Applying a Chemogeographic Strategy for Natural Product Discovery from the Marine Cyanobacterium Moorena bouillonii


 , , , , , and
, , , , , and
Abstract
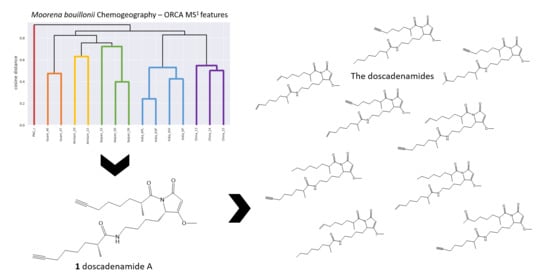
{kind=link}
{kind=link}
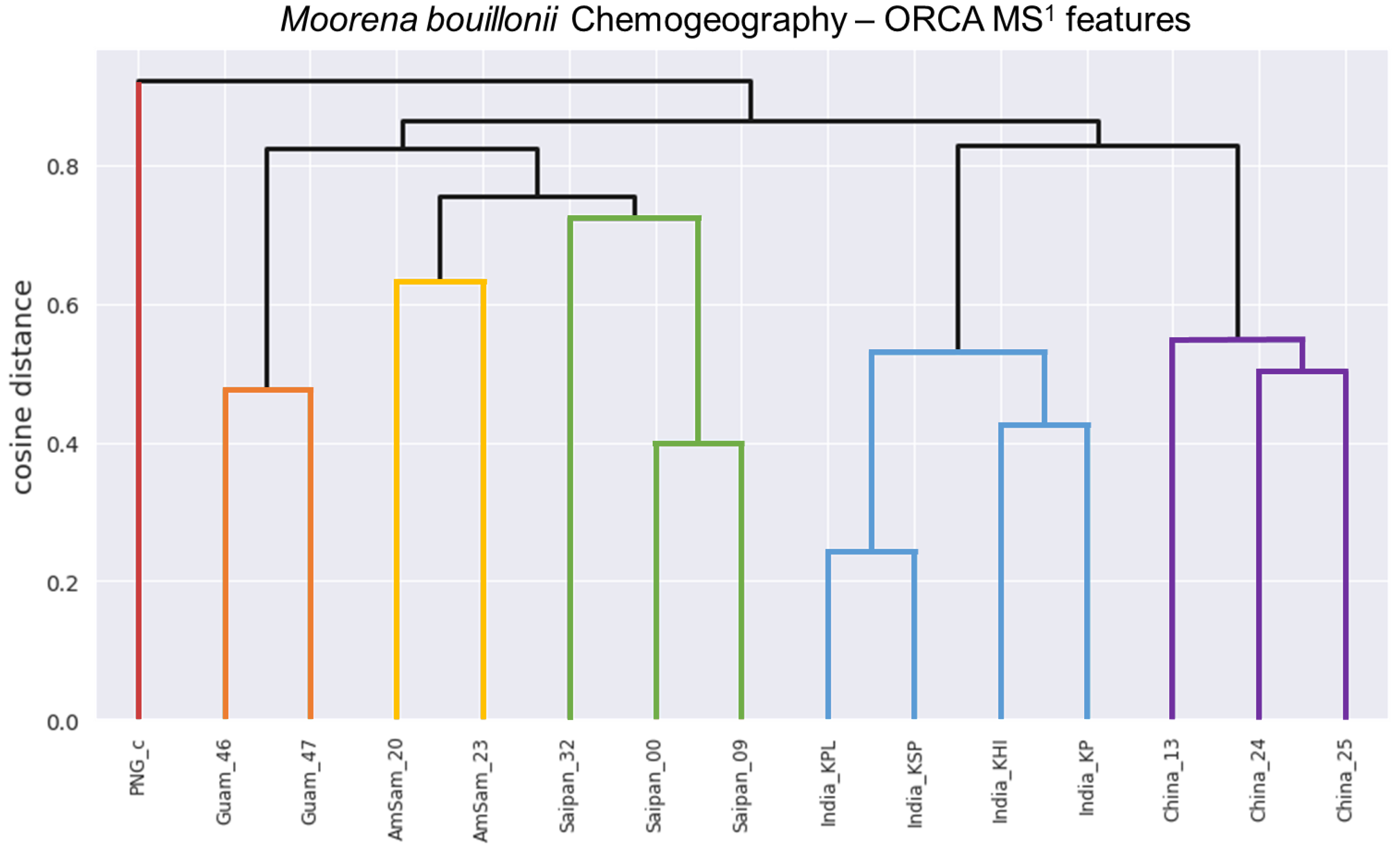{kind=link}
{kind=link}
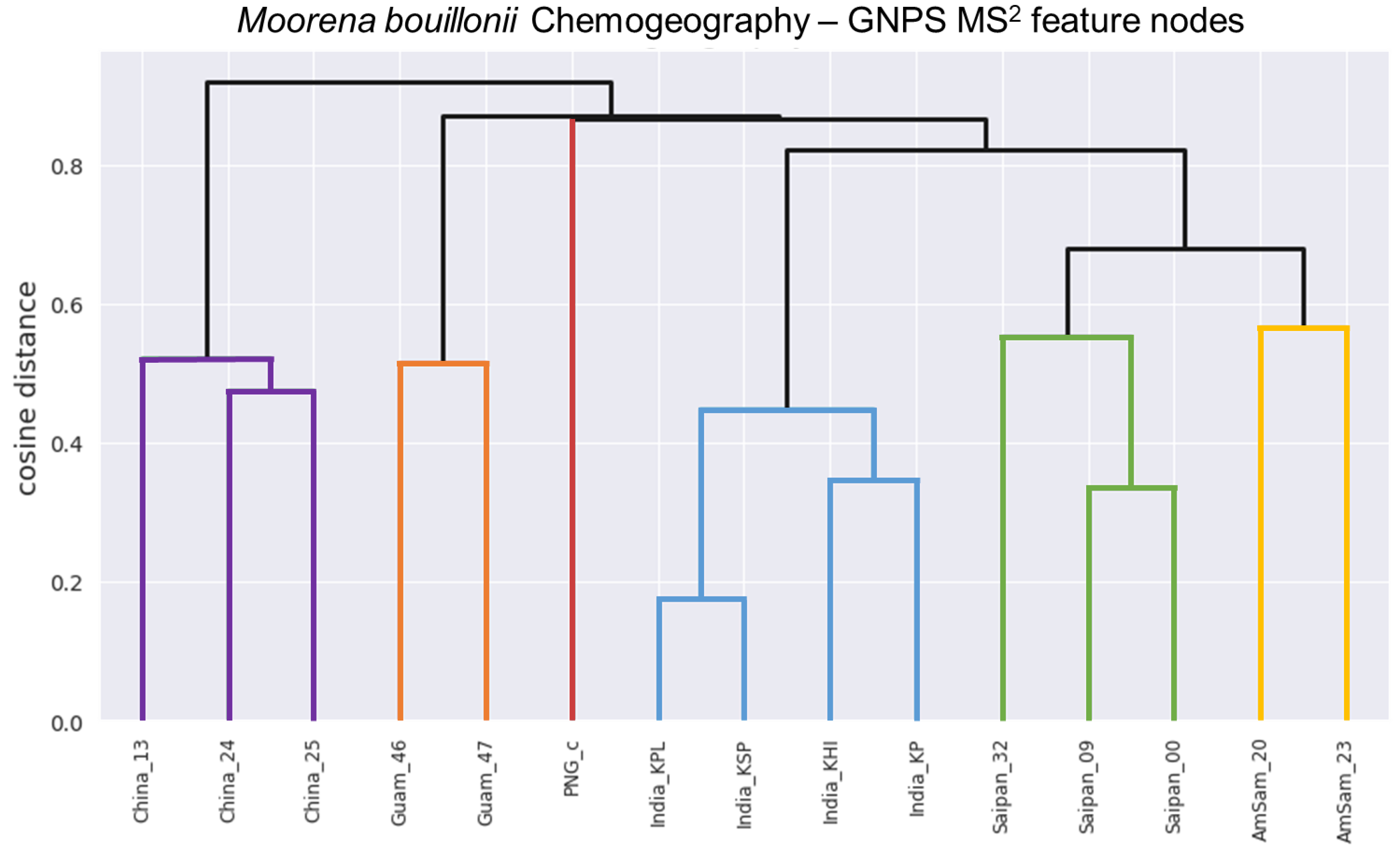{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
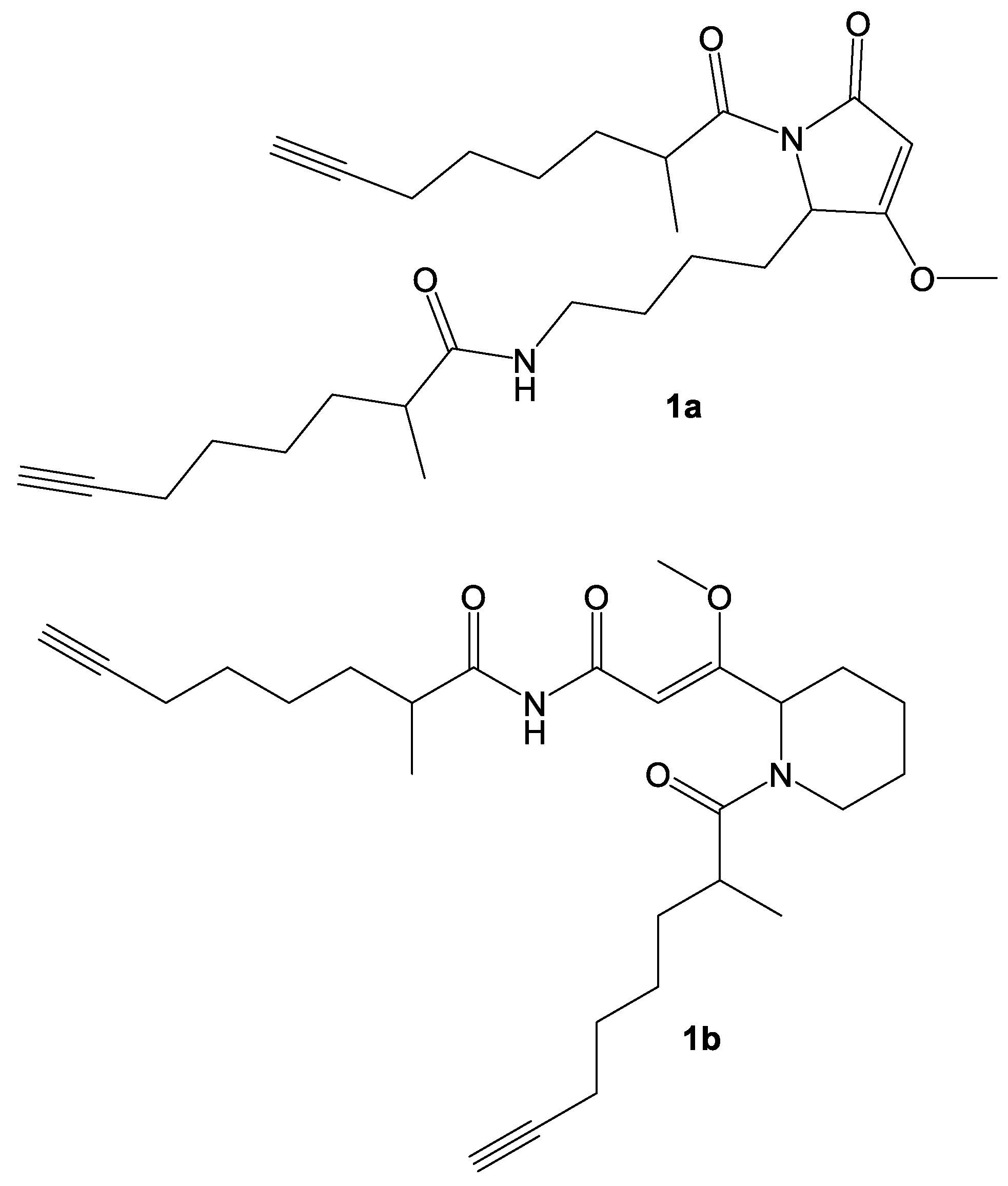
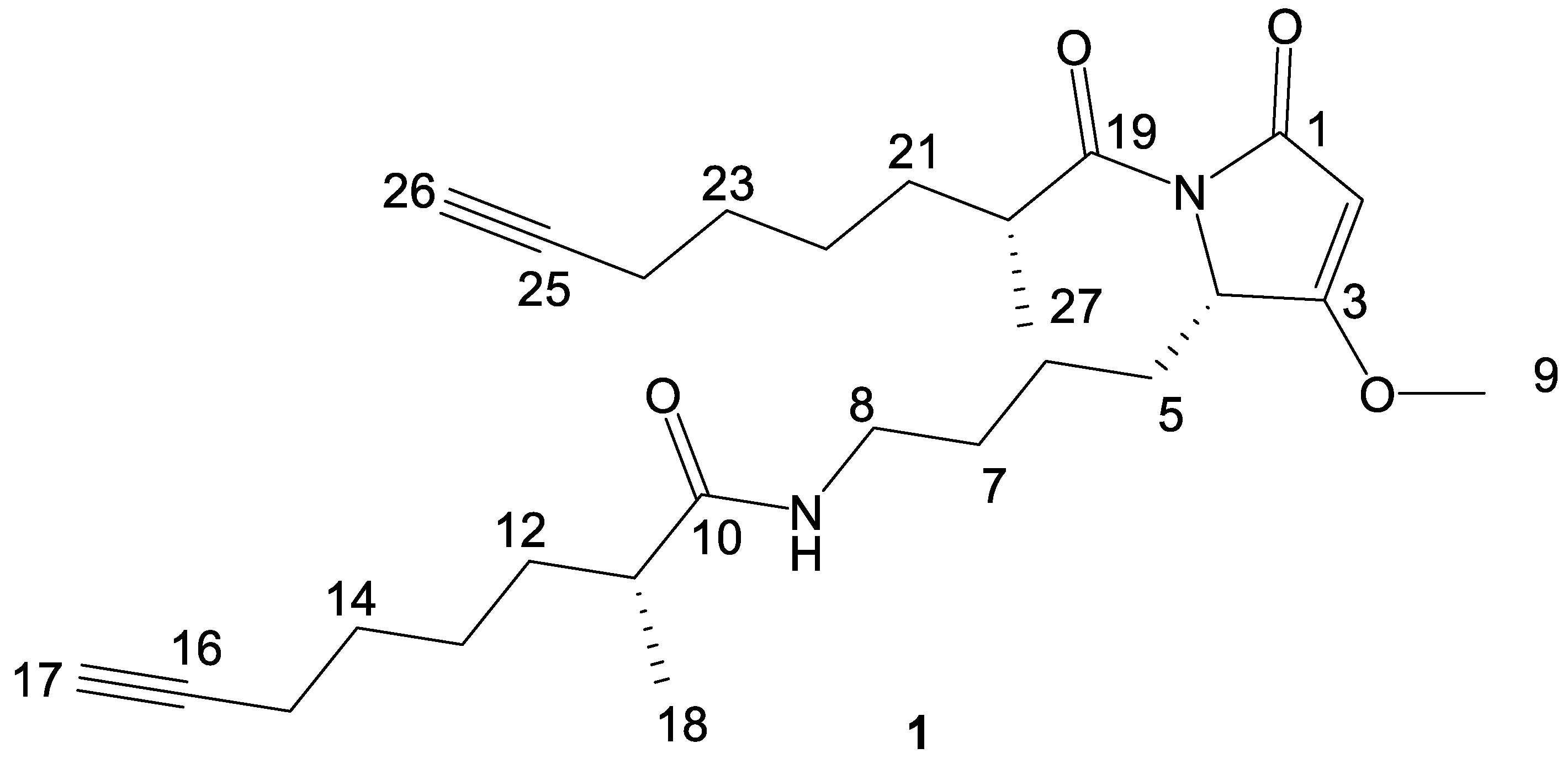
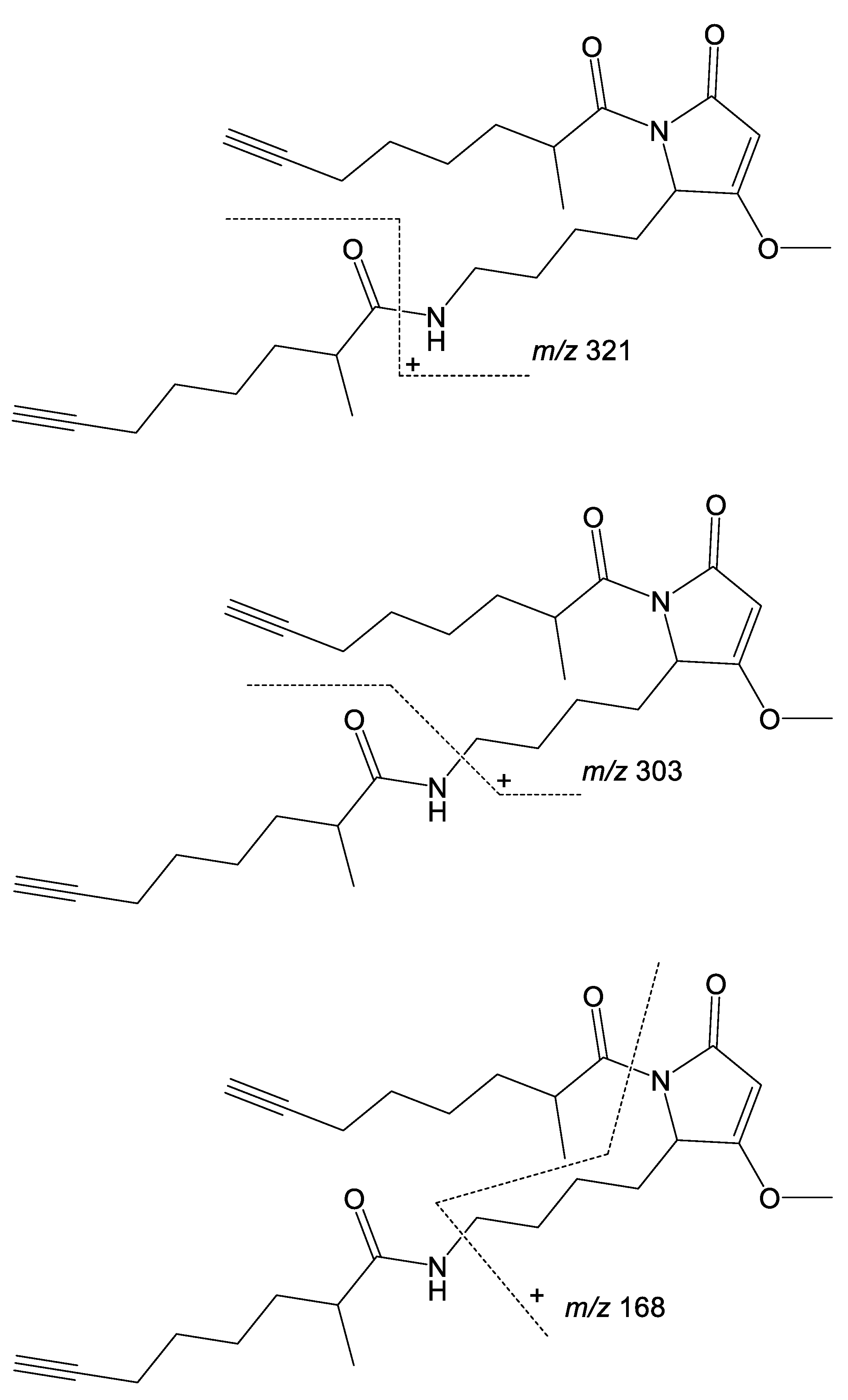
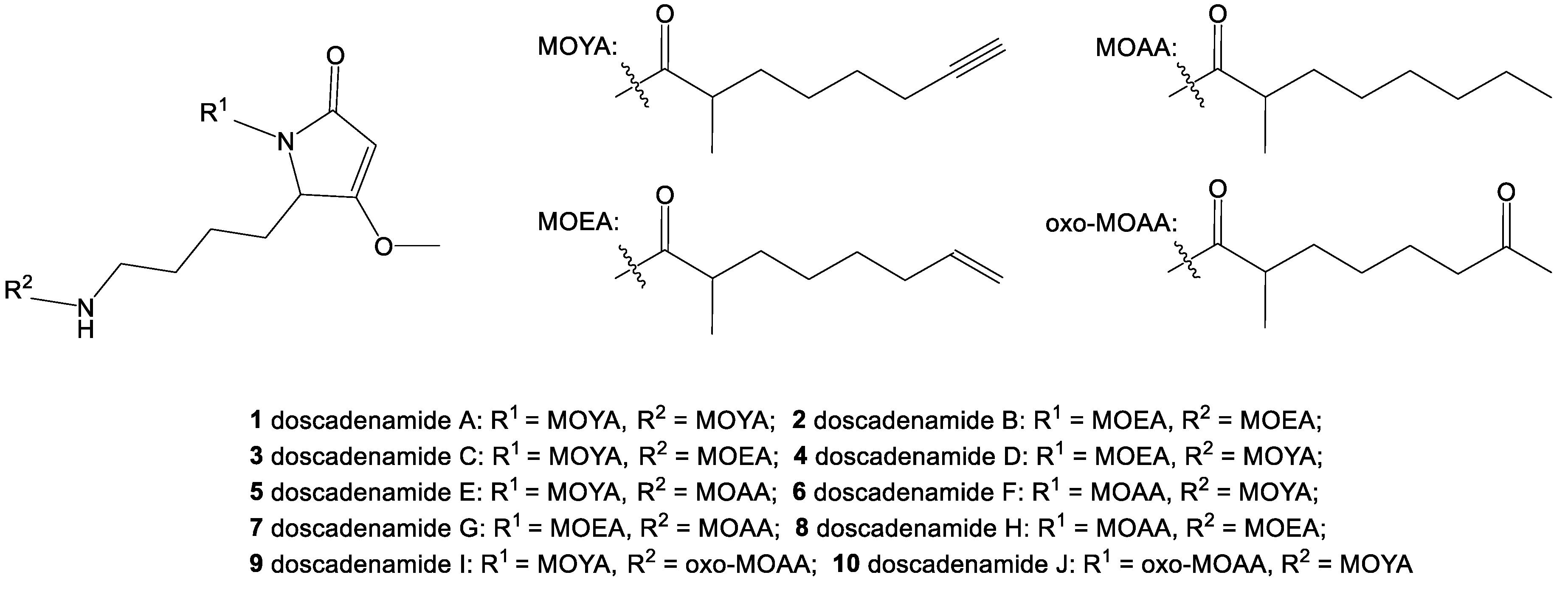
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Sample Collection
3.3. Sample Preparation
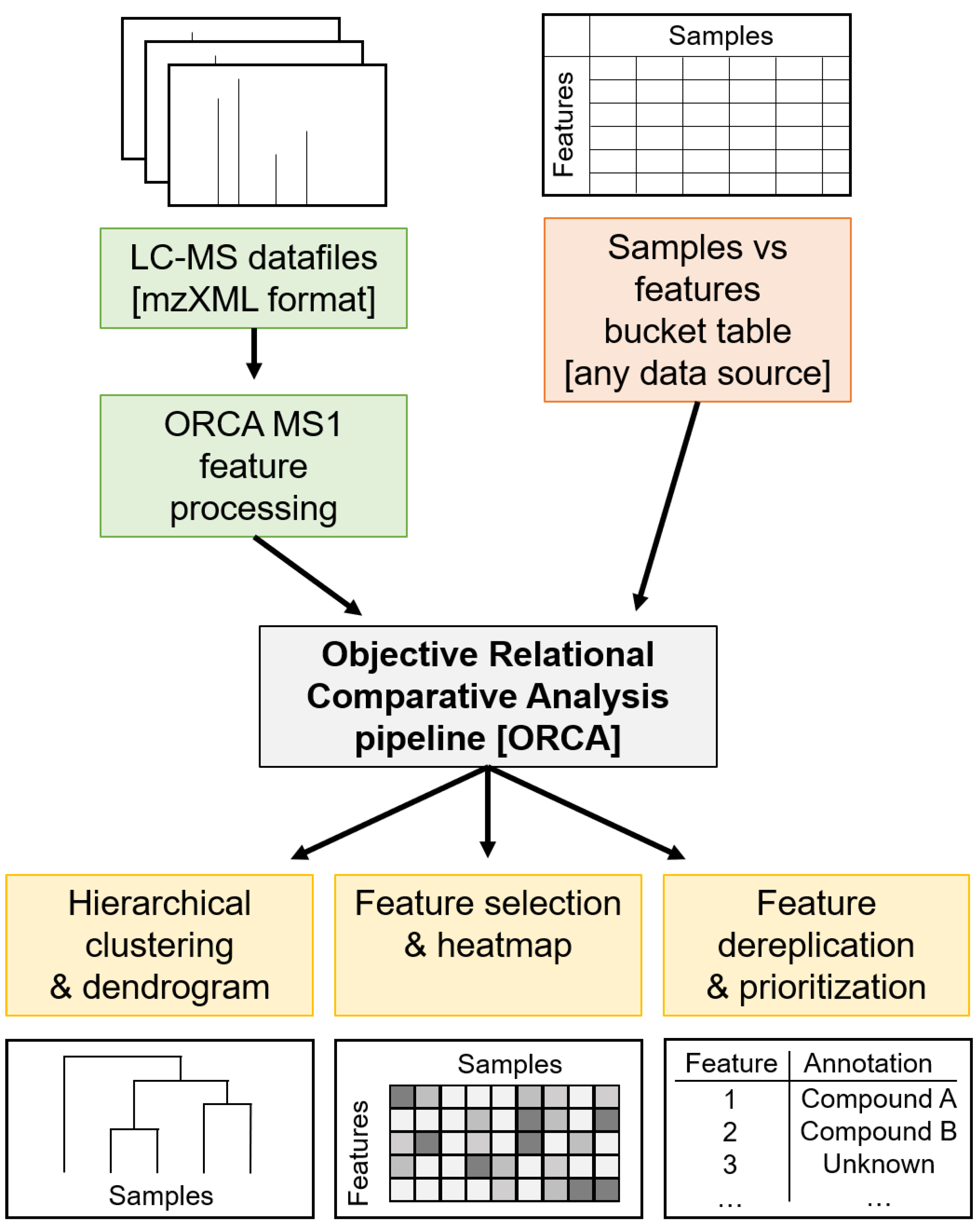
3.4. ORCA Pipeline
3.5. GNPS Classical Molecular Networking
3.6. Compound Isolation
3.7. Planar Structure Characterization
3.8. Structure Elucidation—Standard Preparation and Derivatization for Configurational Characterization
3.8.1. Synthesis of (S)-2-methyloctanoic Acid
3.8.2. Derivatization of 2-methyloctanoic Acid with 2-phenylglycine Methyl Ester
3.8.3. Derivatization of Lysine with Marfey’s Reagent (FDAA)
3.8.4. Derivatization of Compound 1
3.9. ORCA MS2 Auxiliary Pipeline
3.10. Bioassays
3.10.1. Cytotoxicity Assay of Compound 1 with NCI-H460 Cell Line
3.10.2. Griess Assay and Cytotoxicity of Compound 1 in RAW 264.7 Cells
3.10.3. Griess Reaction
3.10.4. In silico Antibiotic Screening
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chanana, S.; Thomas, C.S.; Braun, D.R.; Hou, Y.; Wyche, T.P.; Bugni, T.S. Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR). Metabolites 2017, 7, 34. [Google Scholar] [CrossRef]
- Clark, C.M.; Costa, M.S.; Sanchez, L.M.; Murphy, B.T. Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function. Proc. Natl. Acad. Sci. USA 2018, 115, 4981–4986. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based Molecular Networking in the GNPS Analysis Environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Tronholm, A.; Engene, N. Moorena gen. nov., a valid name for “Moorea Engene & al.” nom. inval. (Oscillatoriaceae, Cyanobacteria). Notulae Algarum 2019, 122, 1–2. [Google Scholar]
- Engene, N.; Rottacker, E.C.; Kaštovský, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komárek, J.; Gerwick, W.H. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Micr. 2012, 62, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Leao, T.; Castelão, G.; Korobeynikov, A.; Monroe, E.A.; Podell, S.; Glukhov, E.; Allen, E.E.; Gerwick, W.H.; Gerwick, L. Comparative genomics uncovers the prolific and distinctive metabolic potential of the cyanobacterial genus Moorea. Proc. Natl. Acad. Sci. USA 2017, 114, 3198–3203. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Continuing Studies on the Cyanobacterium Lyngbya sp.: Isolation and Structure Determination of 15-Norlyngbyapeptin A and Lyngbyabellin D. J. Nat. Prod. 2003, 66, 595–598. [Google Scholar] [CrossRef]
