Structure of the Polysaccharide Secreted by Vibrio alginolyticus CNCM I-5035 (Epidermist 4.0TM)

 ,
,
Abstract
1. Introduction
2. Results and Discussion
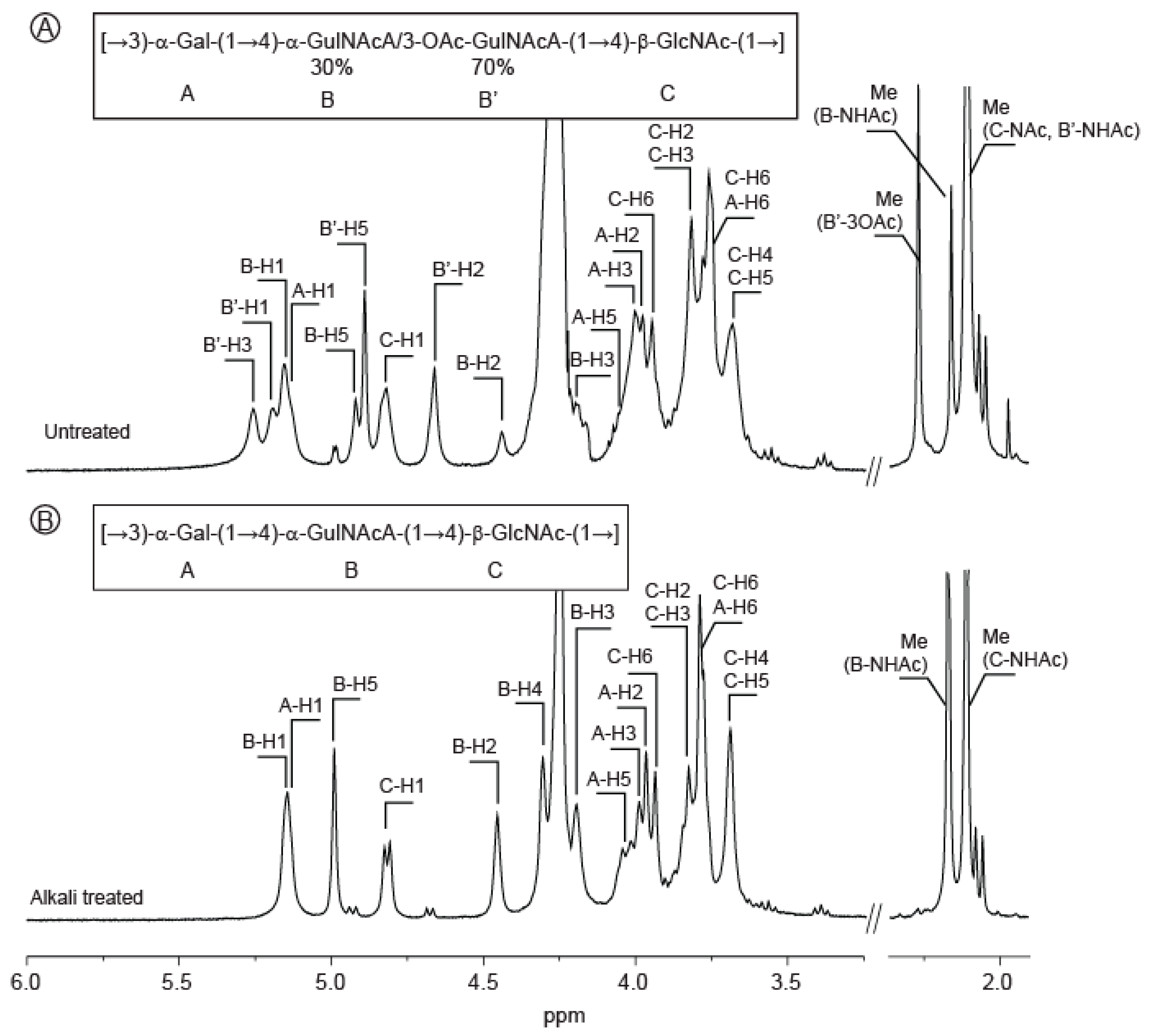
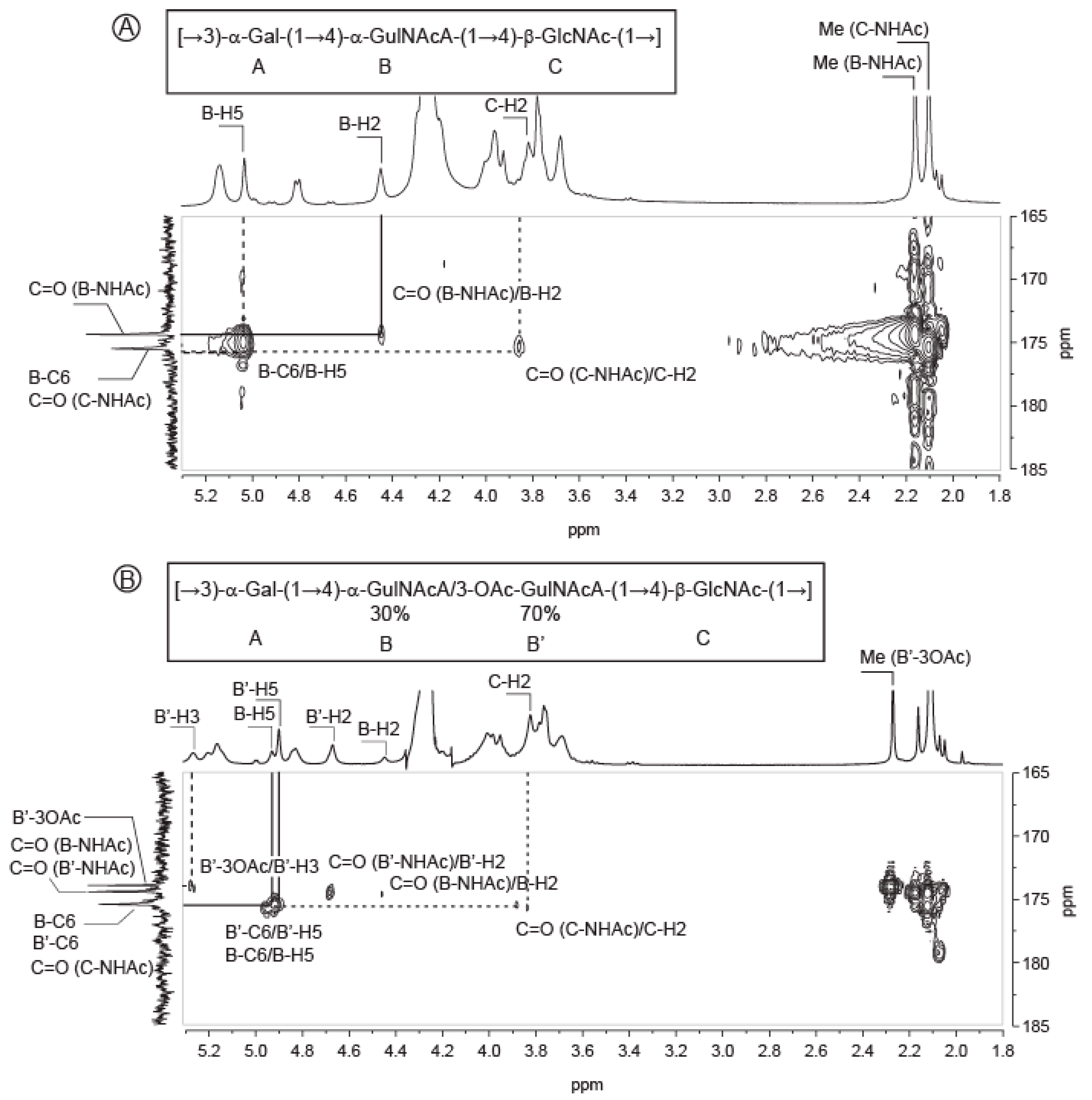
2.1. Composition of the Polysaccharide
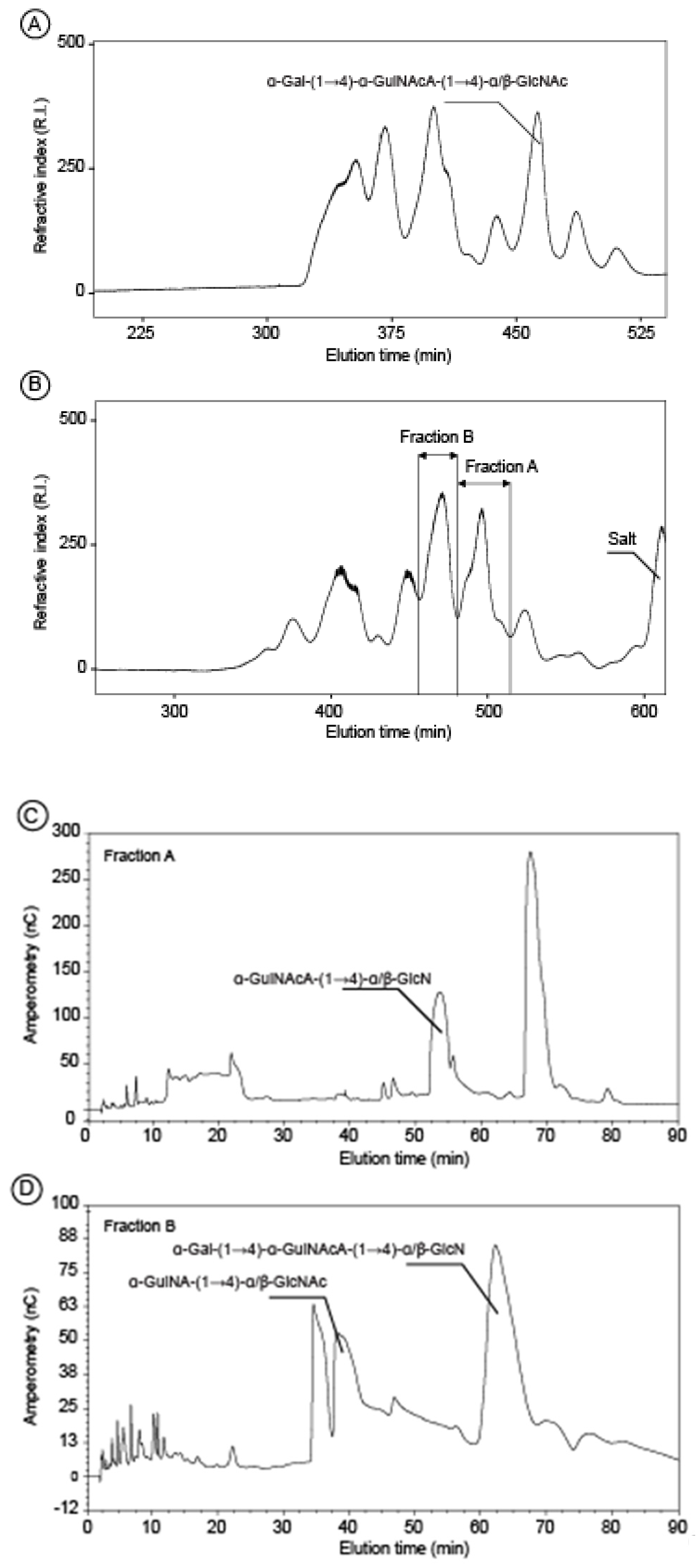
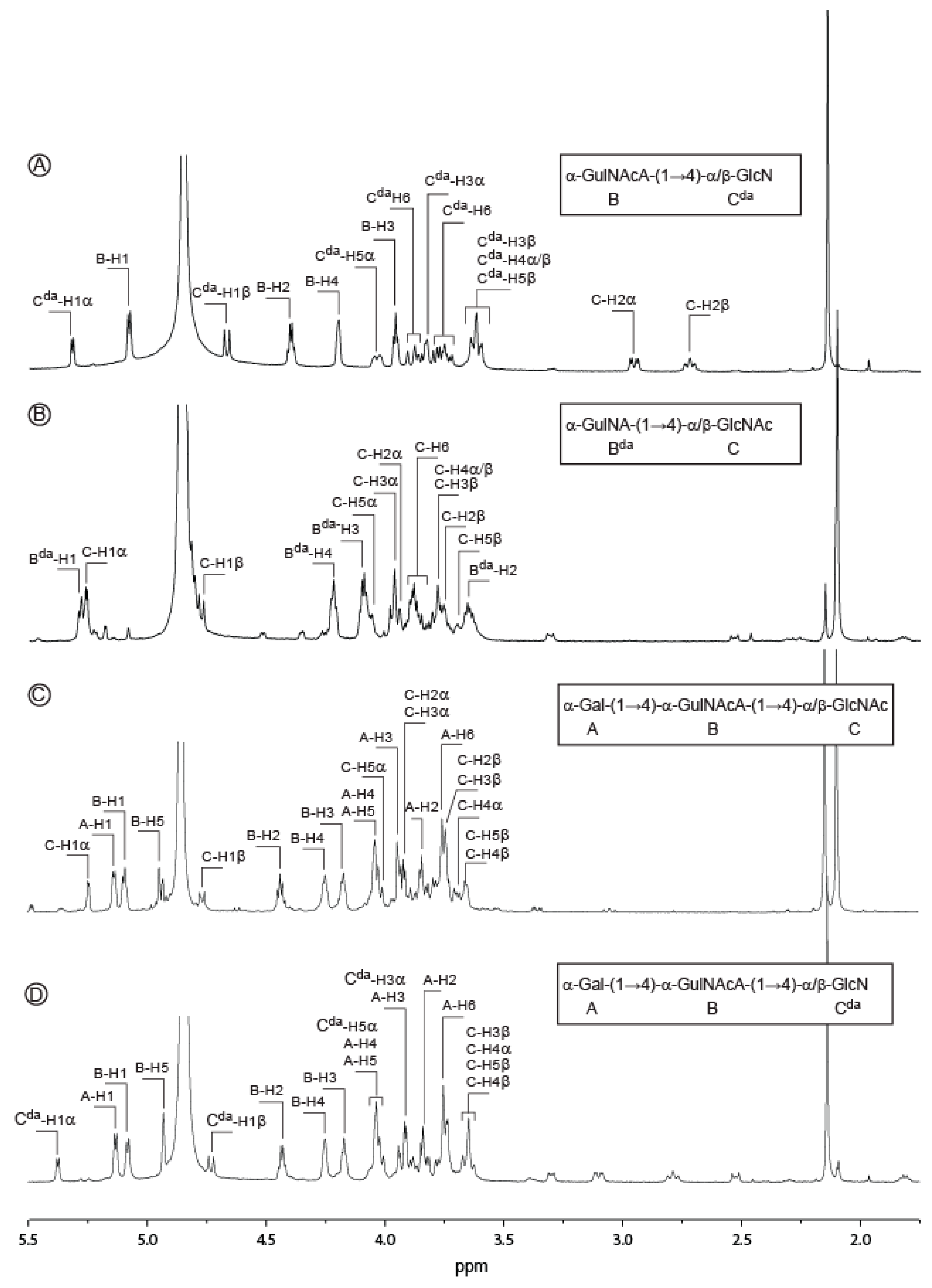
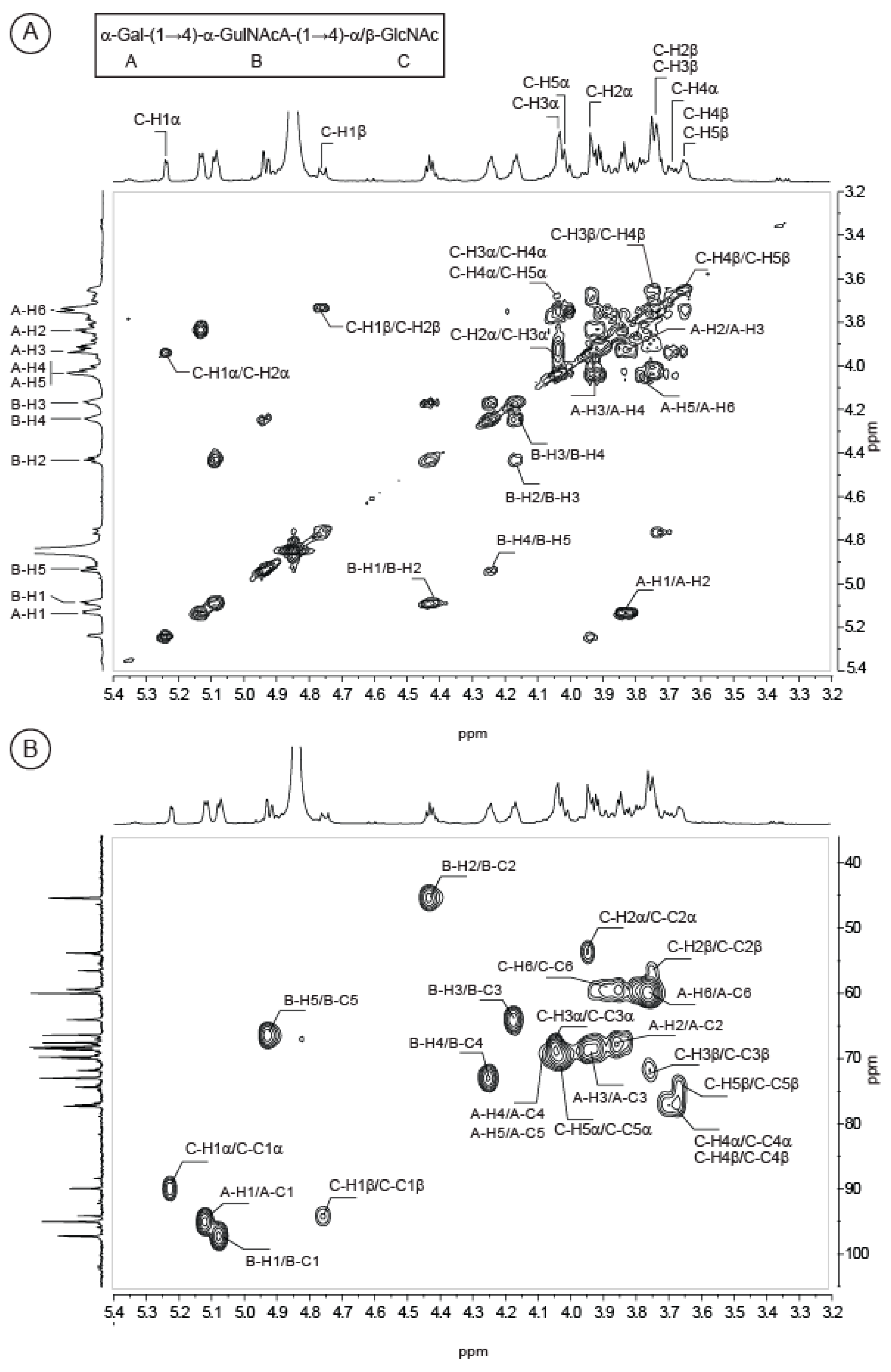
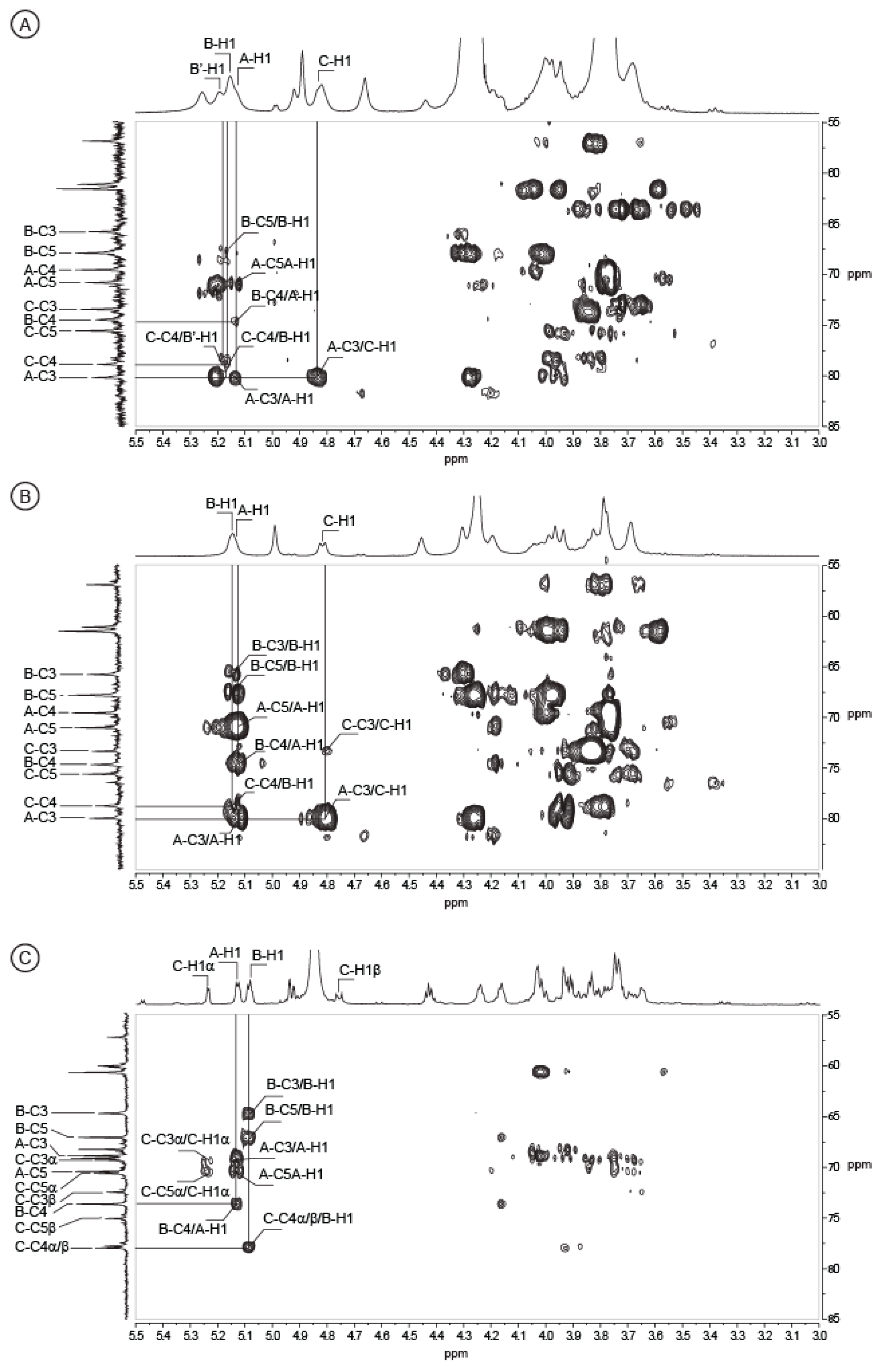
2.2. Structure of Oligosaccharides
2.3. Structure of the Polysaccharide
3. Materials and Methods
3.1. Production, Isolation and Purification of the Vibrio alginolyticus Exopolysaccharide (VA-EPS)
3.2. Monosaccharide Analysis
3.3. Methylation Analysis
3.4. Determination of Absolute Configuration
3.5. Molecular Weight Determination
3.6. Acid Hydrolysis and Oligosaccharides Purification
3.7. NMR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Toukach, P.V.; Egorova, K.S. Carbohydrate structure database merged from bacterial, archaeal, plant and fungal parts. Nucleic Acids Res. 2016, 44, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Toukach, P.; Egorova, K. Carbohydrate Structure Database (CSDB): Examples of Usage. In A Practical Guide to Using Glycomics Databases; Aoki-Kinoshita, K.F., Ed.; Springer: Tokyo, Japan, 2017; pp. 75–113. [Google Scholar]
- Toukach, P.; Egorova, K. Bacterial, Plant, and Fungal Carbohydrate Structure Databases: Daily Usage. In Glycoinformatics; Lütteke, T., Frank, M., Eds.; Springer: New York, NY, USA, 2015; pp. 55–85. [Google Scholar]
