Identification of Conomarphin Variants in the Conus eburneus Venom and the Effect of Sequence and PTM Variations on Conomarphin Conformations




Abstract
1. Introduction
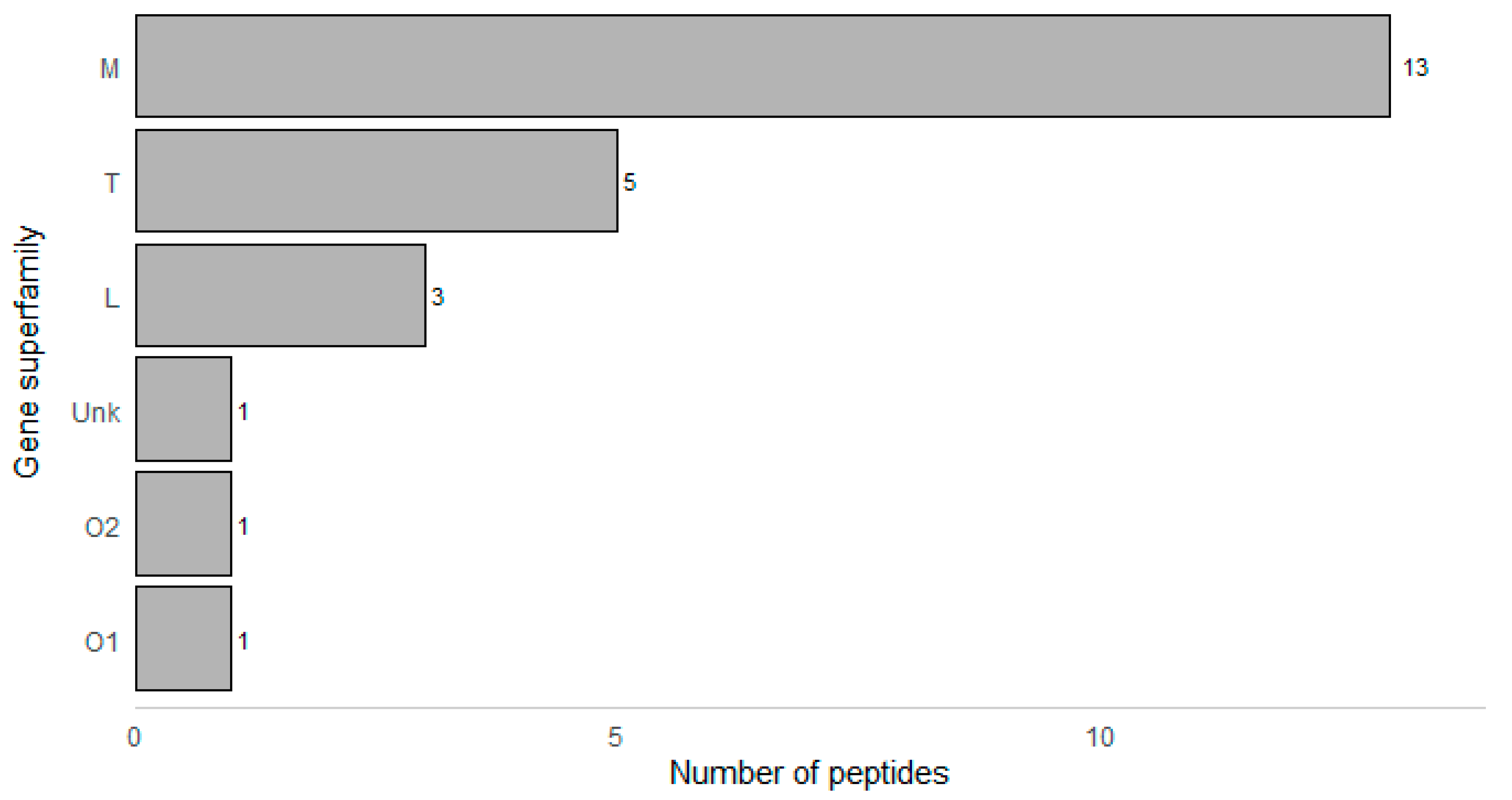
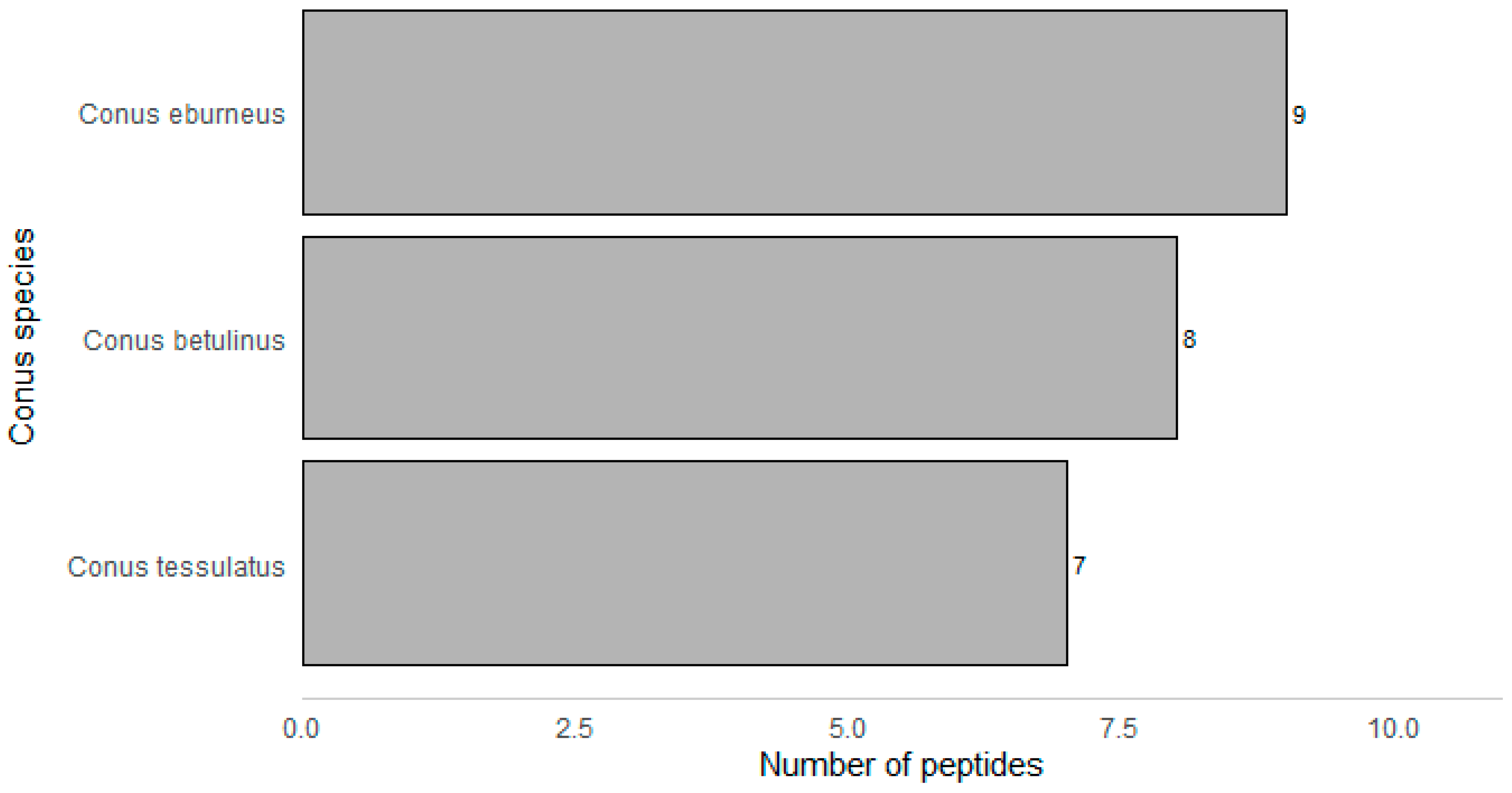
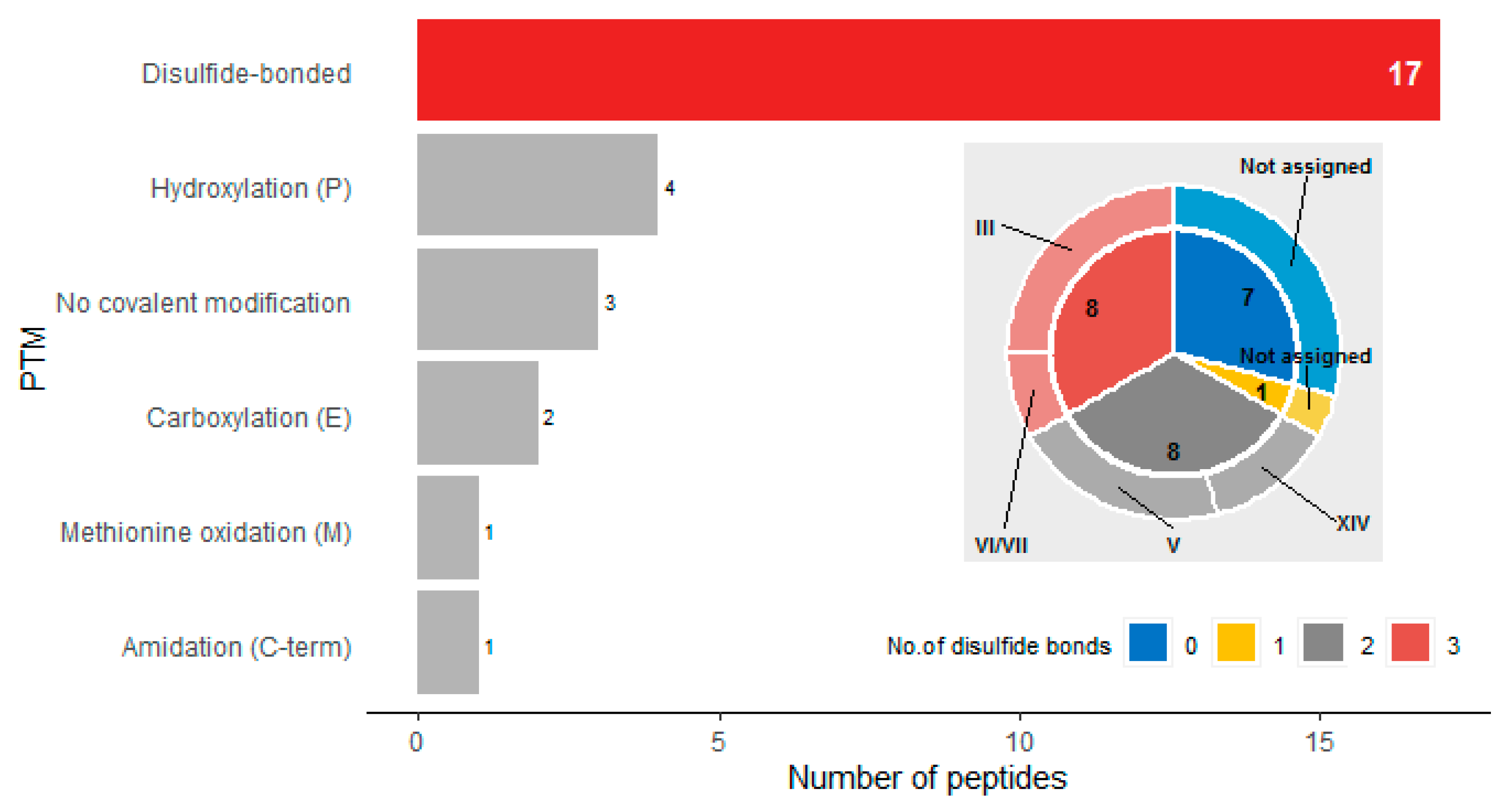
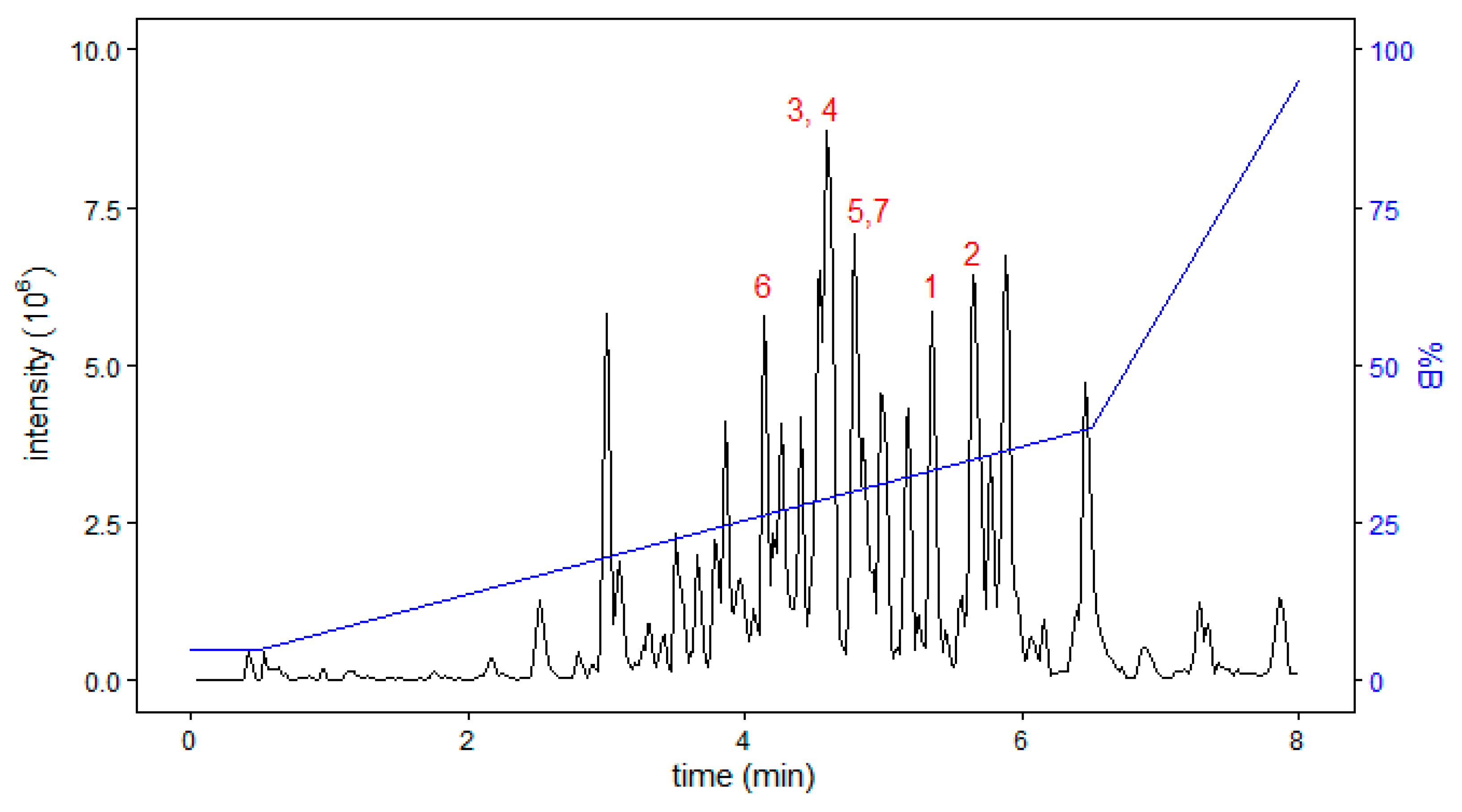
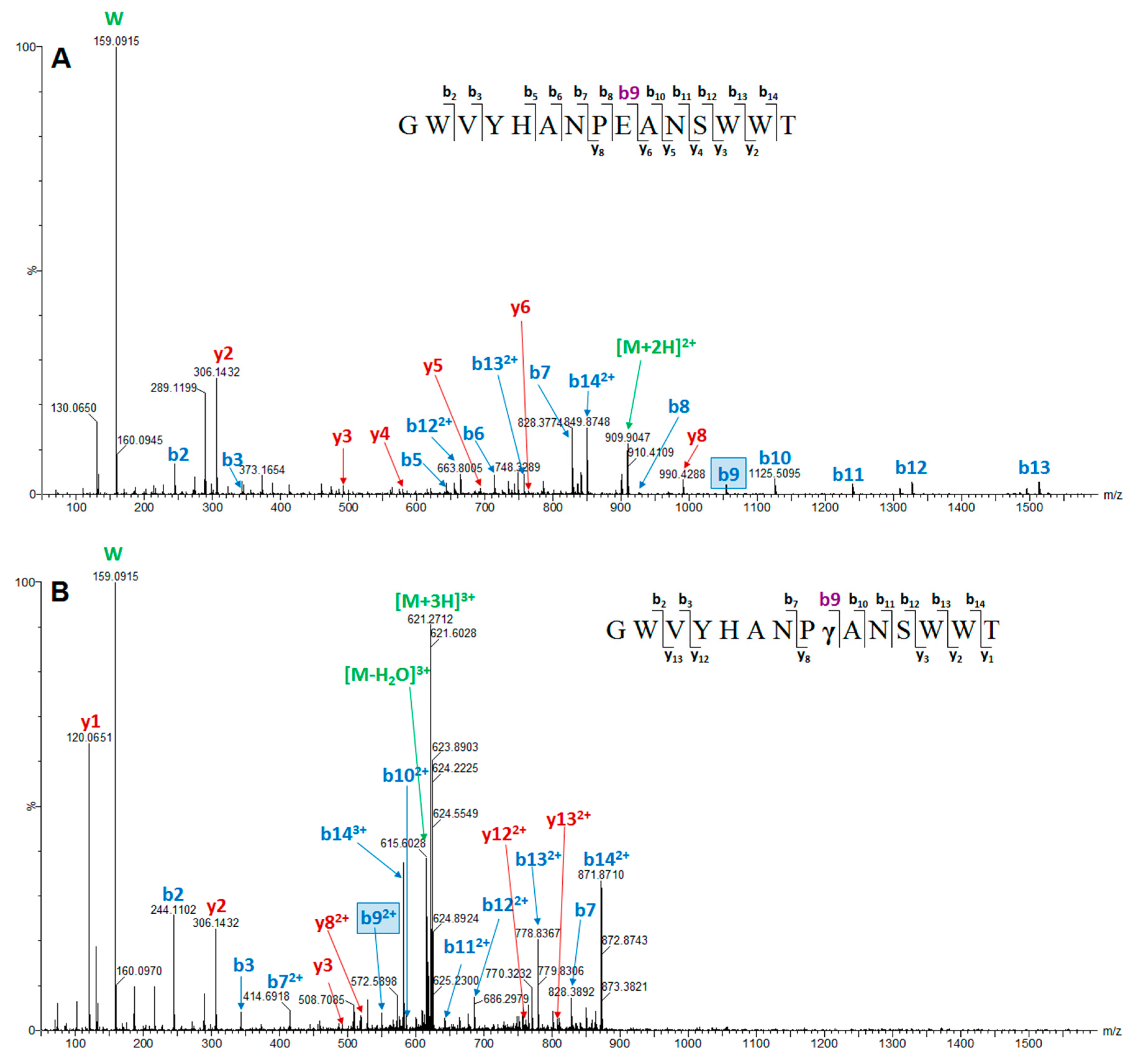
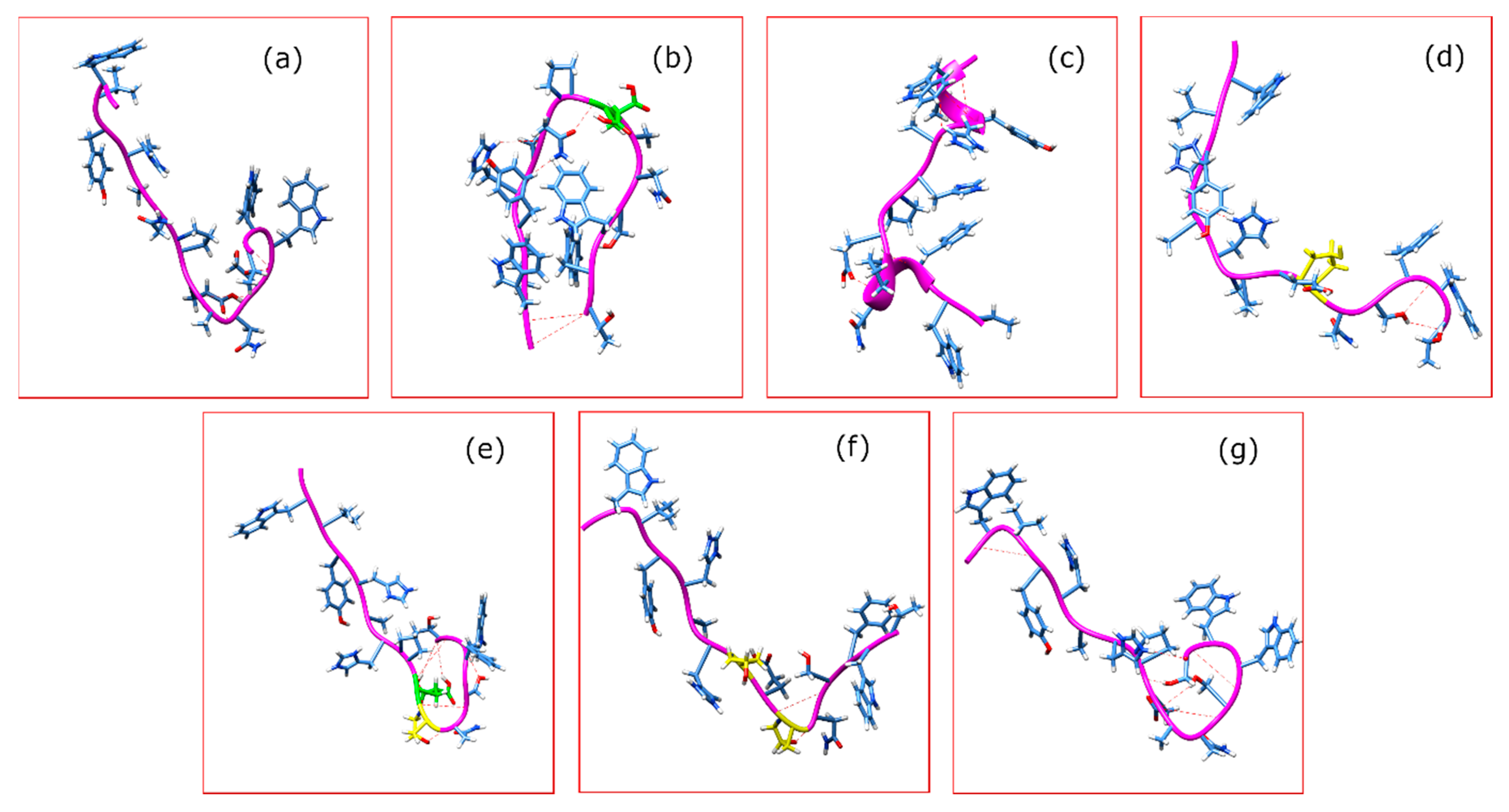
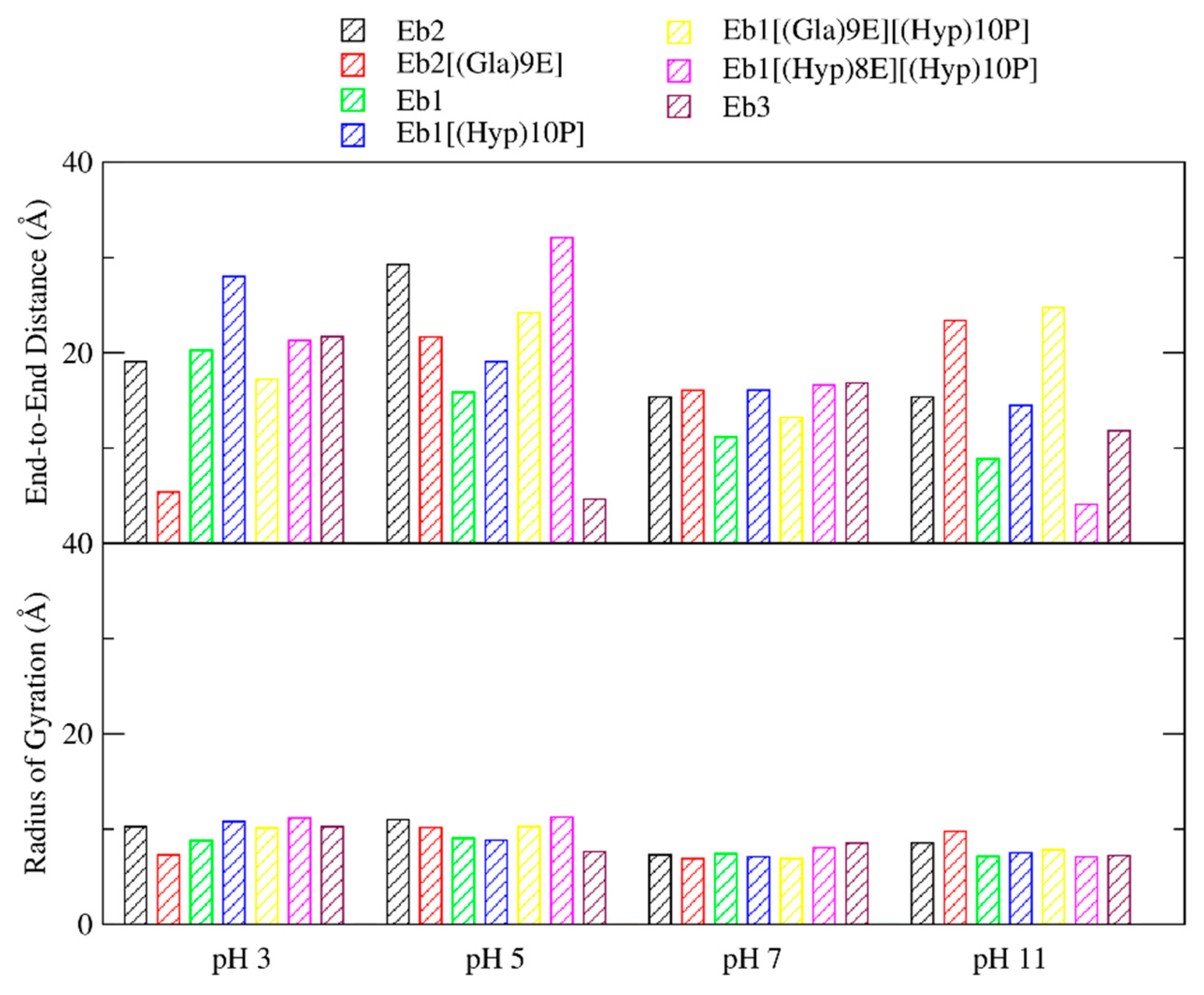
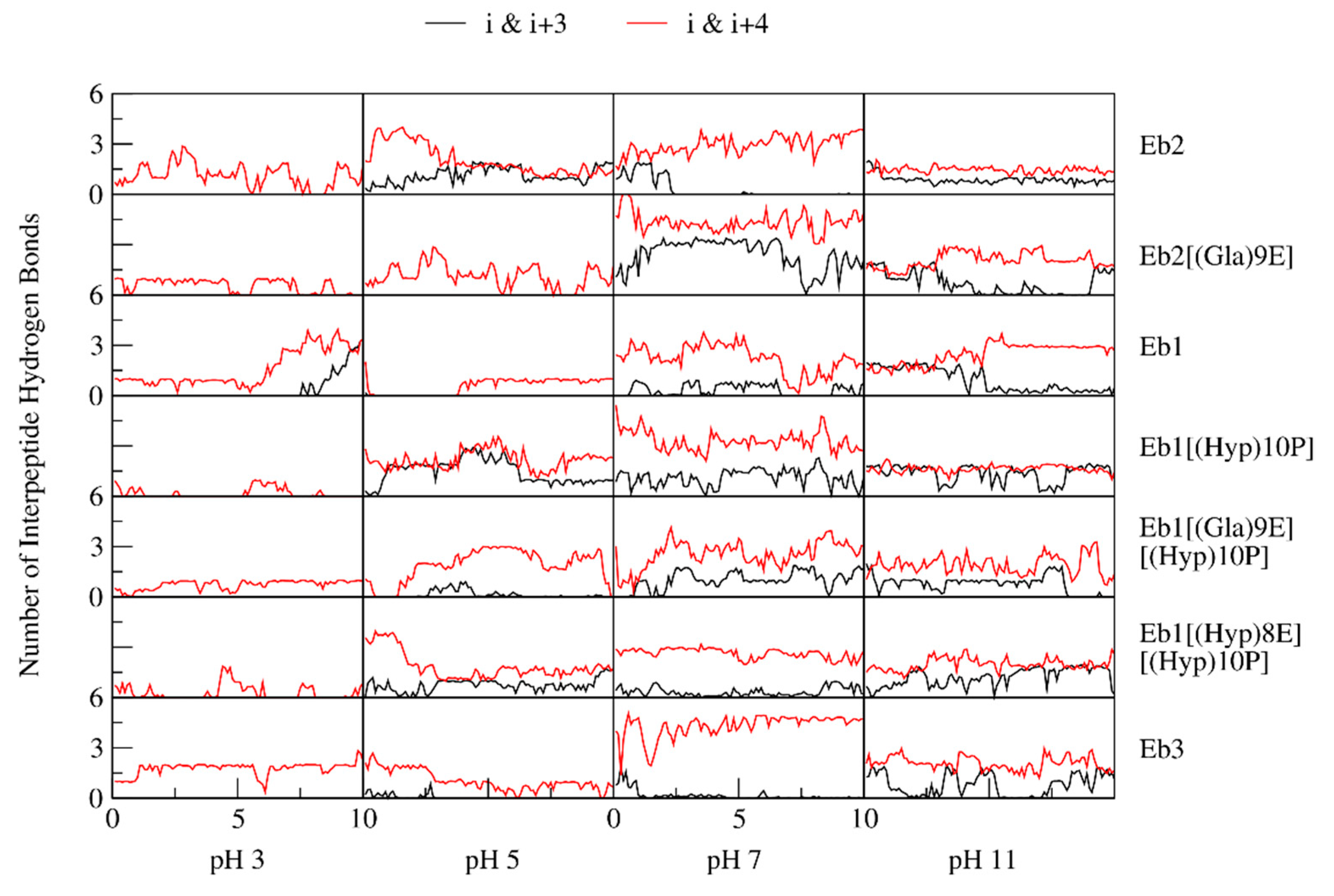
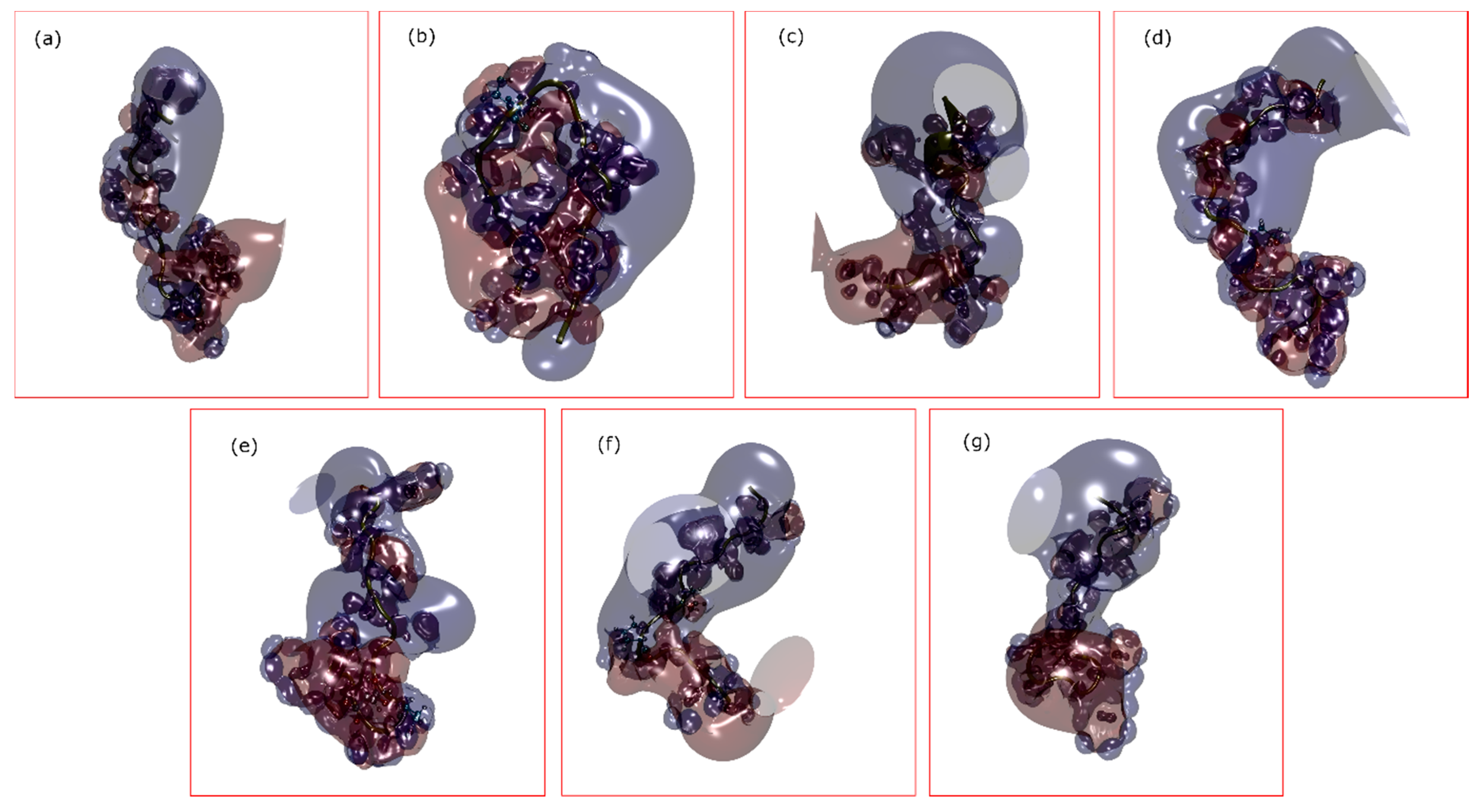
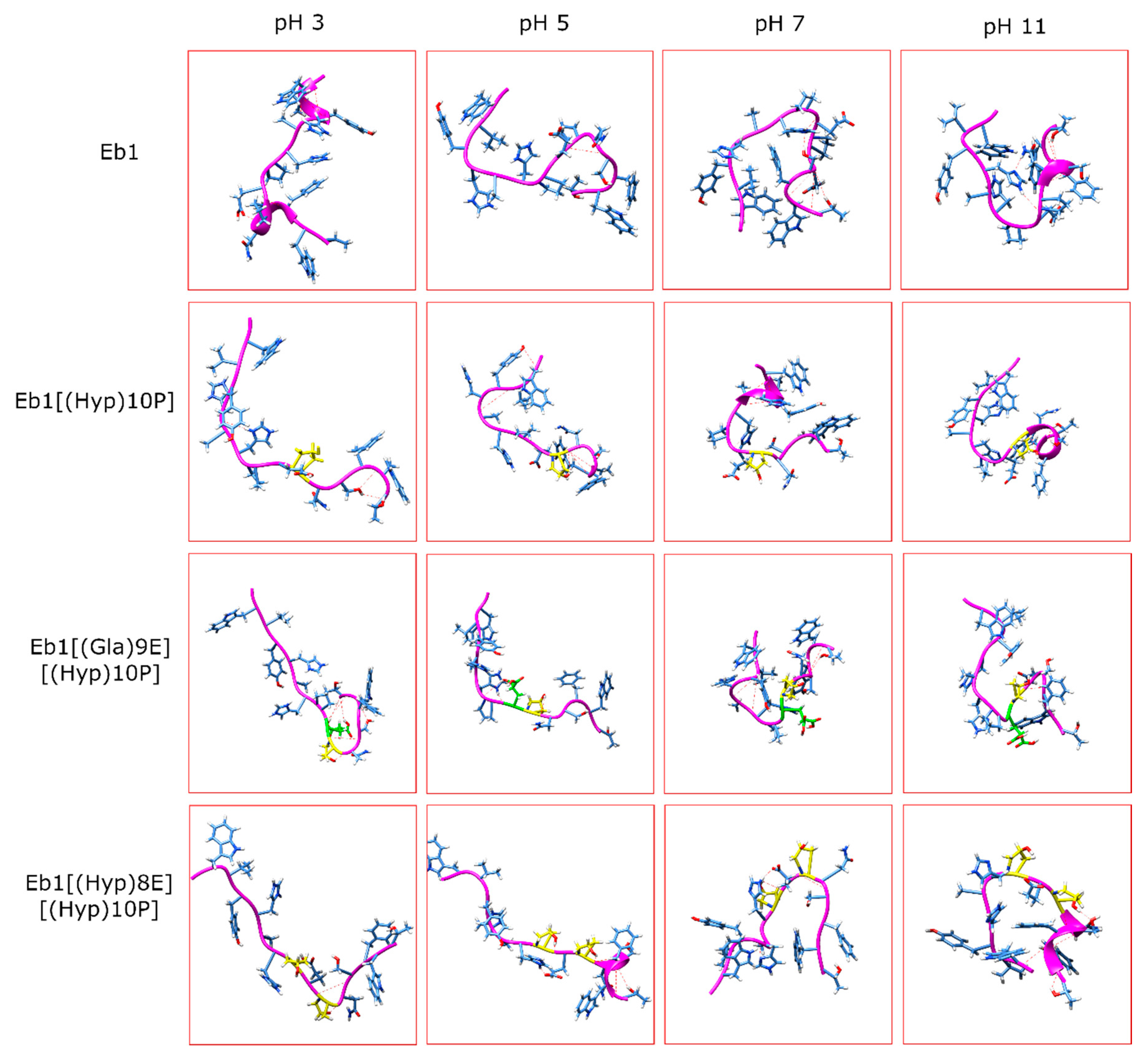
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Venom Extraction
4.2. Peptide Reduction and Alkylation
4.3. Mass Spectrometry
4.4. Mass Spectrometric Data Search and Analysis
4.5. In Silico MD Simulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus Venom Peptide Pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- Daniel, J.; Clark, R. G-Protein Coupled Receptors Targeted by Analgesic Venom Peptides. Toxins 2017, 9, 372. [Google Scholar] [CrossRef]
- Han, Y.; Huang, F.; Jiang, H.; Liu, L.; Wang, Q.; Wang, Y.; Shao, X.; Chi, C.; Du, W.; Wang, C. Purification and structural characterization of a d-amino acid-containing conopeptide, conomarphin, from Conus marmoreus: A d-amino acid-containing conomarphin. FEBS J. 2008, 275, 1976–1987. [Google Scholar] [CrossRef]
