A New Bioassay Platform Design for the Discovery of Small Molecules with Anticancer Immunotherapeutic Activity

 ,
,  , ,
, , 
 , and
, and 
Abstract
1. Introduction
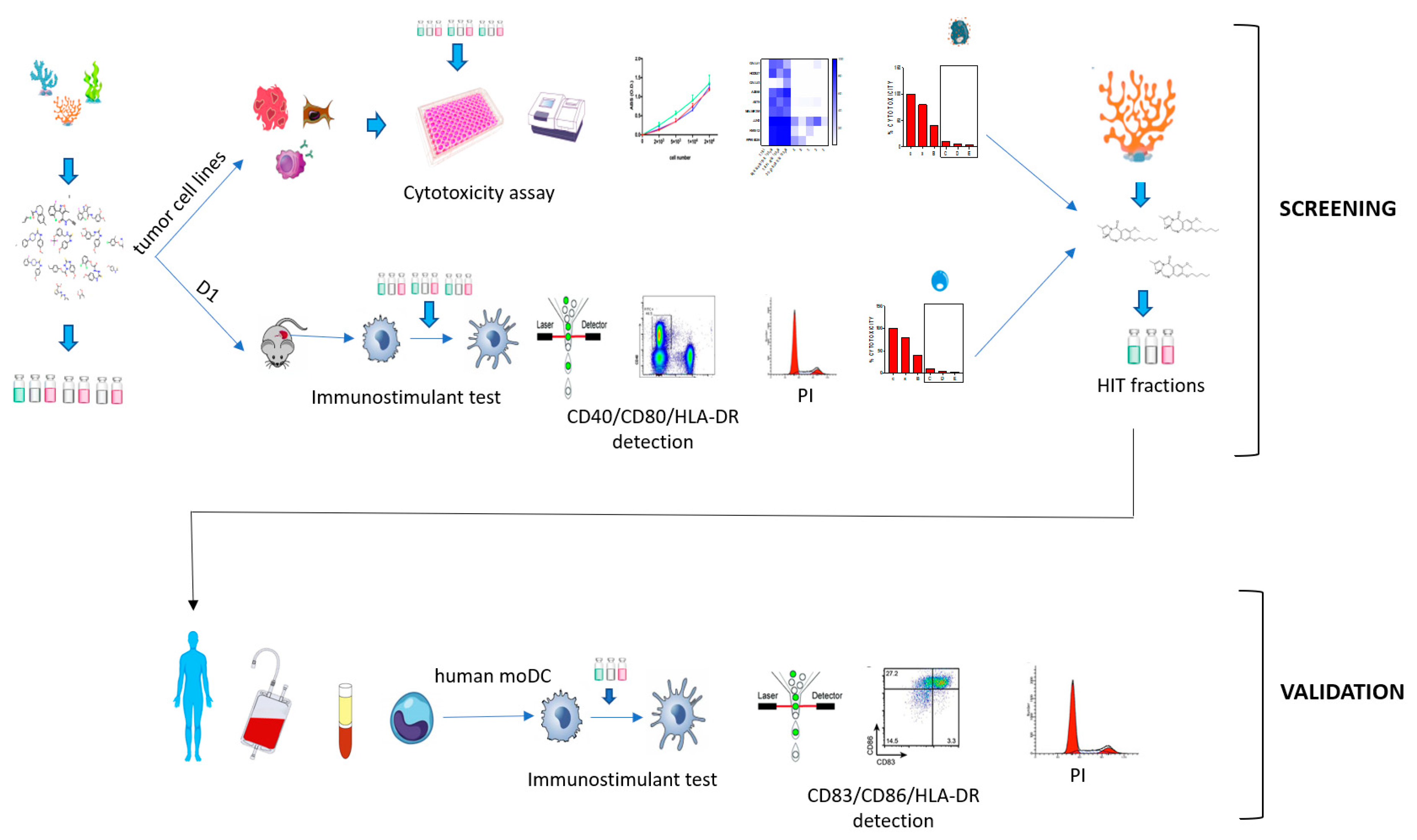
2. Results
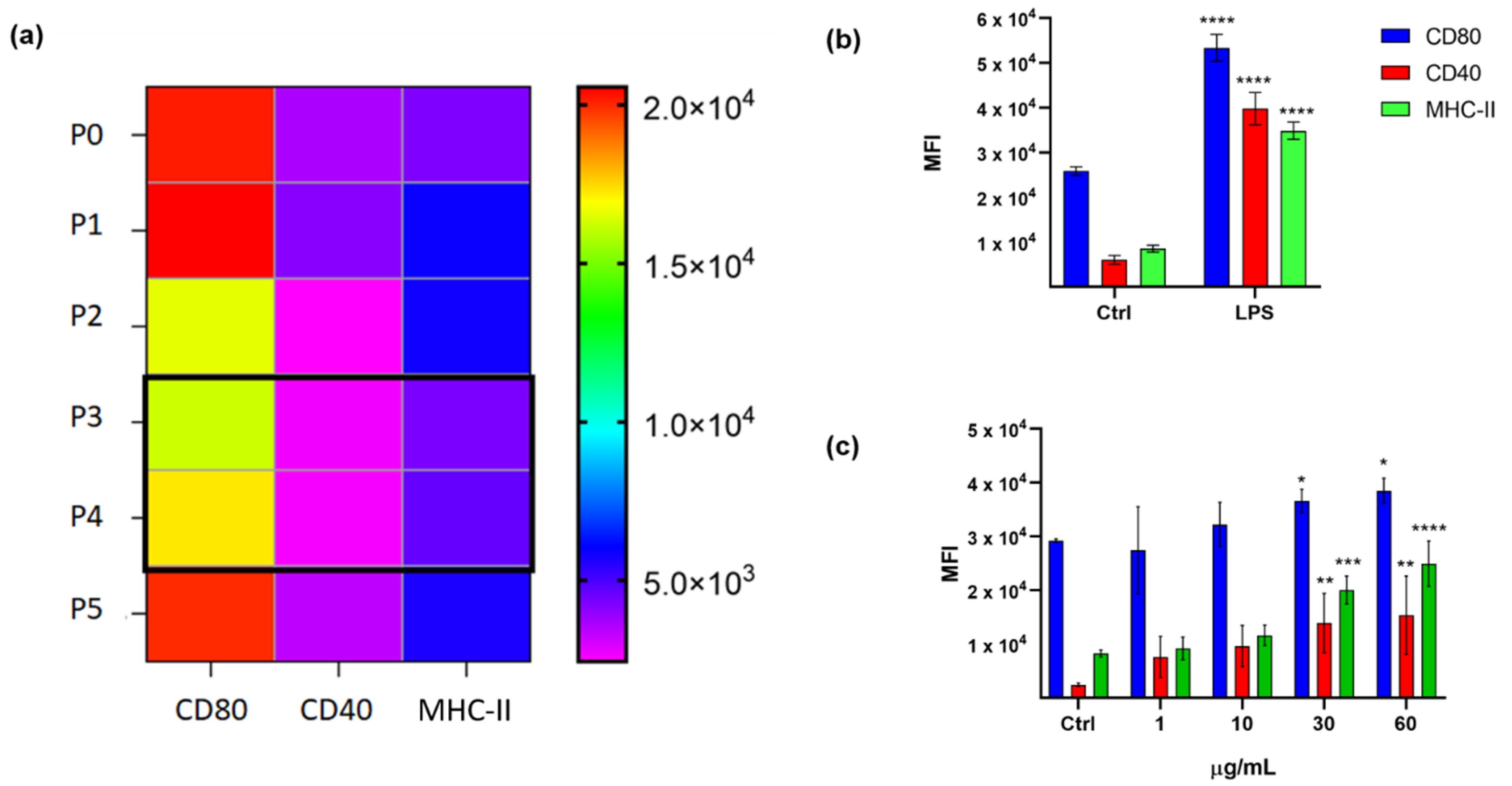
2.1. Immunophenotypic Assay on Dendritic Cells
2.2. Cytotoxicity Assay on Tumor Cell Lines
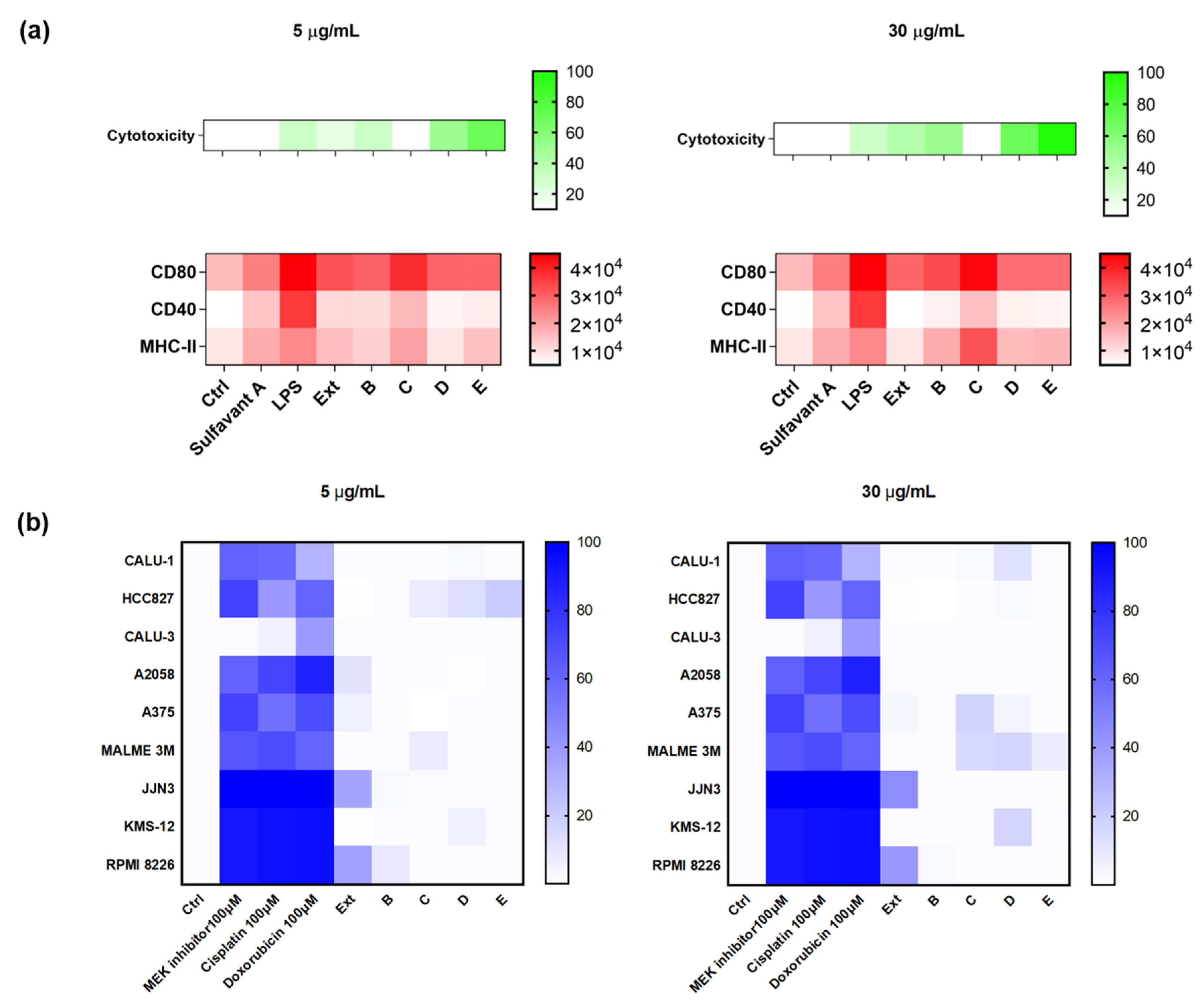
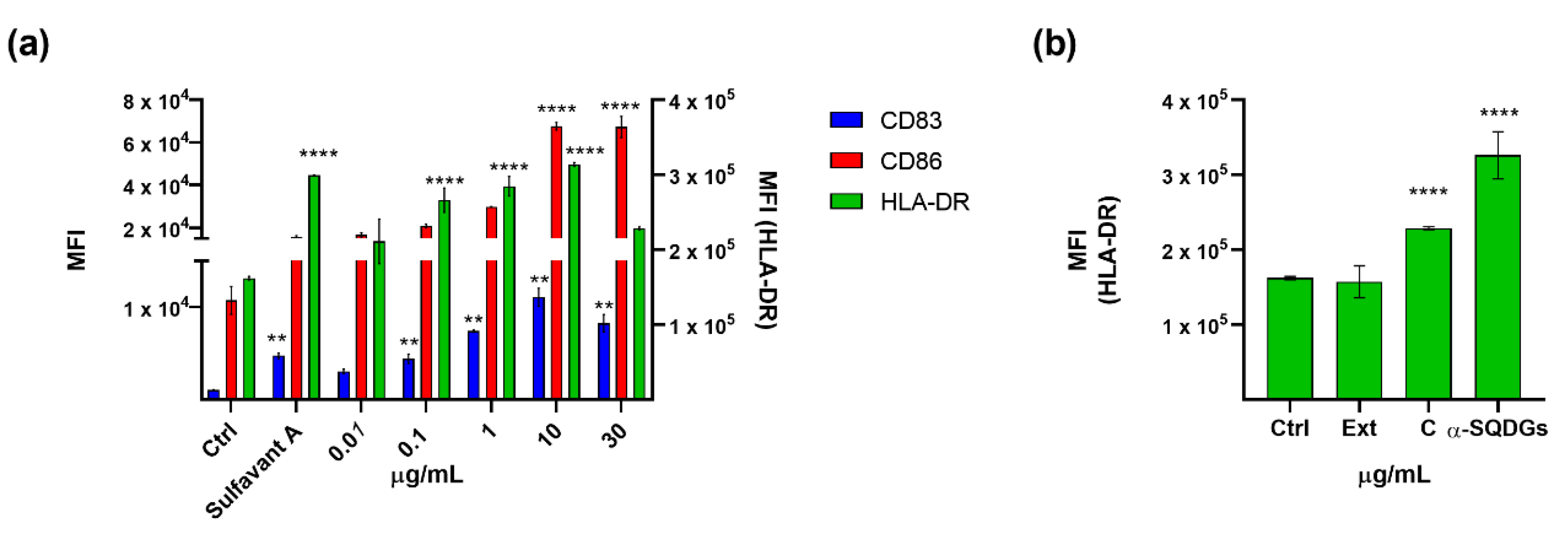
2.3. Validation of the Hits Selected in D1 and Cytotoxic Assay
2.4. Application of the Screening Platform to A Marine Natural Extract
3. Discussion
4. Materials and Methods
4.1. General
4.2. Cell Lines
4.3. Cell Culture Conditions
4.4. Cell Lines Titration and Treatments
4.5. Cytotoxicity Assays
4.6. D1 Cell Assay
4.7. MoDCs Assay
4.8. Diatom Cultivation
4.9. Extraction and Fractionation of T.weissflogii
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer Immunotherapy: Beyond Checkpoint Blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.E.; Ma, A. Molecular Control of Steady-State Dendritic Cell Maturation and Immune Homeostasis. Annu. Rev. Immunol. 2013, 31, 743–791. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Vasievich, E.A.; Huang, L. The Suppressive Tumor Microenvironment: A Challenge in Cancer Immunotherapy. Mol. Pharm. 2011, 8, 635–641. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Perrotta, F.; Barra, G.; Malapelle, U.; Rocco, D.; De Palma, R. Prognostic factors and biomarkers of responses to immune checkpoint inhibitors in lung cancer. Int. J. Mol. Sci. 2019, 20, 4931. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskás, L.G. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int. J. Mol. Sci. 2016, 17, 1958. [Google Scholar] [CrossRef]
- Manzo, E.; Gallo, C.; Fioretto, L.; Nuzzo, G.; Barra, G.; Pagano, D.; Krauss, I.R.; Paduano, L.; Ziaco, M.; DellaGreca, M.; et al. Diasteroselective Colloidal Self-Assembly Affects the Immunological Response of the Molecular Adjuvant Sulfavant. ACS Omega 2019, 4, 7807–7814, corrected in 2019, 4, 10809. [Google Scholar] [CrossRef]