- Matthew, S.; Salvador, L.A.; Schupp, P.J.; Paul, V.J.; Luesch, H. Cytotoxic Halogenated Macrolides and Modified Peptides from the Apratoxin-Producing Marine Cyanobacterium Lyngbya bouillonii from Guam. J. Nat. Prod. 2010, 73, 1544–1552. [Google Scholar] [CrossRef]
- Choi, H.; Mevers, E.; Byrum, T.; Valeriote, F.A.; Gerwick, W.H. Lyngbyabellins K-N from two Palmyra atoll collections of the marine cyanobacterium Moorea bouillonii. Eur. J. Org. Chem. 2012, 2012, 5141–5150. [Google Scholar] [CrossRef]
- Soria-Mercado, I.E.; Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Alotamide A, a novel neuropharmacological agent from the marine cyanobacterium Lyngbya bouillonii. Org. Lett. 2009, 11, 4704–4707. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Apramides A−G, Novel Lipopeptides from the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1106–1112. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total Structure Determination of Apratoxin A, a Potent Novel Cytotoxin from the Marine Cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.A.; McPhail, K.L. Apratoxin H and Apratoxin A Sulfoxide from the Red Sea Cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from guam and palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a Potent Cytotoxic Cyclodepsipeptide from Papua New Guinea Collections of the Marine Cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef]
- Matthew, S.; Schupp, P.J.; Luesch, H. Apratoxin E, a Cytotoxic Peptolide from a Guamanian Collection of the Marine Cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [Google Scholar] [CrossRef]
- Tidgewell, K.; Engene, N.; Byrum, T.; Media, J.; Doi, T.; Valeriote, F.A.; Gerwick, W.H. Evolved Diversification of a Modular Natural Product Pathway: Apratoxins F and G, Two Cytotoxic Cyclic Depsipeptides from a Palmyra Collection of Lyngbya bouillonii. Chembiochem 2010, 11, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Salvador-Reyes, L.A.; Zhang, W.; Chen, Q.Y.; Matthew, S.; Ratnayake, R.; Seo, S.J.; Dolles, S.; Gibson, D.J.; Paul, V.J.; et al. Apratyramide, a Marine-Derived Peptidic Stimulator of VEGF-A and Other Growth Factors with Potential Application in Wound Healing. ACS Chem. Biol. 2018, 13, 91–99. [Google Scholar] [CrossRef]
