Donghaecyclinones A–C: New Cytotoxic Rearranged Angucyclinones from a Volcanic Island-Derived Marine Streptomyces sp.

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
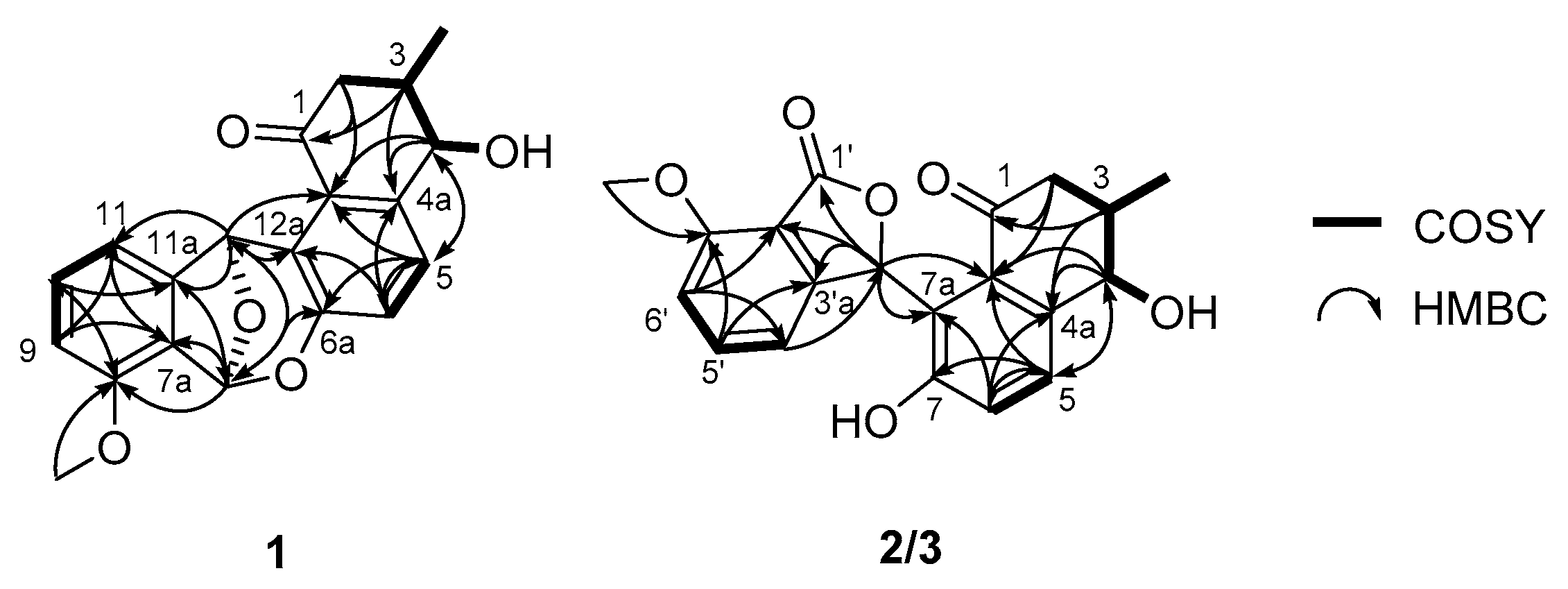
2.1. Structure Elucidation
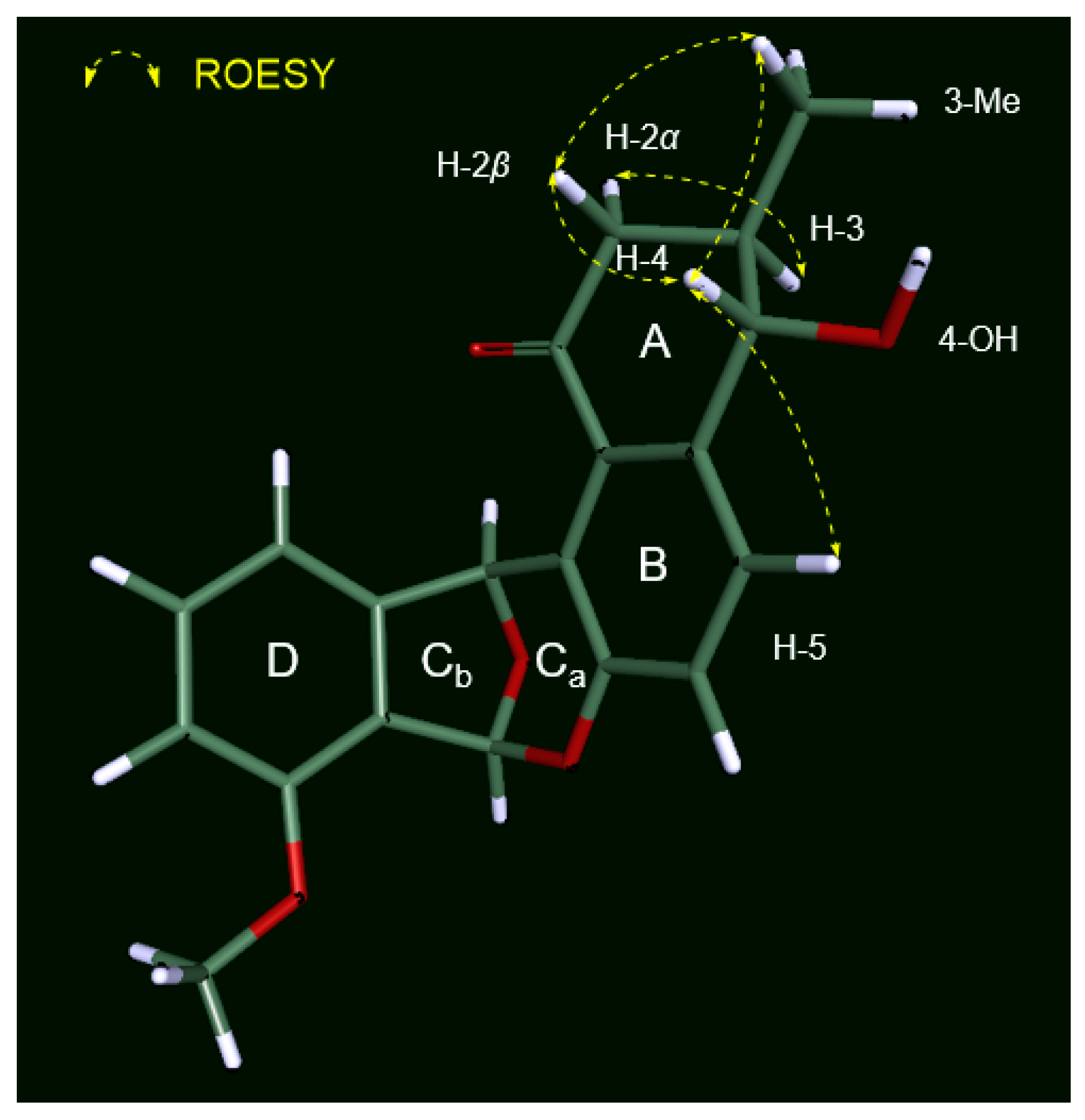
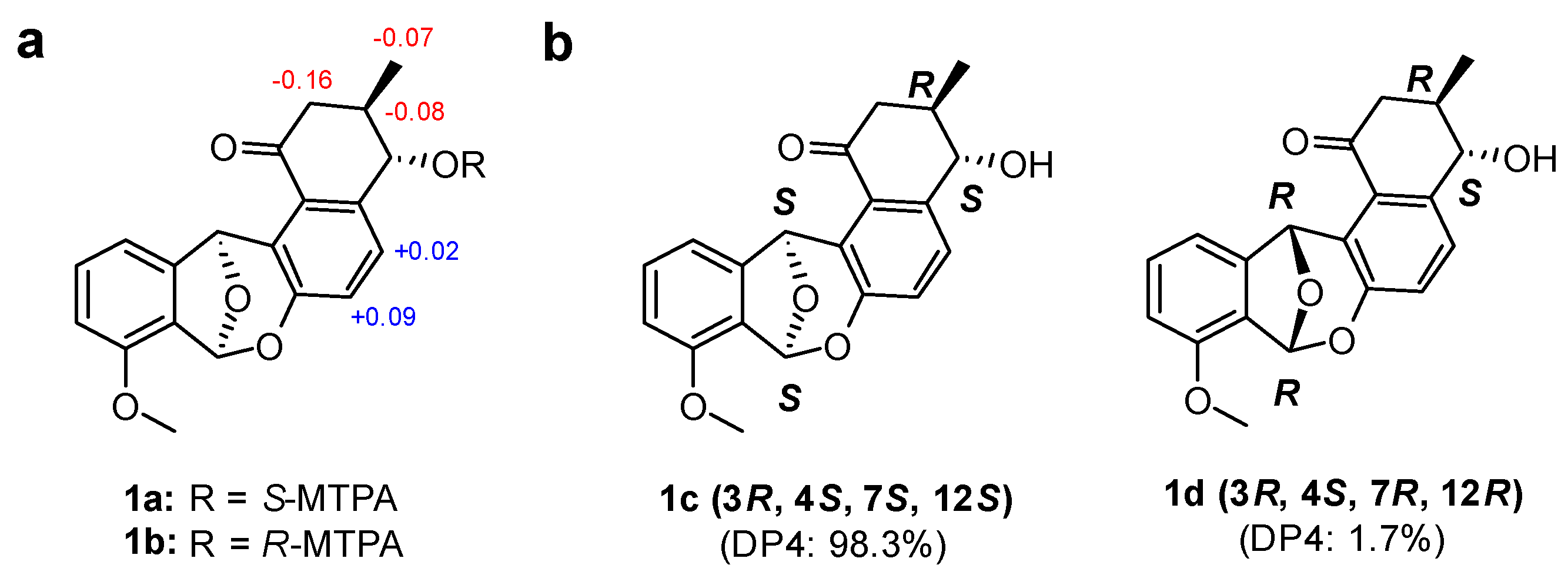
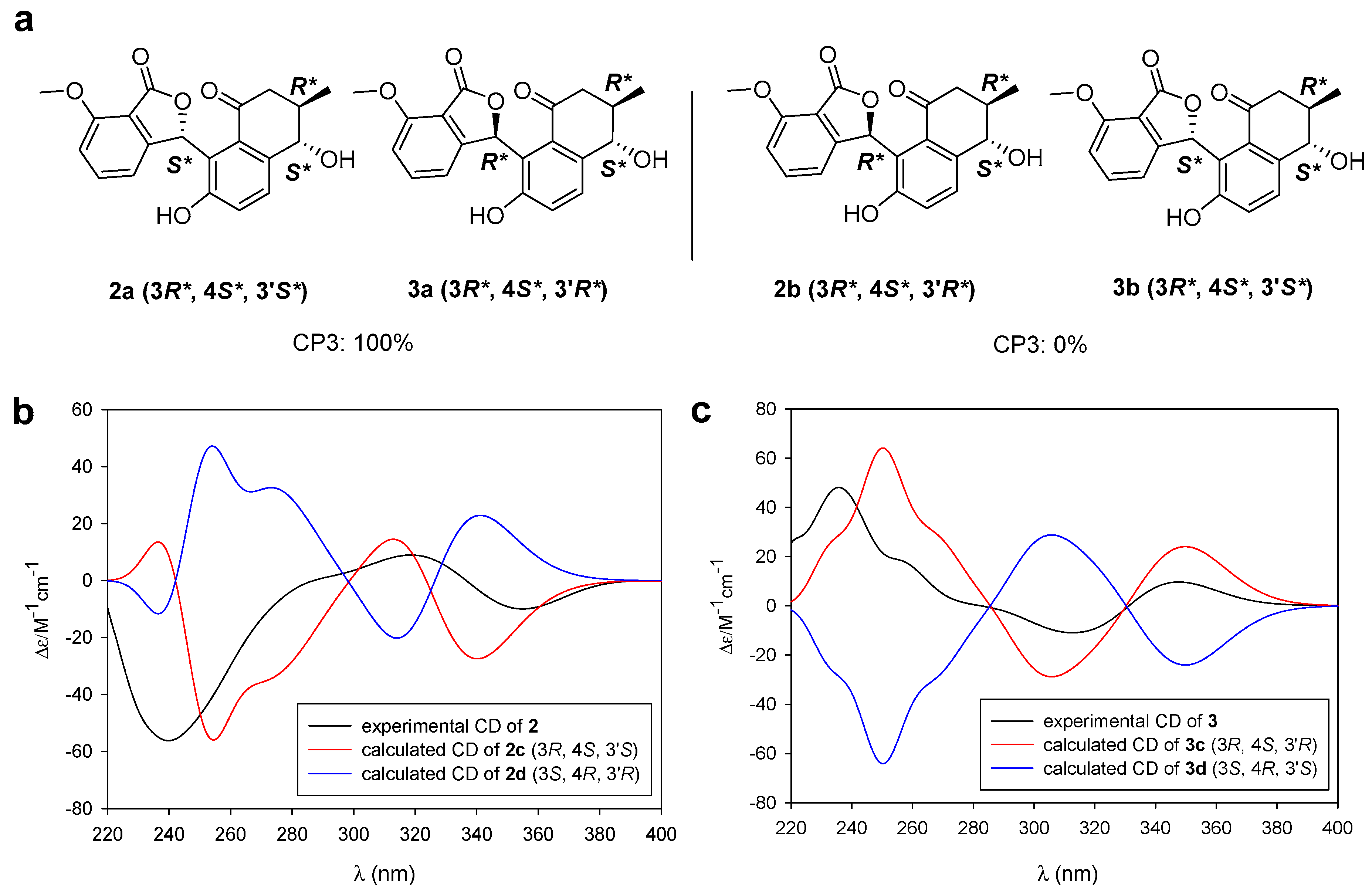
2.2. Determination of the Configurations of Donghaecyclinones A–C
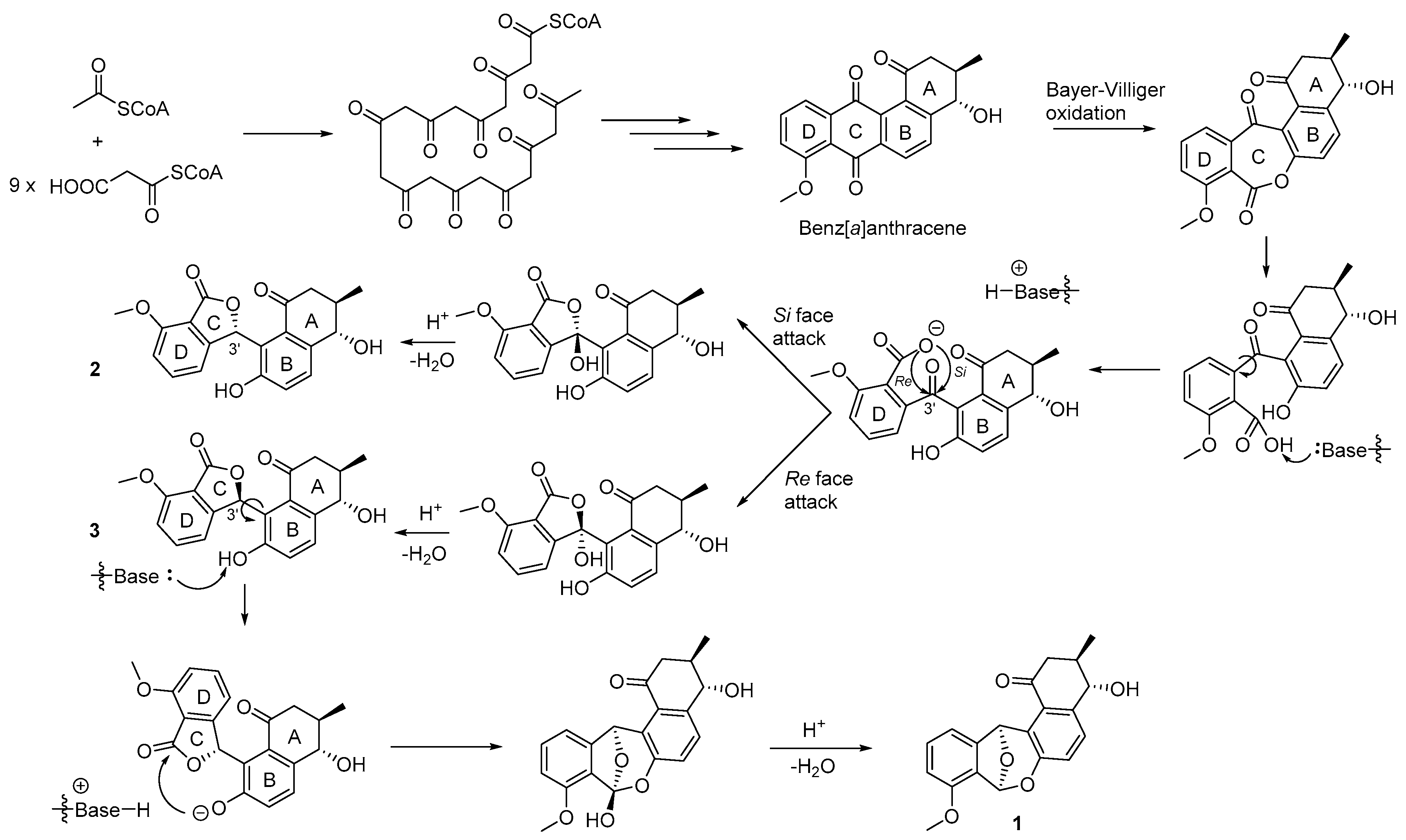
2.3. Proposed Biosynthesis of Donghaecyclinones A–C
2.4. Biological Activities of Donghaecyclinones A–C
3. Experimental
3.1. General Experimental Procedures
3.2. Cultivation and Extraction
3.3. Isolation of Donghaecyclinones A–C (1–3)
3.4. MTPA Esterification of Donghaecylinone A (1)
3.4.1. The S-MTPA Ester (1a) of Donghaecyclinone A (1)
3.4.2. The R-MTPA Ester (1b) of Donghaecyclinone A (1)
3.5. DP4 and CP3 Analyses
3.6. ECD Calculation
3.7. Antibacterial Activity Bioassay
3.8. Antifungal Activity Bioassay
3.9. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3749. [Google Scholar] [CrossRef] [PubMed]