- Manzo, E.; Cutignano, A.; Pagano, D.; Gallo, C.; Barra, G.; Nuzzo, G.; Sansone, C.; Ianora, A.; Urbanek, K.; Fenoglio, D.; et al. A new marine-derived sulfoglycolipid triggers dendritic cell activation and immune adjuvant response. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Winzler, C.; Rovere, P.; Rescigno, M.; Granucci, F.; Penna, G.; Adorini, L.; Zimmermann, V.S.; Davoust, J.; Ricciardi-Castagnoli, P. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 1997, 185, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Mortellaro, A.; Urbano, M.; Citterio, S.; Foti, M.; Granucci, F.; Ricciardi-Castagnoli, P. Generation of Murine Growth Factor-Dependent Long-Term Dendritic Cell Lines to Investigate Host-Parasite Interactions. Methods Mol. Biol. 2009, 531, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Vizzardelli, C.; Virzi, E.; Rescigno, M.; Ricciardi-Castagnoli, P. Transcriptional reprogramming of dendritic cells by differentiation stimuli. Eur. J. Immunol. 2001, 31, 2539–2546. [Google Scholar] [CrossRef]
- Paglia, P.; Girolomoni, G.; Robbiati, F.; Granucci, F.; Ricciardi-Castagnoli, P. Immortalized dendritic cell line fully competent in antigen presentation initiates primary T cell responses in vivo. J. Exp. Med. 1993, 178, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Bigley, V.; Collin, M. Human mononuclear phagocyte system reunited. Semin. Cell Dev. Biol. 2015, 41, 59–69. [Google Scholar] [CrossRef]
- Blanco, P.; Palucka, A.K.; Pascual, V.; Banchereau, J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef]
- Gupta, S.; Chaudhary, K.; Kumar, R.; Gautam, A.; Nanda, J.S.; Dhanda, S.K.; Brahmachari, S.K.; Raghava, G.P.S. Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Sci. Rep. 2016, 6, 23857. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated With Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef]