- Tan, L.T.; Okino, T.; Gerwick, W.H. Bouillonamide: A Mixed Polyketide-Peptide Cytotoxin from the Marine Cyanobacterium Moorea bouillonii. Mar. Drugs 2013, 11, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Rubio, B.K.; Parrish, S.M.; Yoshid, W.; Schupp, P.J.; Schils, T.; Williams, P.G. Depsipeptides from a Guamanian marine cyanobacterium, Lyngbya bouillonii, with selective inhibition of serine proteases. Tetrahedron Lett. 2010, 51, 6718–6721. [Google Scholar] [CrossRef]
- Kleigrewe, K.; Almaliti, J.; Tian, I.Y.; Kinnel, R.B.; Korobeynikov, A.; Monroe, E.A.; Duggan, B.M.; Di Marzo, V.; Sherman, D.H.; Dorrestein, P.C.; et al. Combining Mass Spectrometric Metabolic Profiling with Genomic Analysis: A Powerful Approach for Discovering Natural Products from Cyanobacteria. J. Nat. Prod. 2015, 78, 1671–1682. [Google Scholar] [CrossRef]
- Lopez, J.A.V.; Petitbois, J.G.; Vairappan, C.S.; Umezawa, T.; Matsuda, F.; Okino, T. Columbamides D and E: Chlorinated Fatty Acid Amides from the Marine Cyanobacterium Moorea bouillonii Collected in Malaysia. Org. Lett. 2017, 19, 4231–4234. [Google Scholar] [CrossRef]
- Mehjabin, J.J.; Wei, L.; Petitbois, J.G.; Umezawa, T.; Matsuda, F.; Vairappan, C.S.; Morikawa, M.; Okino, T. Biosurfactants from Marine Cyanobacteria Collected in Sabah, Malaysia. J. Nat. Prod. 2020, 83, 1925–1930. [Google Scholar] [CrossRef]
- Pereira, A.R.; McCue, C.F.; Gerwick, W.H. Cyanolide A, a Glycosidic Macrolide with Potent Molluscicidal Activity from the Papua New Guinea Cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2010, 73, 217–220. [Google Scholar] [CrossRef]
- Liang, X.; Matthew, S.; Chen, Q.Y.; Kwan, J.C.; Paul, V.J.; Luesch, H. Discovery and Total Synthesis of Doscadenamide A: A Quorum Sensing Signaling Molecule from a Marine Cyanobacterium. Org. Lett. 2019, 21, 7274–7278. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Maejima, H.; Kawamura, M.; Arai, D.; Okino, T.; Zhao, M.; Ye, T.; Lee, J.; Chang, Y.; Fusetani, N.; et al. Kakeromamide A, a new cyclic pentapeptide inducing astrocyte differentiation isolated from the marine cyanobacterium Moorea bouillonii. Bioorg. Med. Chem. Lett. 2018, 28, 2206–2209. [Google Scholar] [CrossRef]
- Sweeney-Jones, A.M.; Gagaring, K.; Antonova-Koch, J.; Zhou, H.; Mojib, N.; Soapi, K.; Skolnick, J.; McNamara, C.W.; Kubanek, J. Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens. Mar. Drugs 2020, 18, 167. [Google Scholar] [CrossRef]