- Franco, A.; Kompella, S.N.; Akondi, K.B.; Melaun, C.; Daly, N.L.; Luetje, C.W.; Alewood, P.F.; Craik, D.J.; Adams, D.J.; Marí, F. RegIIA: An α4/7-conotoxin from the venom of Conus regius that potently blocks α3β4 nAChRs. Biochem. Pharmacol. 2012, 83, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.C.V.; Nery, A.A.; Ulrich, H.; Konno, K.; Sciani, J.M.; Pimenta, D.C. α-RgIB: A Novel Antagonist Peptide of Neuronal Acetylcholine Receptor Isolated from Conus regius Venom. Int. J. Pept. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus Peptides: Biodiversity-based Discovery and Exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.M.; Peigneur, S.; Maiti, M.; Mille, B.G.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Discovery of a new subclass of α-conotoxins in the venom of Conus australis. Toxicon 2014, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, C.; Dong, S.; Wu, Y.; Zhangsun, D.; Luo, S. Discovery Methodology of Novel Conotoxins from Conus Species. Mar. Drugs 2018, 16, 417. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Biass, D.; Stöcklin, R.; Favreau, P. Dramatic intraspecimen variations within the injected venom of Conus consors: An unsuspected contribution to venom diversity. Toxicon 2010, 55, 1453–1462. [Google Scholar] [CrossRef]