- Duus, J.Ø.; Gotfredsen, C.H.; Bock, K. Carbohydrate structural determination by NMR spectroscopy: modern methods and limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Ermanis, K.; Parkes, K.E.B.; Agback, T.; Goodman, J.M. The optimal DFT approach in DP4 NMR structure analysis–pushing the limits of relative configuration elucidation. Org. Biomol. Chem. 2019, 17, 5886–5890. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem. 2009, 74, 4597–4607. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Nugroho, A.E.; Morita, H. Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1–10. [Google Scholar] [CrossRef]
- Li, X.-C.; Ferreira, D.; Ding, Y. Determination of absolute configuration of natural products: Theoretical calculation of electronic circular dichroism as a tool. Curr. Org. Chem. 2010, 14, 1678–1697. [Google Scholar] [CrossRef]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 17, 2717–2727. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: Marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef]
- Srinivas, T.N.R.; Anil Kumar, P.; Tank, M.; Sunil, B.; Poorna, M.; Zareena, B.; Shivaji, S. Aquipuribacter nitratireducens sp. nov., isolated from a soil sample of a mud volcano. Int. J. Syst. Evol. Microbiol. 2015, 65, 2391–2396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.-H.; Lee, S.K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Ohmyungsamycins A and B: Cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Jang, J.; Sim, J.; Son, W.S.; Ahn, H.-C.; Kim, T.S.; Shin, Y.-H.; Lim, C.; Lee, S.; An, H.; et al. Conformation-enabled total syntheses of ohmyungsamycins A and B and structural revision of ohmyungsamycin B. Angew. Chem. Int. Ed. 2018, 57, 3069–3073. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Shin, Y.; Lee, H.-M.; Kim, J.K.; Choe, J.H.; Jang, J.-C.; Um, S.; Jin, H.S.; Komatsu, M.; Cha, G.-H.; et al. Ohmyungsamycins promote antimicrobial responses through autophagy activation via AMP-activated protein kinase pathway activation. Sci. Rep. 2017, 7, 3431. [Google Scholar] [CrossRef]
- Kim, E.; Shin, Y.-H.; Kim, T.H.; Byun, W.S.; Cui, J.; Du, Y.E.; Lim, H.-J.; Song, M.C.; Kwon, A.S.; Kang, S.H.; et al. Characterization of the ohmyungsamycin biosynthetic pathway and generation of derivatives with improved antituberculosis activity. Biomolecules 2019, 9, 672. [Google Scholar] [CrossRef]
- Byun, W.S.; Kim, S.; Shin, Y.-H.; Kim, W.K.; Oh, D.-C.; Lee, S.K. Antitumor activity of ohmyungsamycin A through the regulation of the Skp2-p27 axis and MCM4 in human colorectal cancer cells. J. Nat. Prod. 2020, 83, 118–126. [Google Scholar] [CrossRef]