- Laine, R.A. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide―The isomer-barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994, 4, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.W. Microbial polysaccharides from Gram-negative bacteria. Int. Dairy J. 2001, 11, 663–674. [Google Scholar] [CrossRef]
- Freitas, F.; Alves, V.D.; Reis, M.A.M. Advances in bacterial exopolysaccharides: From production to biotechnological applications. Trends Biotechnol. 2011, 29, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Drouillard, S.; Jeacomine, I.; Buon, L.; Boisset, C.; Courtois, A.; Thollas, B.; Morvan, P.-Y.; Vallée, R.; Helbert, W. Structure of the exopolysaccharide secreted by a marine strain Vibrio alginolyticus. Mar. Drugs 2018, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Drouillard, S.; Jeacomine, I.; Buon, L.; Boisset, C.; Courtois, A.; Thollas, B.; Morvan, P.-Y.; Vallée, R.; Helbert, W. Structure of an amino acid-derived decorated exopolysaccharide secreted by a Vibrio alginolyticus strain. Mar. Drugs 2018, 13, 6723. [Google Scholar] [CrossRef] [PubMed]
- Kanlayavattanakul, M.; Lourith, N. Cosmetics: Active Polymers. In Encyclopedia of Polymer Applications; Mishra, M., Ed.; CRC Press Taylor & Francis Group: London, UK, 2019; pp. 705–721. [Google Scholar]