- Bhatia, S.; Kil, Y.J.; Ueberheide, B.; Chait, B.T.; Tayo, L.; Cruz, L.; Lu, B.; Yates, J.R.; Bern, M. Constrained De Novo Sequencing of Conotoxins. J. Proteome Res. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Yang, L.; Xu, S.; Wang, C. Various Conotoxin Diversifications Revealed by a Venomic Study of Conus flavidus. Mol. Cell. Proteomics 2014, 13, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Dutt, M.; Dutertre, S.; Jin, A.-H.; Lavergne, V.; Alewood, P.; Lewis, R. Venomics Reveals Venom Complexity of the Piscivorous Cone Snail, Conus tulipa. Mar. Drugs 2019, 17, 71. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep Venomics Reveals the Mechanism for Expanded Peptide Diversity in Cone Snail Venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.; Lewis, R.J. Venomics-Accelerated Cone Snail Venom Peptide Discovery. Int. J. Mol. Sci. 2018, 19, 788. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, Y.; Wang, L.; Liang, A.; Chen, S.; Xu, A. Identifying novel conopepetides from the venom ducts of Conus litteratus through integrating transcriptomics and proteomics. J. Proteom. 2019, 192, 346–357. [Google Scholar] [CrossRef]
- Biass, D.; Violette, A.; Hulo, N.; Lisacek, F.; Favreau, P.; Stöcklin, R. Uncovering Intense Protein Diversification in a Cone Snail Venom Gland Using an Integrative Venomics Approach. J. Proteome Res. 2015, 14, 628–638. [Google Scholar] [CrossRef]