- Bae, M.; Park, S.; Kwon, Y.; Lee, S.K.; Shin, J.; Nam, J.-W.; Oh, D.-C. QM-HiFSA-aided structure determination of succinilenes A–D, new triene polyols from a marine-derived Streptomyces sp. Mar. Drugs 2017, 15, 38. [Google Scholar] [CrossRef]
- Bae, M.; An, J.S.; Bae, E.S.; Oh, J.; Park, S.H.; Lim, Y.; Ban, Y.H.; Kwon, Y.; Cho, J.-C.; Yoon, Y.J.; et al. Donghaesulfins A and B, dimeric benz[a]anthracene thioethers from volcanic Island derived Streptomyces sp. Org. Lett. 2019, 21, 3635–3639. [Google Scholar] [CrossRef]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef]
- Wu, C.; van der Heul, H.U.; Melnik, A.V.; Lübben, J.; Dorrestein, P.C.; Minaard, A.J.; Choi, Y.H.; van Wezel, G.P. Lugdunomycin, an angucycline-derived molecule with unprecedented chemical architecture. Angew. Chem. Int. Ed. 2019, 58, 2809–2814. [Google Scholar] [CrossRef]
- Hu, Z.; Qin, L.; Wang, Q.; Ding, W.; Chen, Z.; Ma, Z. Angucycline antibiotics and its derivatives from marine-derived actinomycete Streptomyces sp. A6H. Nat. Prod. Res. 2016, 30, 2551–2558. [Google Scholar] [CrossRef]
- Yang, L.; Hou, L.; Li, H.; Li, W. Antibiotic angucycline derivatives from the deepsea-derived Streptomyces lusitanus. Nat. Prod. Res. 2019, 1–7. [Google Scholar] [CrossRef]