- Scagliotti, G.V. The Biology of Epidermal Growth Factor Receptor in Lung Cancer. Clin. Cancer Res. 2004, 10, 4227S–4232S. [Google Scholar] [CrossRef]
- Yu, S.; Sha, H.; Qin, X.; Chen, Y.; Li, X.; Shi, M.; Feng, J. EGFR E746-A750 deletion in lung cancer represses antitumor immunity through the exosome-mediated inhibition of dendritic cells. Oncogene 2020, 39, 2643–2657. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Han, Y.; Li, J.; Chai, R.; Bai, C. Prognostic value of KRAS/TP53/PIK3CA in non-small cell lung cancer. Oncol. Lett. 2019, 7, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Amanuel, B.; Grieu, F.; Kular, J.; Millward, M.; Iacopetta, B. Incidence of BRAF p.Val600Glu and p.Val600Lys mutations in a consecutive series of 183 metastatic melanoma patients from a high incidence region. Pathology 2012, 44, 357–359. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF inhibition causes resilience of melanoma cell lines by inducing the secretion of FGF1. Oncogenesis 2018, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef]
- Chen, K.G.; Leapman, R.D.; Zhang, G.; Lai, B.; Valencia, J.C.; Cardarelli, C.O.; Vieira, W.D.; Hearing, V.J.; Gottesman, M.M. Influence of melanosome dynamics on melanoma drug sensitivity. J. Natl. Cancer Inst. 2009, 101, 1259–1271. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 2019, 8, 665. [Google Scholar] [CrossRef]
- Patel, A.G.; Kaufmann, S.H. How does doxorubicin work? eLife 2012, 1, e00387. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Udi, J.; Schüler, J.; Wider, D.; Ihorst, G.; Catusse, J.; Waldschmidt, J.; Schnerch, D.; Follo, M.; Wäsch, R.; Engelhardt, M. Potent in vitro and in vivo activity of sorafenib in multiple myeloma: Induction of cell death, CD138-downregulation and inhibition of migration through actin depolymerization. Br. J. Haematol. 