- Sumimoto, S.; Iwasaki, A.; Ohno, O.; Sueyoshi, K.; Teruya, T.; Suenaga, K. Kanamienamide, an Enamide with an Enol Ether from the Marine Cyanobacterium Moorea bouillonii. Org. Lett. 2016, 18, 4884–4887. [Google Scholar] [CrossRef]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Demoulin, V. Laingolide, a novel 15-membered macrolide from Lyngbya bouillonii (cyanophyceae). Tetrahedron Lett. 1996, 37, 7519–7520. [Google Scholar] [CrossRef]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Castillo, G.; Demoulin, V. Madangolide and Laingolide A, Two Novel Macrolides from Lyngbya bouillonii (Cyanobacteria). J. Nat. Prod. 1999, 62, 934–936. [Google Scholar] [CrossRef]
- Tan, L.T.; Márquez, B.L.; Gerwick, W.H. Lyngbouilloside, a Novel Glycosidic Macrolide from the Marine Cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2002, 65, 925–928. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, Structure Determination, and Biological Activity of Lyngbyabellin A from the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and Structure of the Cytotoxin Lyngbyabellin B and Absolute Configuration of Lyngbyapeptin A from the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1437–1439. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Structurally diverse new alkaloids from Palauan collections of the apratoxin-producing marine cyanobacterium Lyngbya sp. Tetrahedron 2002, 58, 7959–7966. [Google Scholar] [CrossRef]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Demoulin, V. Lyngbyaloside, a Novel 2,3,4-Tri-O-methyl-6-deoxy-α-mannopyranoside Macrolide from Lyngbya bouillonii (Cyanobacteria). J. Nat. Prod. 1997, 60, 1057–1059. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Harrigan, G.G.; Doom, J.P.; Moore, R.E.; Paul, V.J. Lyngbyaloside B, a New Glycoside Macrolide from a Palauan Marine Cyanobacterium, Lyngbya sp. J. Nat. Prod. 2002, 65, 1945–1948. [Google Scholar] [CrossRef]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Castillo, G.; Demoulin, V. Lyngbyapeptin A, a modified tetrapeptide from Lyngbya bouillonii (Cyanophyceae). Tetrahedron Lett. 1999, 40, 695–696. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Lyngbyastatin 2 and Norlyngbyastatin 2, Analogues of Dolastatin G and Nordolastatin G from the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1999, 62, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Mevers, E.; Matainaho, T.; Allara, M.; Di Marzo, V.; Gerwick, W.H. Mooreamide A: A cannabinomimetic lipid from the marine cyanobacterium Moorea bouillonii. Lipids 2014, 49, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A−F, New β-Amino Acid-Containing Cyclodepsipeptides from Palauan Collections of the Marine Cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.A. Alkaloid-bearing plants: An ecogeographic perspective. Am. Nat. 1976, 110, 261–284. [Google Scholar] [CrossRef]