- Ma, M.; Rateb, M.E.; Teng, Q.; Yang, D.; Rudolf, J.D.; Zhu, X.; Huang, Y.; Zhao, L.-X.; Jiang, Y.; Li, X.; et al. Angucyclines and angucyclinones from Streptomyces sp. CB01913 featuring C-ring cleavage and expansion. J. Nat. Prod. 2015, 78, 2471–2480. [Google Scholar] [CrossRef]
- Chen, C.; Song, F.; Guo, H.; Abdel-Mageed, W.M.; Bai, H.; Dai, H.; Liu, X.; Wang, J.; Zhang, L. Isolation and characterization of LS1924A, a new analog of emycins. J. Antibiot. 2012, 65, 433–435. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Guo, L.; Xie, Z.; Yang, Q.; Gong, S. Preparing Method and Application of Furamycins I and II. China Patent CN 108084126, 29 May 2018. [Google Scholar]
- Seco, J.M.; Quiñoá, E.; Riguera, R. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron: Asymmetry 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Bringmann, G.; Tasler, S.; Endress, H.; Kraus, J.; Messer, K.; Wohlfarth, M.; Lobin, W. Murrastifoline-F: First total synthesis, atropo-enantiomer resolution, and stereoanalysis of an axially chiral N,C-coupled biaryl alkaloid. J. Am. Chem. Soc. 2001, 123, 2703–2711. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 a | 2 b | 3 b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C/H | δH c | mult (J in Hz) | δC c | C/H | δH c | mult (J in Hz) | δC c | δH c | mult (J in Hz) | δC c |