2013, 161, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Jin, H.-O.; Seo, S.-K.; Woo, S.H.; Choe, T.-B.; An, S.; Hong, S.-I.; Lee, S.-J.; Lee, K.-H.; Park, I.-C. Sorafenib induces apoptotic cell death in human non-small cell lung cancer cells by down-regulating mammalian target of rapamycin (mTOR)-dependent survivin expression. Biochem. Pharmacol. 2011, 82, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of Cisplatin-Induced Cell Death In Vitro and In Vivo on Cyclin-Dependent Kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef]
- Cheong, C.; Matos, I.; Choi, J.-H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial Stimulation Fully Differentiates Monocytes to DC-SIGN/CD209+ Dendritic Cells for Immune T Cell Areas. Cell 2010, 143, 416–429. [Google Scholar] [CrossRef]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef]
- Lauritano, C.; Andersen, J.H.; Hansen, E.; Albrigtsen, M.; Escalera, L.; Esposito, F.; Helland, K.; Hanssen, K.Ø.; Romano, G.; Ianora, A. Bioactivity Screening of Microalgae for Antioxidant, Anti-Inflammatory, Anticancer, Anti-Diabetes, and Antibacterial Activities. Front. Mar. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Samarakoon, K.W.; Ko, J.-Y.; Shah, M.M.R.; Lee, J.-H.; Kang, M.-C.; Kwon, O.-N.; Lee, J.-B.; Jeon, Y.-J. In vitro studies of anti-inflammatory and anticancer activities of organic solvent extracts from cultured marine microalgae. ALGAE 2013, 28, 111–119. [Google Scholar] [CrossRef]
- Manzo, E.; Gallo, C.; Sartorius, R.; Nuzzo, G.; Sardo, A.; De Berardinis, P.; Fontana, A.; Cutignano, A. Immunostimulatory Phosphatidylmonogalactosyldiacylglycerols (PGDG) from the Marine Diatom Thalassiosira weissflogii: Inspiration for a Novel Synthetic Toll-Like Receptor 4 Agonist. Mar. Drugs 2019, 17, 103. [Google Scholar] [CrossRef]