- Pantophlet, R.; Haseley, S.R.; Vinogradov, E.V.; Brade, L.; Holst, O.; Brade, H. Chemical and antigenic structure of the O-polysaccharide of the lipopolysaccharides from two Acinetobacter haemolyticus strains differing only in the anomeric configuration of one glycosyl residue in their O-antigens. Eur. J. Biochem. 1999, 263, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Lanzetta, R.; Scognamiglio, R.; Alfieri, F.; Izzo, V.; Di Donato, A.; Parrilli, M.; Holst, O.; Molinaro, A. The structure of the O-specific polysaccharide from the lipopolysaccharide of Pseudomonas sp. OX1 cultivated in the presence of the azo dye Orange II. Carbohydr. Res. 2008, 343, 674–684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yildiz, F.; Fong, J.; Sadovskaya, I.; Grard, T.; Vinogradov, E. Structural Characterization of the Extracellular Polysaccharide from Vibrio cholerae O1 El-Tor. PLoS ONE 2014, 9, e86751. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, G.M.; Shashkov, A.S.; Knirel, Y.A.; Vinogradov, E.V.; Kochetkov, N.K. A computer-assisted structural analysis of regular polysaccharides on the basis of 13C-n.m.r. data. Carbohydr. Res. 1988, 175, 59–75. [Google Scholar] [CrossRef]
- Kondakova, A.N.; Novototskaya-Vlasova, K.A.; Drutskaya, M.S.; Senchenkova, S.N.; Shcherbakova, V.A.; Shashkov, A.S.; Gilichinsky, D.A.; Nedospasov, S.A.; Knirel, Y.A. Structure of the O-polysaccharide chain of the lipopolysaccharide of Psychrobacter muricolla 2pST isolated from overcooled water brines within permafrost. Carbohydr. Res. 