- Coley, P.D.; Aide, T.M. Comparison of herbivory and plant defenses in temperate and tropical broad-leaved forests. In Plant–Animal Interaction: Evolutionary Ecology in Tropical and Temperate Regions; Wiley-Interscience: New York, NY, USA, 1991; pp. 25–49. [Google Scholar]
- Coley, P.D.; Barone, J.A. Herbivory and plant defenses in tropical forests. Annu. Rev. Ecol. Syst. 1996, 27, 305–335. [Google Scholar] [CrossRef]
- Rasmann, S.; Agrawal, A.A. Latitudinal patterns in plant defense: Evolution of cardenolides, their toxicity and induction following herbivory. Ecol. Lett. 2011, 14, 476–483. [Google Scholar] [CrossRef]
- Bakus, G.J.; Green, G. Toxicity in sponges and holothurians: A geographic pattern. Science 1974, 185, 951–953. [Google Scholar] [CrossRef]
- Hay, M.E.; Fenical, W. Marine plant-herbivore interactions: The ecology of chemical defense. Annu. Rev. Ecol. Syst. 1988, 19, 111–145. [Google Scholar] [CrossRef]
- Bolser, R.C.; Hay, M.E. Are tropical plants better defended? Palatability and defenses of temperate vs tropical seaweeds. Ecology 1996, 77, 2269–2286. [Google Scholar] [CrossRef]
- Anstett, D.N.; Nunes, K.A.; Baskett, C.; Kotanen, P.M. Sources of Controversy Surrounding Latitudinal Patterns in Herbivory and Defense. Trends Ecol. Evol. 2016, 31, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kooyers, N.J.; Blackman, B.K.; Holeski, L.M. Optimal defense theory explains deviations from latitudinal herbivory defense hypothesis. Ecology 2017, 98, 1036–1048. [Google Scholar] [CrossRef]
- Shang, Z.; Winter, J.M.; Kauffman, C.A.; Yang, I.; Fenical, W. Salinipeptins: Integrated Genomic and Chemical Approaches Reveal D-Amino Acid-Containing Ribosomally Synthesized and Post-Translationally Modified Peptides from a Great Salt Lake Streptomyces sp. ACS Chem. Biol. 2019, 14, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Marcolefas, E.; Leung, T.; Okshevsky, M.; McKay, G.; Hignett, E.; Hamel, J.; Aguirre1, G.; Blenner-Hassett, O.; Boyle, B.; Lévesque, R.C.; et al. Culture-Dependent Bioprospecting of Bacterial Isolates From the Canadian High Arctic Displaying Antibacterial Activity. Front. Microbiol. 2019, 10, 1836. [Google Scholar] [CrossRef] [PubMed]
- Bory, A.; Shilling, A.J.; Allen, J.; Azhari, A.; Roth, A.; Shaw, L.N.; Kyle, D.E.; Adams, J.H.; Amsler, C.D.; McClintock, J.B.; et al. Bioactivity of Spongian Diterpenoid Scaffolds from the Antarctic Sponge Dendrilla antarctica. Mar. Drugs 2020, 18, 327. [Google Scholar] [CrossRef]
- Zhou, H.; He, Y.; Tian, Y.; Cong, B.; Yang, H. Bacilohydrin A, a New Cytotoxic Cyclic Lipopeptide of Surfactins Class Produced by Bacillus sp. SY27F from the Indian Ocean Hydrothermal Vent. Nat. Prod. Commun. 2019, 14, 141–146. [Google Scholar] [CrossRef]