- Degueldre, M.; Verdenaud, M.; Legarda, G.; Minambres, R.; Zuniga, S.; Leblanc, M.; Gilles, N.; Ducancel, F.; De Pauw, E.; Quinton, L. Diversity in sequences, post-translational modifications and expected pharmacological activities of toxins from four Conus species revealed by the combination of cutting-edge proteomics, transcriptomics and bioinformatics. Toxicon 2017, 130, 116–125. [Google Scholar] [CrossRef]
- Vijayasarathy, M.; Balaram, P. Mass spectrometric identification of bromotryptophan containing conotoxin sequences from the venom of C. amadis. Toxicon 2018, 144, 68–74. [Google Scholar] [CrossRef]
- Biass, D.; Dutertre, S.; Gerbault, A.; Menou, J.-L.; Offord, R.; Favreau, P.; Stöcklin, R. Comparative proteomic study of the venom of the piscivorous cone snail Conus consors. J. Proteom. 2009, 72, 210–218. [Google Scholar] [CrossRef]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.M.; Dutertre, S.; Lewis, R.J.; Marí, F. Intraspecific variations in Conus purpurascens injected venom using LC/MALDI-TOF-MS and LC-ESI-TripleTOF-MS. Anal. Bioanal. Chem. 2015, 407, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.W.A.; Jin, A.