- Cutignano, A.; Nuzzo, G.; Ianora, A.; Luongo, E.; Romano, G.; Gallo, C.; Sansone, C.; Aprea, S.; Mancini, F.; D’Oro, U.; et al. Development and Application of a Novel SPE-Method for Bioassay-Guided Fractionation of Marine Extracts. Mar. Drugs 2015, 13, 5736–5749. [Google Scholar] [CrossRef]
- Cutignano, A.; Luongo, E.; Nuzzo, G.; Pagano, D.; Manzo, E.; Sardo, A.; Fontana, A. Profiling of complex lipids in marine microalgae by UHPLC/tandem mass spectrometry. Algal Res. 2016, 17, 348–358. [Google Scholar] [CrossRef]
- Kerr, W.G.; Chisholm, J.D. The Next Generation of Immunotherapy for Cancer: Small Molecules Could Make Big Waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Tang, C.; Niu, Q.; Ma, T.; Du, Y.; Fu, H. HTiP: High-Throughput Immunomodulator Phenotypic Screening Platform to Reveal IAP Antagonists as Anti-cancer Immune Enhancers. Cell Chem. Biol. 2019, 26, 331–339.e3. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Mitsiades, C.S. High-throughput approaches to discover novel immunomodulatory agents for cancer. Oncoimmunology 2012, 1, 1406–1408. [Google Scholar] [CrossRef][Green Version]
- Del Palacio, J.P.; Díaz, C.; de la Cruz, M.; Annang, F.; Martín, J.; Pérez-Victoria, I.; González-Menéndez, V.; de Pedro, N.; Tormo, J.R.; Algieri, F.; et al. High-Throughput Screening Platform for the Discovery of New Immunomodulator Molecules from Natural Product Extract Libraries. J. Biomol. Screen. 2016, 21, 567–578. [Google Scholar] [CrossRef] [PubMed]
- El-Murr, T.; Patel, A.; Sedlak, C.; D’Souza-Lobo, B. Evaluating dendritic cells as an in vitro screening tool for immunotherapeutic formulations. J. Immunol. Methods 2018, 459, 55–62. [Google Scholar] [CrossRef]
- Mizumoto, N.; Gao, J.; Matsushima, H.; Ogawa, Y.; Tanaka, H.; Takashima, A. Discovery of novel immunostimulants by dendritic-cell-based functional screening. Blood 2005, 106, 3082–3089. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Scheicher, C.; Mehlig, M.; Zecher, R.; Reske, K. Dendritic cells from mouse bone marrow: In vitro differentiation using low doses of recombinant granulocyte-macrophage colony-stimulating factor. J. Immunol. Methods 1992, 154, 253–264. [Google Scholar] [CrossRef]