- Zhang, S.; Gui, C.; Shao, M.; Kumar, P.S.; Huang, H.; Ju, J. Antimicrobial tunicamycin derivatives from the deep sea-derived Streptomyces xinghaiensis SCSIO S15077. Nat. Prod. Res. 2020, 34, 1499–1504. [Google Scholar] [CrossRef]
- Luzzatto-Knaan, T.; Garg, N.; Wang, M.; Glukhov, E.; Peng, Y.; Ackermann, G.; Amir, A.; Duggan, B.M.; Ryazanov, S.; Gerwick, L.; et al. Digitizing mass spectrometry data to explore the chemical diversity and distribution of marine cyanobacteria and algae. eLife 2017, 6, e24214. [Google Scholar] [CrossRef]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A.; et al. Integrating molecular networking and biological assays to target the isolation of a cytotoxic cyclic octapeptide, samoamide A, from an American Samoan marine cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef]
- Crnkovic, C.M.; May, D.S.; Orjala, J. The impact of culture conditions on growth and metabolomic profiles of freshwater cyanobacteria. J. Appl. Phycol. 2018, 30, 375–384. [Google Scholar] [CrossRef]
- Pallarés, N.; Tolosa, J.; Mañes, J.; Ferrer, E. Occurrence of Mycotoxins in Botanical Dietary Supplement Infusion Beverages. J. Nat. Prod. 2019, 82, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Tronholm, A.; Paul, V.J. Uncovering cryptic diversity of Lyngbya: The new tropical marine cyanobacterial genus Dapis (Oscillatoriales). J. Phycol. 2018, 54, 435–446. [Google Scholar] [CrossRef]
- Jayaram, B.; Klawonn, F. Can unbounded distance measures mitigate the curse of dimensionality? Int. J. Data Min. Model. Manag. 2012, 4, 361–383. [Google Scholar] [CrossRef]
- Milligan, K.E.; Márquez, B.; Williamson, R.T.; Davies-Coleman, M.; Gerwick, W.H. Two New Malyngamides from a Madagascan Lyngbya majuscula. J. Nat. Prod. 2000, 63, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Clark, B.R.; Trimble, E.E.; Almanza, A.; Ureña, L.D.; Kyle, D.E.; Gerwick, W.H. Antimalarial Peptides from Marine Cyanobacteria: Isolation and Structural Elucidation of Gallinamide A. J. Nat. Prod. 2009, 72, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E.; Tan, Z.; Liang, B.; Novak, T. An efficient and general route to reduced polypropionates via Zr-catalyzed asymmetric C-C bond formation. Proc. Natl. Acad. Sci. USA 2004, 101, 5782–5787. [Google Scholar] [CrossRef]
- Pereira, A.; Etzbach, L.; Engene, N.; Müller, R.; Gerwick, W.H. Molluscicidal Metabolites from an Assemblage of Palmyra Atoll Cyanobacteria. J. Nat. Prod. 2011, 74, 1175–1181. [Google Scholar] [CrossRef]