| 1 | 198.9, s | 1 | 201.2, s | 200.4, s | ||||||
| 2α | 2.81 | dd (16.0, 4.5), 1H | 44.3, t | 2α | 2.81 | dd (15.0, 4.5), 1H | 46.8, t | 2.99 | dd (15.0, 4.5), 1H | 45.6, t |
| 2β | 2.32 | dd (16.0, 12.0), 1H | 2β | 2.64 | dd (15.0, 10.0), 1H | 2.38 | dd (15.0, 10.0), 1H | |||
| 3 | 2.08 | m, 1H | 37.0, d | 3 | 2.29 | m, 1H | 39.6, d | 2.24 | m, 1H | 38.2, d |
| 3- | 1.04 | d (6.5), 3H | 17.6, q | 3- | 1.20 | d (6.5), 3H | 18.9, q | 1.18 | d (6.5), 3H | 18.0, q |
| Me | Me | |||||||||
| 4 | 4.22 | dd (7.5, 6.0), 1H | 71.4, d | 4 | 4.55 | d (6.5), 1H | 74.4, d | 4.43 | d (6.5), 1H | 73.4, d |
| 4- | 5.61 | d (6.0), 1H | 4- | 4.69 | brs, 1H | 4.74 | brs, 1H | |||
| OH | OH | |||||||||
| 4a | 140.4, s | 4a | 139.9, s | 140.1, s | ||||||
| 5 | 7.49 | d (8.5), 1H | 127.6, d | 5 | 7.59 | d (8.5), 1H | 130.5, d | 7.63 | d (8.5), 1H | 129.4, d |
| 6 | 7.00 | d (8.5), 1H | 122.2, d | 6 | 7.03 | d (8.5), 1H | 122.0, d | 7.05 | d (8.5), 1H | 121.9, d |
| 6a | 148.5, s | 7 | 157.5, s | 157.3, s | ||||||