- Inaba, K.; Swiggard, W.J.; Steinman, R.M.; Romani, N.; Schuler, G. Isolation of Dendritic Cells. Curr. Protoc. Immunol. 2009, 25, 3.7.1–3.7.15. [Google Scholar] [CrossRef] [PubMed]
- Marraco, S.A.F.; Grosjean, F.; Duval, A.; Rosa, M.; Lavanchy, C.; Ashok, D.; Haller, S.; Otten, L.A.; Steiner, Q.-G.; Descombes, P.; et al. Novel murine dendritic cell lines: A powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012, 3, 331–356. [Google Scholar] [CrossRef]
- Ebihara, S.; Endo, S.; Ito, K.; Ito, Y.; Akiyama, K.; Obinata, M.; Takai, T. Immortalized Dendritic Cell Line with Efficient Cross-Priming Ability Established from Transgenic Mice Harboring the Temperature-Sensitive SV40 Large T-Antigen Gene. J. Biochem. 2004, 136, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Gaugler, B.; Olive, D. Generation of Leukemic Dendritic Cells from Patients with Acute Myeloid Leukemia. In Cytokines and Colony Stimulating Factors; Humana Press: New Jersey, NJ, USA, 2003; pp. 463–472. [Google Scholar]
- Ruiz, S.; Beauvillain, C.; Mévélec, M.-N.; Roingeard, P.; Breton, P.; Bout, D.; Dimier-Poisson, I. A novel CD4-CD8α+CD205+CD11b- murine spleen dendritic cell line: Establishment, characterization and functional analysis in a model of vaccination to toxoplasmosis. Cell. Microbiol. 2005, 7, 1659–1671. [Google Scholar] [CrossRef] [PubMed]
- Van Helden, S.F.G.; van Leeuwen, F.N.; Figdor, C.G. Human and murine model cell lines for dendritic cell biology evaluated. Immunol. Lett. 2008, 117, 191–197. [Google Scholar] [CrossRef]
- Santegoets, S.J.A.M.; van den Eertwegh, A.J.M.; van de Loosdrecht, A.A.; Scheper, R.J.; de Gruijl, T.D. Human dendritic cell line models for DC differentiation and clinical DC vaccination studies. J. Leukoc. Biol. 2008, 84, 1364–1373. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mutations | Disease |