2012, 349, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Gorshkova, R.P.; Nazarenko, E.L.; Zubkov, V.A.; Ivanova, E.P.; Gorshkova, N.M.; Isakov, V.V. Structure of O-specific polysaccharide from Pseudoalteromonas nigrifaciens strain KMM 161. Biochemistry 2002, 67, 672–675. [Google Scholar] [PubMed]
- Knirel, Y.A.; Vinogradov, E.V.; Kocharova, N.A.; Shashkov, A.S.; Dmitriev, B.A.; Kochetkov, N.K. Synthesis and 13C-N.m.r. spectra of methyl 2,3-diacetamido-2,3-dideoxy-α-d-hexopyranosides and 2,3-diacetamido-2,3-dideoxy-d-hexoses. Carbohydr. Res. 1983, 122, 181–188. [Google Scholar] [CrossRef]
- Michon, F.; Brisson, J.R.; Roy, R.; Ashton, F.E.; Jennings, H.J. Sructural determination of the capsular polysaccharide of Neisseria-meningitidis group-I: A two-dimensional NMR analysis. Biochemistry 1985, 24, 5592–5598. [Google Scholar] [CrossRef]
- Kamerling, J.P.; Gerwig, G.J.; Vliegenthart, J.F.; Clamp, J.R. Characterization by gas-liquid chromatography-mass spectrometry and proton-magnetic-resonance spectroscopy of pertrimethylsilyl methyl glycosides obtained in the methanolysis of glycoproteins and glycopeptides. Biochem. J. 1975, 151, 491–495. [Google Scholar] [CrossRef]
- Montreuil, J.; Bouquelet, S.; Debray, H.; Fournet, B.; Spik, G.; Strecker, G. Glycoproteines. In Carbohydrates Analysis: A Practical Approach; Chaplin, M.F., Kennedy, J.K., Eds.; IRL Press: Oxford, UK, 1986; pp. 143–204. [Google Scholar]