- Boudreau, P.D.; Byrum, T.; Liu, W.T.; Dorrestein, P.C.; Gerwick, W.H. Viequeamide A, a Cytotoxic Member of the Kulolide Superfamily of Cyclic Depsipeptides from a Marine Button Cyanobacterium. J. Nat. Prod. 2012, 75, 1560–1570. [Google Scholar] [CrossRef]
- Hooper, G.J.; Orjala, J.; Schatzman, R.C.; Gerwick, W.H. Carmabins A and B, New Lipopeptides from the Caribbean Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1998, 61, 529–533. [Google Scholar] [CrossRef]
- Moss, N.A.; Seiler, G.; Leão, T.F.; Castro-Falcón, G.; Gerwick, L.; Hughes, C.C.; Gerwick, W.H. Nature’s Combinatorial Biosynthesis Produces Vatiamides A–F. Angew. Chem. Int. Ed. 2019, 58, 9027–9031. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Tan, L.T.; Sitachitta, N. Nitrogen-containing metabolites from marine cyanobacteria. In The Alkaloids: Chemistry and Biology, 1st ed.; Cordell, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2001; Volume 57, pp. 75–184. [Google Scholar] [CrossRef]
- Tidgewell, K.; Clark, B.R.; Gerwick, W.H. The Natural Products Chemistry of Cyanobacteria. In Comprehensive Natural Products II: Chemistry and Biology, 1st ed.; Mander, L.N., Liu, H.-W., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 2, pp. 144–188. [Google Scholar] [CrossRef]
- Zhang, C.; Idelbayev, Y.; Roberts, N.; Tao, Y.; Nannapaneni, Y.; Duggan, B.M.; Min, J.; Lin, E.C.; Gerwick, E.C.; Cottrell, G.W.; et al. Small Molecule Accurate Recognition Technology (SMART) to Enhance Natural Products Research. Sci. Rep. 2017, 7, 14243. [Google Scholar] [CrossRef]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Van Soest, R.; Andersen, R.J. Sintokamides A to E, Chlorinated Peptides from the Sponge Dysidea sp. that Inhibit Transactivation of the N-Terminus of the Androgen Receptor in Prostate Cancer Cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef]
- Lowery, C.A.; Park, J.; Gloeckner, C.; Meijler, M.M.; Mueller, R.S.; Boshoff, H.I.; Ulrich, R.L.; Barry, C.E.; Bartlett, D.H.; Kravchenko, V.V.; et al. Defining the Mode of Action of Tetramic Acid Antibacterials Derived from Pseudomonas aeruginosa Quorum Sensing Signals. J. Am. Chem. Soc. 2009, 131, 14473–14479. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 181, 475–483. [Google Scholar] [CrossRef]
- Moss, N.A.; Leao, T.; Glukhov, E.; Gerwick, L.; Gerwick, W.H. Collection, Culturing, and Genome Analyses of Tropical Marine Filamentous Benthic Cyanobacteria. In Methods in Enzymology, 1st ed.; Tawfik, D.S., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 604, pp. 3–43. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual, Release 2.0.1; PythonLabs: Amsterdam, The Netherlands, 1995; Available online: https://docs.python.org/2.0/ref/ref.html (accessed on 14 October 2020).
