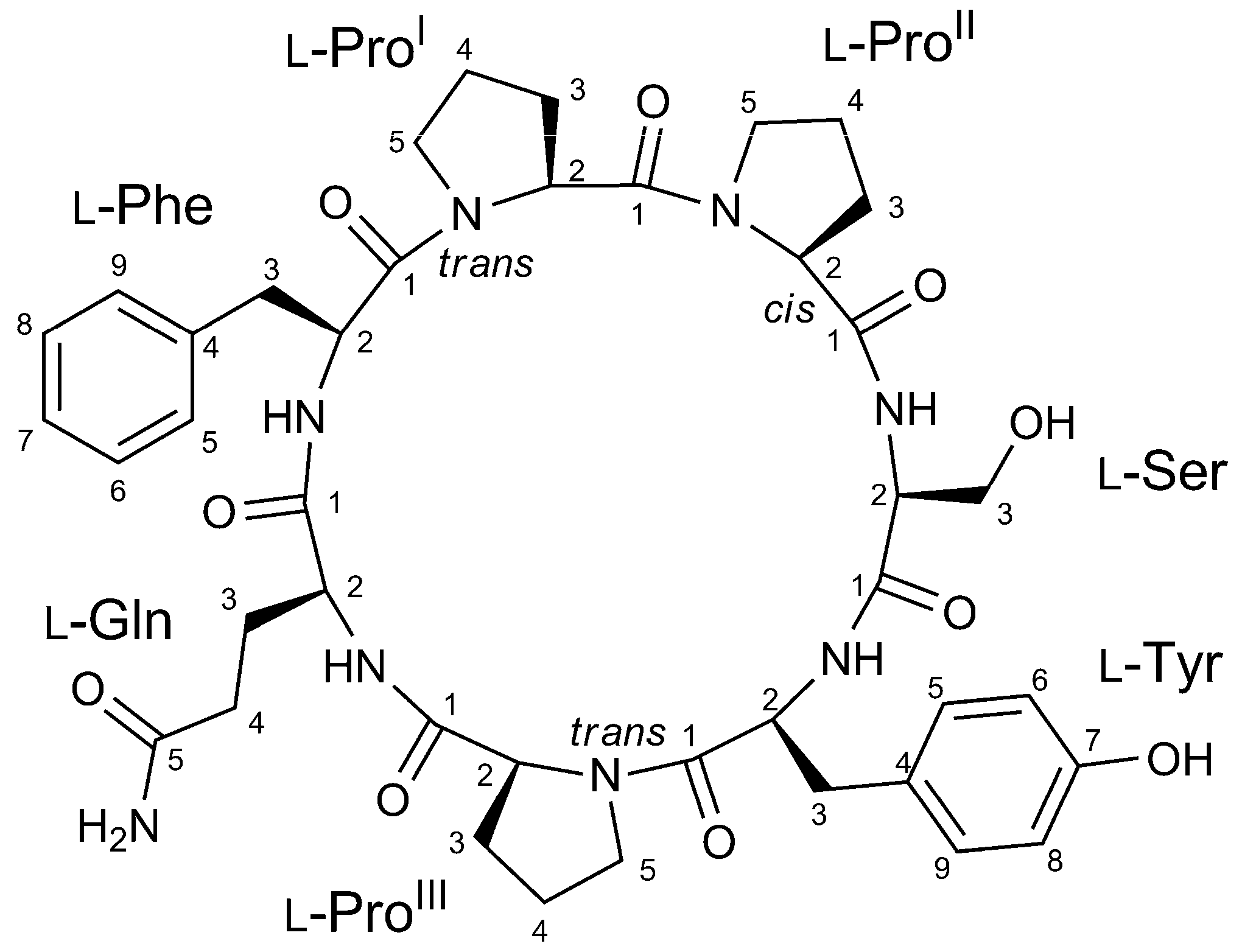
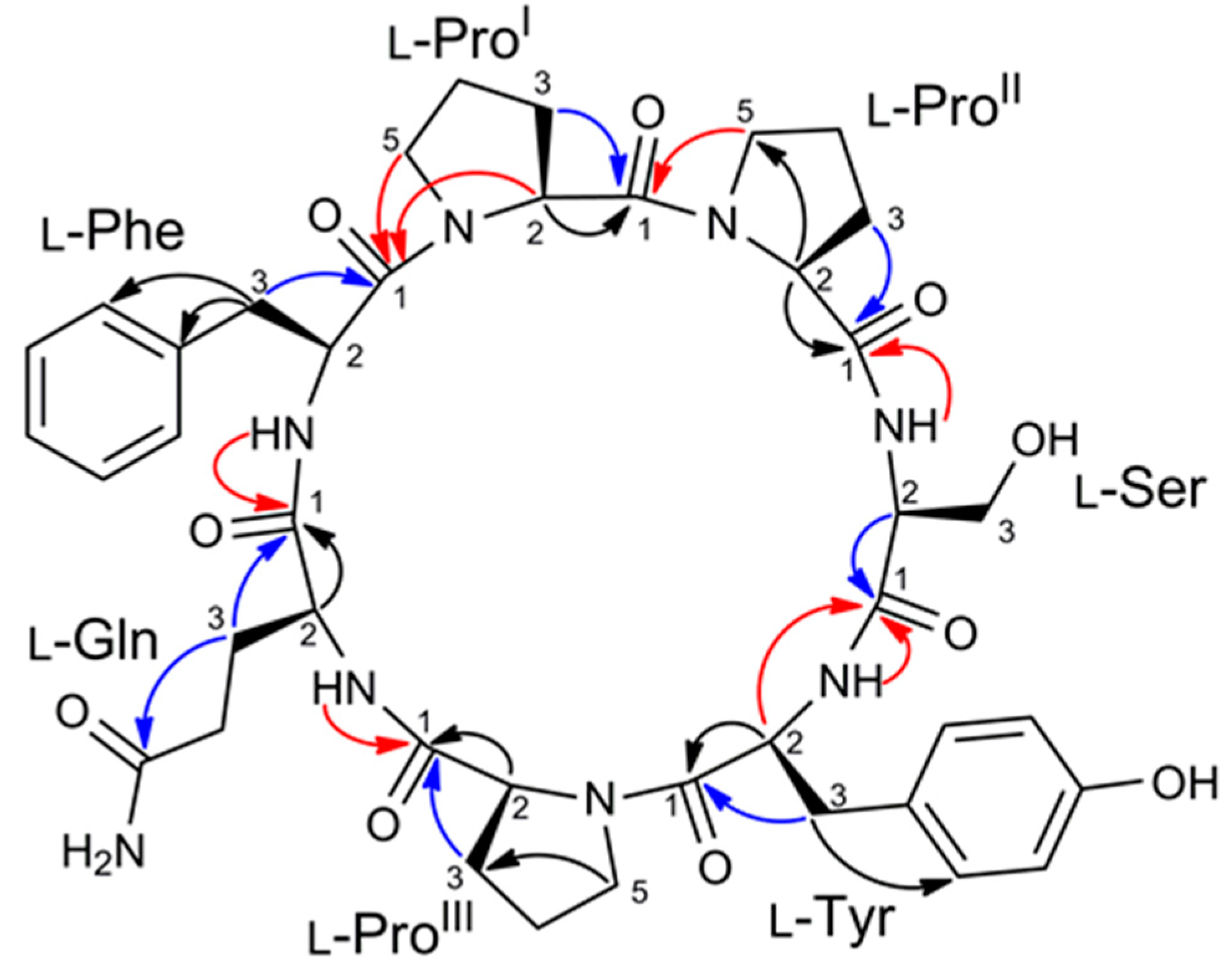
2.2. Structure Elucidation of Stylissamide L (1)
2.3. Cell Proliferation and Migration Assays
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Collection, Extraction and Isolation
3.3. LC-HRMS and LC-HRMS2
3.4. LC-HRMS2 Data Processing and Molecular Networking
3.5. Advanced Marfey’s Analysis
3.6. Cell Proliferation and Migration Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kong, D.-X.; Jiang, Y.-Y.; Zhang, H.-Y. Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discov. Today 2010, 15, 884–886. [Google Scholar] [CrossRef]
- Shang, J.; Hu, B.; Wang, J.; Zhu, F.; Kang, Y.; Li, D.; Sun, H.; Kong, D.-X.; Hou, T. Cheminformatic Insight into the Differences between Terrestrial and Marine Originated Natural Products. J. Chem. Inf. Model. 2018, 58, 1182–1193. [Google Scholar] [CrossRef]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides A and B, chlorinated peptide/polyketide hybrids containing a dolapyrrolidinone unit from the Caribbean sponge Smenospongia aurea. Evaluation of their role as leads in antitumor drug research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Marteinsson, V.T.; Longeon, A.; Klonowski, A.M.; Groben, R.; Bourguet-Kondracki, M.-L.L.; Costantino, V.; Mangoni, A. Thermoactinoamide A, an Antibiotic Lipophilic Cyclopeptide from the Icelandic Thermophilic Bacterium Thermoactinomyces vulgaris. J. Nat. Prod. 2017, 80, 2530–2535. [Google Scholar] [CrossRef]
- Esposito, G.; Della Sala, G.; Teta, R.; Caso, A.; Bourguet-Kondracki, M.L.; Pawlik, J.R.; Mangoni, A.; Costantino, V. Chlorinated Thiazole-Containing Polyketide-Peptides from the Caribbean Sponge Smenospongia conulosa: Structure Elucidation on Microgram Scale. Eur. J. Org. Chem. 2016, 2016, 2871–2875. [Google Scholar] [CrossRef]
- Ito, T.; Masubuchi, M. Dereplication of microbial extracts and related analytical technologies. J. Antibiot. (Tokyo) 2014, 67, 353–360. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 Data-Preprocessing to Enhance Molecular Networking Reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef] [PubMed]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based Molecular Networking in the GNPS Analysis Environment. bioRxiv 2019, 812404. [Google Scholar] [CrossRef]
- Available online: https://gnps.ucsd.edu/ (accessed on 5 August 2020).