| 7 | 6.86 | s, 1H | 98.7, d | 7- | 8.83 | brs, 1H | 8.80 | brs, 1H | ||
| 7a | 122.8, s | OH | ||||||||
| 8 | 153.6, s | 7a | 122.9, s | 122.8, s | ||||||
| 7b | 133.9, s | 132.7, s | ||||||||
| 8- | 3.85 | s, 3H | 55.6, q | 1′ | 169.3, s | 169.3, s | ||||
| OMe | 3′ | 7.43 | s, 1H | 76.4, d | 7.41 | s, 1H | 76.5, d | |||
| 9 | 6.94 | t (8.5), 1H | 111.5, d | 3′a | 153.9, s | 153.8, s | ||||
| 10 | 7.27 | t (8.5), 1H | 131.5, d | 4′ | 6.92 | d (8.5), 1H | 115.1, d | 6.93 | d (8.5), 1H | 115.0, d |
| 11 | 7.10 | d (8.5), 1H | 111.7, d | 5′ | 7.54 | t (8.5), 1H | 136.2, d | 7.54 | t (8.5), 1H | 136.1, d |
| 11a | 148.3, s | 6′ | 7.04 | d (8.5), 1H | 111.3, d | 7.04 | d (8.5), 1H | 111.2, d | ||
| 12 | 6.68 | s, 1H | 77.3, d | 7′ | 158.9, s | 158.8, s | ||||
| 12a | 125.9, s | 7′a | 116.0, s | 115.8, s | ||||||
| 12b | 126.3, s | 7′- | 3.93 | s, 3H | 56.1, q | 3.94 | s, 3H | 56.0, q | ||
| OMe | ||||||||||
| Cytotoxicity (IC50 μM) | |||||
|---|---|---|---|---|---|
| HCT116 | MDA-MB231 | SNU638 | A549 | SK-HEP1 | |
| 1 | 28.9 | 20.0 | 16.1 | 22.9 | 14.2 |
| 2 | 27.3 | 19.3 | 19.6 | 19.0 | 9.6 |
| 3 | 8.0 | 6.7 | 9.5 | 9.6 | 6.0 |