|---|---|---|
| LUNG | ||
| HCC827 | EGFR | Adenocarcinoma |
| CALU-1 | KRAS; P53 | grade III, epidermoid carcinoma |
| CALU-3 | P53 | Adenocarcinoma |
| MELANOMA | ||
| A375 | BRAF; CDKN2A; TERT | malignant melanoma |
| A2058 | BRAF; TERT; TP53 | Melanoma |
| MALME-3M | BRAF; CDKN2A; TERT | malignant melanoma |
| MYELOMA | ||
| KMS-12 | TP53 | multiple myeloma |
| RPMI8226 | EGFR; KRAS; TP53 | Plasmacytoma |
| JJN-3 | - | plasma cell leukemia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, C.; Barra, G.; Saponaro, M.; Manzo, E.; Fioretto, L.; Ziaco, M.; Nuzzo, G.; d’Ippolito, G.; De Palma, R.; Fontana, A. A New Bioassay Platform Design for the Discovery of Small Molecules with Anticancer Immunotherapeutic Activity. Mar. Drugs 2020, 18, 604. https://doi.org/10.3390/md18120604
Gallo C, Barra G, Saponaro M, Manzo E, Fioretto L, Ziaco M, Nuzzo G, d’Ippolito G, De Palma R, Fontana A. A New Bioassay Platform Design for the Discovery of Small Molecules with Anticancer Immunotherapeutic Activity. Marine Drugs. 2020; 18(12):604. https://doi.org/10.3390/md18120604
Chicago/Turabian StyleGallo, Carmela, Giusi Barra, Marisa Saponaro, Emiliano Manzo, Laura Fioretto, Marcello Ziaco, Genoveffa Nuzzo, Giuliana d’Ippolito, Raffaele De Palma, and Angelo Fontana. 2020. "A New Bioassay Platform Design for the Discovery of Small Molecules with Anticancer Immunotherapeutic Activity" Marine Drugs 18, no. 12: 604. https://doi.org/10.3390/md18120604
APA StyleGallo, C., Barra, G., Saponaro, M., Manzo, E., Fioretto, L., Ziaco, M., Nuzzo, G., d’Ippolito, G., De Palma, R., & Fontana, A. (2020). A New Bioassay Platform Design for the Discovery of Small Molecules with Anticancer Immunotherapeutic Activity. Marine Drugs, 18(12), 604. https://doi.org/10.3390/md18120604