- Hakomori, S.I. A rapid permethylation of glycolipid, and polysaccharide catalyzed by methylsulfinyl carbanion in dimethyl sulfoxide. J. Biochem. 1964, 55, 205–208. [Google Scholar] [PubMed]
- Taylor, R.L.; Conrad, H.E. Stoichiometric depolymerization of polyuronides and glycosaminoglycans to monosaccharides following reduction of their carbodiimide activated carboxyl groups. Biochemistry 1972, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
- Kvernheim, A.L. Methylation analysis of polysaccharides with butyllithium in dimethyl sulfoxide. Acta Chemica Scandinavia 1987, B41, 150–152. [Google Scholar] [CrossRef][Green Version]
- Gerwig, G.J.; Kamerling, J.P.; Vliegenthart, F.G. Determination of the D and L configuration of neutral polysaccharides by high resolution capillary G.L.C. Carbohydr. Res. 1978, 62, 349–357. [Google Scholar] [CrossRef]
- Gerwig, G.J.; Kamerling, J.P.; Vliegenthart, F.G. Determination of the absolute configuration of monosaccharides in complex carbohydrate by capillary G.L.C. Carbohydr. Res. 1979, 77, 1–7. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sugar Residue | 1 | 2 | 3 | 4 | 5 | 6(6a,6b) | NHAc (CH3,CO) | OAc (CH3,CO) | |
|---|---|---|---|---|---|---|---|---|---|
| EPS | |||||||||
| →3)-〈-d-Gal-(1→ | 1H | 5.13 | 3.95 | 3.99 | 4.26 | 4.05 | 3.77,3.93 | ||
| 13C | 96.66 | 67.62 | 80.19 | 69.58 | 70.82 | 61.58 | |||
| →4)-〈-l-GulNAcA-(1→ | 1H | 5.14 | 4.44 | 4.18 | 4.30 | 4.92 | 2.16 | ||