-H.; Dutertre, S.; Giacomotto, J.; Mohialdeen, H.; Vetter, I.; Alewood, P.F.; Lewis, R.J. Comparative Venomics Reveals the Complex Prey Capture Strategy of the Piscivorous Cone Snail Conus catus. J. Proteome Res. 2015, 14, 4372–4381. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Dutertre, S.; Dutt, M.; Lavergne, V.; Jones, A.; Lewis, R.; Alewood, P. Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis. Mar. Drugs 2019, 17, 177. [Google Scholar] [CrossRef] [PubMed]
- Prator, C.A.; Murayama, K.M.; Schulz, J.R. Venom Variation during Prey Capture by the Cone Snail, Conus textile. PLoS ONE 2014, 9, e98991. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Tran, N.H.; Zhang, X.; Xin, L.; Shan, B.; Li, M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. USA 2017, 114, 8247–8252. [Google Scholar] [CrossRef]
- Mendoza, C.B.; Masacupan, D.J.M.; Batoctoy, D.C.R.; Yu, E.T.; Lluisma, A.O.; Salvador-Reyes, L.A. Conomarphins cause paralysis in mollusk: Critical and tunable structural elements for bioactivity. J. Pept. Sci. 2019, 25. [Google Scholar] [CrossRef]
- Aman, J.W.; Imperial, J.S.; Ueberheide, B.; Zhang, M.-M.; Aguilar, M.; Taylor, D.; Watkins, M.; Yoshikami, D.; Showers-Corneli, P.; Safavi-Hemami, H.; et al. Insights into the origins of fish hunting in venomous cone snails from studies of Conus tessulatus. Proc. Natl. Acad. Sci. USA 2015, 112, 5087–5092. [Google Scholar] [CrossRef]