| Etoposide | 0.4 | 0.5 | 0.4 | 0.5 | 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, M.; An, J.S.; Hong, S.-H.; Bae, E.S.; Chung, B.; Kwon, Y.; Hong, S.; Oh, K.-B.; Shin, J.; Lee, S.K.; et al. Donghaecyclinones A–C: New Cytotoxic Rearranged Angucyclinones from a Volcanic Island-Derived Marine Streptomyces sp. Mar. Drugs 2020, 18, 121. https://doi.org/10.3390/md18020121
Bae M, An JS, Hong S-H, Bae ES, Chung B, Kwon Y, Hong S, Oh K-B, Shin J, Lee SK, et al. Donghaecyclinones A–C: New Cytotoxic Rearranged Angucyclinones from a Volcanic Island-Derived Marine Streptomyces sp. Marine Drugs. 2020; 18(2):121. https://doi.org/10.3390/md18020121
Chicago/Turabian StyleBae, Munhyung, Joon Soo An, Seong-Heon Hong, Eun Seo Bae, Beomkoo Chung, Yun Kwon, Suckchang Hong, Ki-Bong Oh, Jongheon Shin, Sang Kook Lee, and et al. 2020. "Donghaecyclinones A–C: New Cytotoxic Rearranged Angucyclinones from a Volcanic Island-Derived Marine Streptomyces sp." Marine Drugs 18, no. 2: 121. https://doi.org/10.3390/md18020121
APA StyleBae, M., An, J. S., Hong, S.-H., Bae, E. S., Chung, B., Kwon, Y., Hong, S., Oh, K.-B., Shin, J., Lee, S. K., & Oh, D.-C. (2020). Donghaecyclinones A–C: New Cytotoxic Rearranged Angucyclinones from a Volcanic Island-Derived Marine Streptomyces sp. Marine Drugs, 18(2), 121. https://doi.org/10.3390/md18020121