| 13C | 98.83 | 46.99 | 65.79 | 74.49 | 67.92 | 175.40 | 22.59, 174.47 | ||
| →4)-〈-3OAc-l-GulNAcA-(1→ | 1H | 5.18 | 4.66 | 5.26 | 4.31 | 4.89 | 2.11 | 2.27 | |
| 13C | 97.1 | 45.62 | 67.84 | 71.68 | 68.1 | 175.75 | 22.59, 174.47 | 21.20, 174.00 | |
| →4)-®-d-GlcNAc-(1→ | 1H | 4.81 | 3.81 | 3.79 | 3.68 | 3.68 | 3.77,3.94 | 2.11 | |
| 13C | 103.19 | 56.84 | 73.45 | 78.89 | 75.58 | 61.13 | 22.99, 175.50 | ||
| Deacetylated EPS | |||||||||
| →3)-〈-d-Gal-(1→ | 1H | 5.14 | 3.95 | 3.95 | 4.25 | 4.00 | 3.77,3.93 | ||
| 13C | 97.01 | 67.81 | 79.95 | 69.56 | 71.02 | 61.49 | |||
| →4)-〈-l-GulNAcA-(1→ | 1H | 5.14 | 4.45 | 4.19 | 4.30 | 4.99 | 2.17 | ||
| 13C | 99.04 | 47.06 | 65.76 | 74.66 | 67.81 | 175.00 | 22.59, 174.47 | ||
| →4)-®-d-GlcNAc-(1→ | 1H | 4.81 | 3.81 | 3.81 | 3.68 | 3.68 | 3.77,3.93 | 2.11 | |
| 13C | 103.1 | 56.92 | 73.33 | 78.71 | 75.59 | 61.08 | 22.99, 175.59 | ||
| Sugar Residue | 1 | 2 | 3 | 4 | 5 | 6(6a, 6b) | NHAc (CH3,CO) | |
|---|---|---|---|---|---|---|---|---|
| Trisaccharide ABC | ||||||||
| 〈-d-Gal-(1→ | 1H | 5.13 | 3.84 | 3.93 | 4.04 | 4.02 | 3.74, 3.74 | |
| 13C | 96.84 | 69.42 | 70.31 | 70.07 | 71.61 | 61.83 | ||
| →4)-〈-l-GulNAcA-(1→ | 1H | 5.08 | 4.43 | 4.17 | 4.24 | 4.93 | 2.14 | |
| 13C | 99.06 | 47.28 | 65.89 | 74.82 | 68.25 | 175.89 | 22.93, 175.13 | |
| →4)-〈-d-GlcNAc | 1H | 5.24 | 3.93 | 4.03 | 3.69 | 4.02 | 3.76, 3.85 | 2.09 |
| 13C | 91.76 | 55.66 | 70.48 | 79.15 | 71.79 | 61.20 | 23.05, 175.66 | |