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference (SciPy2010), Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar] [CrossRef]
- Pandas-Dev/Pandas, Version v0.25.2, 2018, Zenodo. Available online: https://doi.org/10.5281/zenodo.3509135 (accessed on 14 October 2020).
- Oliphant, T.E. A Guide to NumPy, 2nd ed.; Trelgol Publishing: USA, 2006; Available online: https://web.mit.edu/dvp/Public/numpybook.pdf (accessed on 14 October 2020).
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Goloborodko, A.A.; Levitsky, L.I.; Ivanov, M.V.; Gorshkov, M.V. Pyteomics—A Python Framework for Exploratory Data Analysis and Rapid Software Prototyping in Proteomics. J. Am. Soc. Mass Spectr. 2013, 24, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, L.I.; Klein, J.; Ivanov, M.V.; Gorshkov, M.V. Pyteomics 4.0: Five years of development of a Python proteomics framework. J. Proteome Res. 2019, 18, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods. 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Mwaskom/Seaborn, v0.9.0. 2018. Available online: https://doi.org/10.5281/zenodo.1313201 (accessed on 14 October 2020).
- Pérez, F.; Granger, B.E. IPython: A System for Interactive Scientific Computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; Loizides, F., Scmidt, B., Eds.; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar] [CrossRef]
- Holman, J.D.; Tabb, D.L.; Mallick, P. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr. Protoc. Bioinform. 2014, 46, 1–9. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Iwasaki, A.; Ohno, O.; Sumimoto, S.; Ogawa, H.; Nguyen, K.A.; Suenaga, K. Jahanyne, an Apoptosis-Inducing Lipopeptide from the Marine Cyanobacterium Lyngbya sp. Org. Lett. 2015, 17, 652–655. [Google Scholar] [CrossRef]
- Linington, R.G.; González, J.; Ureña, L.D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial Constituents of the Panamanian Marine Cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef]
- Yoshimura, A.; Kishimoto, S.; Nishimura, S.; Otsuka, S.; Sakai, Y.; Hattori, A.; Kakeya, H. Prediction and Determination of the Stereochemistry of the 1,3,5-Trimethyl-Substituted Alkyl Chain in Verucopeptin, a Microbial Metabolite. J. Org. Chem. 2014, 79, 6858–6867. [Google Scholar] [CrossRef]
- Tao, Y.; Li, P.; Zhang, D.; Glukhov, E.; Gerwick, L.; Zhang, C.; Murray, T.F.; Gerwick, W.H. Samholides, Swinholide-Related Metabolites from a Marine Cyanobacterium cf. Phormidium sp. J. Org. Chem. 2018, 83, 3034–3046. [Google Scholar] [CrossRef]
- Choi, H.; Mascuch, S.J.; Villa, F.A.; Byrum, T.; Teasdale, M.E.; Smith, J.E.; Preskitt, L.B.; Rowley, D.C.; Gerwick, L.; Gerwick, W.H. Honaucins A-C, potent inhibitors of inflammation and bacterial quorum sensing: Synthetic derivatives and structure-activity relationships. Chem. Biol. 2012, 19, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [N-15]-labeled nitrate in biological-fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leber, C.A.; Naman, C.B.; Keller, L.; Almaliti, J.; Caro-Diaz, E.J.E.; Glukhov, E.; Joseph, V.; Sajeevan, T.P.; Reyes, A.J.; Biggs, J.S.; et al. Applying a Chemogeographic Strategy for Natural Product Discovery from the Marine Cyanobacterium Moorena bouillonii. Mar. Drugs 2020, 18, 515. https://doi.org/10.3390/md18100515
Leber CA, Naman CB, Keller L, Almaliti J, Caro-Diaz EJE, Glukhov E, Joseph V, Sajeevan TP, Reyes AJ, Biggs JS, et al. Applying a Chemogeographic Strategy for Natural Product Discovery from the Marine Cyanobacterium Moorena bouillonii. Marine Drugs. 2020; 18(10):515. https://doi.org/10.3390/md18100515
Chicago/Turabian StyleLeber, Christopher A., C. Benjamin Naman, Lena Keller, Jehad Almaliti, Eduardo J. E. Caro-Diaz, Evgenia Glukhov, Valsamma Joseph, T. P. Sajeevan, Andres Joshua Reyes, Jason S. Biggs, and et al. 2020. "Applying a Chemogeographic Strategy for Natural Product Discovery from the Marine Cyanobacterium Moorena bouillonii" Marine Drugs 18, no. 10: 515. https://doi.org/10.3390/md18100515
APA StyleLeber, C. A., Naman, C. B., Keller, L., Almaliti, J., Caro-Diaz, E. J. E., Glukhov, E., Joseph, V., Sajeevan, T. P., Reyes, A. J., Biggs, J. S., Li, T., Yuan, Y., He, S., Yan, X., & Gerwick, W. H. (2020). Applying a Chemogeographic Strategy for Natural Product Discovery from the Marine Cyanobacterium Moorena bouillonii. Marine Drugs, 18(10), 515. https://doi.org/10.3390/md18100515