- Robinson, S.; Norton, R. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Bouchet, P.; Duda, T.F.; Kauferstein, S.; Kohn, A.J.; Olivera, B.M.; Watkins, M.; Meyer, C. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 2014, 78, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Yao, G.; Gao, B.-M.; Fan, C.-X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. GigaScience 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.M.; Tytgat, J. In the picture: Disulfide-poor conopeptides, a class of pharmacologically interesting compounds. J. Venom. Anim. Toxins Trop. Dis. 2016, 22, 30. [Google Scholar] [CrossRef]
- Huang, F.; Du, W.; Wang, B. Solution Structure of Conomarphin, a Novel Conopeptide Containing D-Amino Acid at pH 5. Acta Phys. Chim. Sin. 2008, 24, 1558–1562. [Google Scholar] [CrossRef]
- Phuong, M.A.; Mahardika, G.N.; Alfaro, M.E. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genom. 2016, 17, 401. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins Struct. Funct. Bioinforma. 2006, 63, 928–938. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.; Darden, T. AMBER 2019; University of California: San Francisco, CA, USA, 2019. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Thompson, J.P.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput. 2013, 9, 5653–5674. [Google Scholar] [CrossRef]
- Khoury, G.A.; Smadbeck, J.; Tamamis, P.; Vandris, A.C.; Kieslich, C.A.; Floudas, C.A. Forcefield_NCAA: Ab Initio Charge Parameters to Aid in the Discovery and Design of Therapeutic Proteins and Peptides with Unnatural Amino Acids and Their Application to Complement Inhibitors of the Compstatin Family. ACS Synth. Biol. 2014, 3, 855–869. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Gaza, J.T.; Sampaco, A.-R.B.; Custodio, K.K.S.; Nellas, R.B. Conformational dynamics of $$\alpha $$α-conotoxin PnIB in complex solvent systems. Mol. Divers. 2019. [Google Scholar] [CrossRef]
- Huston, S.E.; Marshall, G.R. α/310-Helix transitions in α-methylalanine homopeptides: Conformational transition pathway and potential of mean force. Biopolymers 1994, 34, 75–90. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name (in C. eburneus venom duCt transCriptome) a | Peptide Name (in ConoServer Database) | ConoServer ID | Peptide SequenCe † | Gene Super-Family | Cysteine Frame-Work | Number of Disulfide Bonds |
|---|---|---|---|---|---|---|
| CE102 | ErVIA | P06773 | CAGIGSFCGLPGLVDCCSGRCFIVCLP | O1 | VI/VII | 3 |
| CE030 | * | TALEDADMKTEKGVLSGIMSNLGTVGNMVGGFCCTVYSGCCAE | T | V | 2 | |
| CE031 | * | AALEDADMKTAKGILSNIMGNLGNIGNMAGSFCCSVYSGCCPE | T | V | 2 | |