| →4)-®-d-GlcNAc | 1H | 4.76 | 3.73 | 3.74 | 3.65 | 3.64 | 3.76, 3.85 | 2.09 |
| 13C | 95.97 | 58.37 | 73.65 | 78.97 | 76.24 | 61.33 | 23.33, 175.89 | |
| Trisaccharide ABCda | ||||||||
| 〈-d-Gal-(1→ | 1H | 5.13 | 3.84 | 3.93 | 4.04 | 4.03 | 3.75, 375 | |
| 13C | 96.78 | 69.36 | 70.24 | 69.98 | 71.55 | 61.75 | ||
| →4)-〈-l-GulNAcA-(1→ | 1H | 5.08 | 4.43 | 4.17 | 4.25 | 4.93 | 2.14 | |
| 13C | 99.03 | 47.19 | 65.78 | 74.74 | 68.19 | 176.88 | 22.86, 175.08 | |
| →4)-〈-d-GlcN | 1H | 5.37 | 3.10 | 3.91 | 3.65 | 4.04 | 3.76, 3.87 | |
| 13C | 91.73 | 56.18 | 71.36 | 78.69 | 71.91 | 61.24 | ||
| →4)-®-d-GlcN | 1H | 4.73 | 2.79 | 3.66 | 3.65 | 3.65 | 3.76, 3.87 | |
| 13C | 96.46 | 58.76 | 74.47 | 78.69 | 76.29 | 61.05 | ||
| Disaccharide BdaC | ||||||||
| 〈-l-GulNA-(1→ | 1H | 5.27 | 3.64 | 4.10 | 4.21 | 4.82 | ||
| 13C | 97.85 | 48.17 | 69.56 | 70.97 | 68.93 | 177.22 | ||
| →4)-〈-d-GlcNAc | 1H | 5.25 | 3.96 | 3.97 | 3.77 | 4.06 | 3.87, 3.92 | 2.09 |
| 13C | 91.63 | 55.41 | 70.31 | 79.28 | 71.33 | 61.80 | 23.05, 175.70 | |
| →4)-®-d-GlcNAc | 1H | 4.77 | 3.75 | 3.77 | 3.77 | 3.68 | 3.87, 3.92 | 2.09 |
| 13C | 95.92 | 58.11 | 73.54 | 79.15 | 75.83 | 61.91 | 23.33, 175.96 | |
| Disaccharide BCda | ||||||||
| 〈-l-GulNAcA-(1→ | 1H | 5.08 | 4.40 | 3.96 | 4.20 | 4.85 | 2.14 | |
| 13C | 99.10 | 47.10 | 70.34 | 71.29 | 68.75 | 177.48 | 22.84, 175.05 | |
| →4)-〈-d-GlcN | 1H | 5.31 | 2.95 | 3.82 | 3.62 | 4.03 | 3.74, 3.87 | |
| 13C | 92.47 | 56.29 | 72.41 | 78.86 | 71.73 | 61.25 | ||