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the t-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Caso, A.; Esposito, G.; Della Sala, G.; Pawlik, J.R.; Teta, R.; Mangoni, A.; Costantino, V. Fast Detection of Two Smenamide Family Members Using Molecular Networking. Mar. Drugs 2019, 17, 618. [Google Scholar] [CrossRef] [PubMed]
- Grauso, L.; Yegdaneh, A.; Sharifi, M.; Mangoni, A.; Zolfaghari, B.; Lanzotti, V. Molecular Networking-Based Analysis of Cytotoxic Saponins from Sea Cucumber Holothuria atra. Mar. Drugs 2019, 17, 86. [Google Scholar] [CrossRef]
- Della Sala, G.; Mangoni, A.; Costantino, V.; Teta, R. Identification of the Biosynthetic Gene Cluster of Thermoactinoamides and Discovery of New Congeners by Integrated Genome Mining and MS-Based Molecular Networking. Front. Chem. 2020, 8, 397. [Google Scholar] [CrossRef]
- Mohammed, R.; Peng, J.; Kelly, M.; Hamann, M.T. Cyclic heptapeptides from the Jamaican sponge Stylissa caribica. J. Nat. Prod. 2006, 69, 1739–1744. [Google Scholar] [CrossRef]
- Grube, A.; Maier, T.; Köck, M. MS-guided Fractionation as a Fast Way to the Identification of New Natural Products—MALDI-TOF-MS Screening of the Marine Sponge Stylissa caribica. Z. Für Nat. B 2007, 62, 600–604. [Google Scholar] [CrossRef]
- Schmidt, G.; Grube, A.; Köck, M. Stylissamides A-D-New proline-containing cyclic heptapeptides from the marine sponge Stylissa caribica. Eur. J. Org. Chem. 2007, 2, 4103–4110. [Google Scholar] [CrossRef]
- Cychon, C.; Köck, M. Stylissamides E and F, Cyclic Heptapeptides from the Caribbean Sponge Stylissa caribica. J. Nat. Prod. 2010, 73, 738–742. [Google Scholar] [CrossRef]
- Wang, X.; Morinaka, B.I.; Molinski, T.F. Structures and solution conformational dynamics of stylissamides G and H from the Bahamian Sponge Stylissa caribica. J. Nat. Prod. 2014, 77, 625–630. [Google Scholar] [CrossRef]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Teta, R.; Della Sala, G.; Esposito, G.; Via, C.W.; Mazzoccoli, C.; Piccoli, C.; Bertin, M.J.; Costantino, V.; Mangoni, A. A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front. 2019, 6, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yamano, M.; Fujita, M.; Setiawan, A.; Kobayashi, M. Stylissamide X, a new proline-rich cyclic octapeptide as an inhibitor of cell migration, from an Indonesian marine sponge of Stylissa sp. Bioorg. Med. Chem. Lett. 2012, 22, 1818–1821. [Google Scholar] [CrossRef]
- Teta, R.; Della Sala, G.; Renga, B.; Mangoni, A.; Fiorucci, S.; Costantino, V. Chalinulasterol, a chlorinated steroid disulfate from the caribbean sponge Chalinula molitba. Evaluation of its role as PXR receptor modulator. Mar. Drugs 2012, 10, 1383–1390. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Pos. | δC, Type | δH, Mult (J in Hz) | AA | Pos. | δC, Type | δH, Mult (J in Hz) | ||