| →4)-β-d-GlcN | 1H | 4.66 | 2.72 | 3.62 | 3.62 | 3.64 | 3.74, 3.87 | |
| 13C | 97.09 | 58.81 | 74.93 | 78.95 | 76.19 | 61.05 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drouillard, S.; Chambon, R.; Jeacomine, I.; Buon, L.; Boisset, C.; Courtois, A.; Thollas, B.; Morvan, P.-Y.; Vallée, R.; Helbert, W. Structure of the Polysaccharide Secreted by Vibrio alginolyticus CNCM I-5035 (Epidermist 4.0TM). Mar. Drugs 2020, 18, 509. https://doi.org/10.3390/md18100509
Drouillard S, Chambon R, Jeacomine I, Buon L, Boisset C, Courtois A, Thollas B, Morvan P-Y, Vallée R, Helbert W. Structure of the Polysaccharide Secreted by Vibrio alginolyticus CNCM I-5035 (Epidermist 4.0TM). Marine Drugs. 2020; 18(10):509. https://doi.org/10.3390/md18100509
Chicago/Turabian StyleDrouillard, Sophie, Rémi Chambon, Isabelle Jeacomine, Laurine Buon, Claire Boisset, Anthony Courtois, Bertrand Thollas, Pierre-Yves Morvan, Romuald Vallée, and William Helbert. 2020. "Structure of the Polysaccharide Secreted by Vibrio alginolyticus CNCM I-5035 (Epidermist 4.0TM)" Marine Drugs 18, no. 10: 509. https://doi.org/10.3390/md18100509
APA StyleDrouillard, S., Chambon, R., Jeacomine, I., Buon, L., Boisset, C., Courtois, A., Thollas, B., Morvan, P.-Y., Vallée, R., & Helbert, W. (2020). Structure of the Polysaccharide Secreted by Vibrio alginolyticus CNCM I-5035 (Epidermist 4.0TM). Marine Drugs, 18(10), 509. https://doi.org/10.3390/md18100509