|---|---|---|---|---|---|---|---|---|---|
| ProI | 1 | 170.3, C | ProIII | 1 | 171.9, C | ||||
| 2 | 59.1, CH | 4.34, dd (5.1, 8.6) | 2 | 63.1, CH | 4.06, t (8.7) | ||||
| 3 | 28.1, CH2 | a | 2.15, m | 3 | 28.7, CH2 | a | 2.22 m | ||
| b | 1.75, m | b | 1.81, m | ||||||
| 4 | 24.3, CH2 | 1.87, m | 4 | 25.0, CH2 | a | 2.11, m | |||
| 5 | 46.7, CH2 | a | 3.45, m | b | 1.98, m | ||||
| b | 3.36, m | 5 | 46.9, CH2 | a | 3.93, ddd (6.8, 9.8, 9.8) | ||||
| ProII | 1 | 171.8, C | b | 3.82, m | |||||
| 2 | 60.1, CH | 4.28, dd (1.5, 8.8) | Gln | NH | 8.17, d (7,0) | ||||
| 3 | 31.8, CH2 | a | 2.16, m | 1 | 170.7, C | ||||
| b | 2.00, m | 2 | 52.8, CH | 4.05, ddd (4.3, 7.0, 10.0) | |||||
| 4 | 21.7, CH2 | a | 1.77, m | 3 | 25.9, CH2 | a | 1.85, m | ||
| b | 1.57, m | b | 1.73, m | ||||||
| 5 | 46.8, CH2 | a | 3.60, ddd (1.5, 8.4, 10.8) | 4 | 31.5, CH2 | a | 2.13, ddd (7.2, 15.7, 7.2) | ||
| b | 3.33, ddd (10.8, 10.8, 7.1) | b | 2.04, ddd (7.2, 15.7, 7.2) | ||||||
| Ser | NH | 7.65, d (5.9) | 5 | 174.5, C | |||||
| 1 | 167.7, C | 5-NH2 | 6.92, s | ||||||
| 2 | 60.0, CH | 3.85, ddd (3.6, 5.9, 10.2) | Phe | NH | 7.11, d (7.2) | ||||
| 3 | 60.9, CH2 | a | 3.46, dd (10.2, 11.9) | 1 | 167.5, C | ||||
| b | 3.14, dd (11.9, 3.6) | 2 | 51.5, CH | 4.69, ddd (5.8,7.2, 8.0) | |||||
| Tyr | NH | 7.34, d (9.1) | 3 | 36.9, CH2 | a | 3.18, dd (8.0, 14.2) | |||
| 1 | 171.5 C | b | 2.71, dd (5.8, 14.2) | ||||||
| 2 | 51.5 CH | 4.88 ddd (3.2, 9.1,10.9) | 4 | 138.0, C | |||||
| 3 | 37.0 CH2 | a | 3.35, dd (3.2,13.5) | 5/9 | 128.9, CH | 7.16, d (7.5) | |||
| b | 2.42, dd (10.9, 13.5) | 6/8 | 126.0, CH | 7.18, t (7.3) | |||||
| 4 | 126.6 C | 7 | 128.0, CH | 7.22, t (7.5) | |||||
| 5/9 | 130.5 CH | 7.08, d (8.5) | |||||||
| 6/8 | 114.9 CH | 6.66, d (8.5) | |||||||
| 7 | 156.0 C | ||||||||
| 7-OH | 7.42, s |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpato, S.; Teta, R.; Della Sala, G.; Pawlik, J.R.; Costantino, V.; Mangoni, A. New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica. Mar. Drugs 2020, 18, 443. https://doi.org/10.3390/md18090443
Scarpato S, Teta R, Della Sala G, Pawlik JR, Costantino V, Mangoni A. New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica. Marine Drugs. 2020; 18(9):443. https://doi.org/10.3390/md18090443
Chicago/Turabian StyleScarpato, Silvia, Roberta Teta, Gerardo Della Sala, Joseph R. Pawlik, Valeria Costantino, and Alfonso Mangoni. 2020. "New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica" Marine Drugs 18, no. 9: 443. https://doi.org/10.3390/md18090443
APA StyleScarpato, S., Teta, R., Della Sala, G., Pawlik, J. R., Costantino, V., & Mangoni, A. (2020). New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica. Marine Drugs, 18(9), 443. https://doi.org/10.3390/md18090443