| CE103 | * | FLGLIGPITSIAGKLCCTVSVSFCCNE | T | V | 2 | |
| CE120 | * | TLQRHWAKFLCCPEDDWCC | T | V | 2 | |
| CE123 | * | DLCPHCPNGCHVDRTCI | L | XIV | 2 | |
| CE133 | * | LCPPMCRSCSNC | L | XIV | 2 | |
| CE133 [(MOx)5M] | * | LCPP(MOx)CRSCSNC | L | XIV | 2 | |
| Conomarphin-Bt1 | P05978 | GWVYHANPEANSWWT | M | Not assigned | 0 | |
| Conomarphin-Eb2 | P08992 | GWVYHANP(Gla)ANSWWT | M | Not assigned | 0 | |
| Conomarphin-Bt2 | P05979 | GWVYHAHPEPNSFWT | M | Not assigned | 0 | |
| Conomarphin-Eb1 | P08991 | GWVYHAHPEONSFWT | M | Not assigned | 0 | |
| Conomarphin-Bt2 [(Gla)9E][(Hyp)10E] | P05979 | GWVYHAHP(Gla)ONSFWT | M | Not assigned | 0 | |
| Conomarphin-Bt2 [(Hyp)9E] [(Hyp)10E] | P05979 | GWVYHAHOEONSFWT | M | Not assigned | 0 | |
| CE019 | Conomarphin-Bt3 | P05980 | GWVYHAHPDANSWWS | M | Not assigned | 0 |
| CE138 | Contryphan-Bt1 [(Hyp)3E] | P05977 | GCOPGLWC(Nh2) | O2 | Not assigned | 1 |
| Eu3.5 | P04637 | CCVVCNAGCSGNCCP | M | III | 3 | |
| CE135 | Ts-011 | P02712 | GCCEDKTCCFI | T | V | 2 |
| CE128 | Ts3.3 | P03167 | CCSRYCYICIPCCPN | M | III | 3 |
| Ts3-SGN01 | P05089 | CCVVCNAGCSGNCCS | M | III | 3 | |
| CE119 | TsIIIA | P07525 | GCCRWPCPSRCGMARCCSS | M | III | 3 |
| TsMMSK-021 | P03154 | CCDWPCTIGCVPCCLP | M | III | 3 | |
| TsVIA | P06849 | CAAFGSFCGLPGLVDCCSGRCFIVCLL | Unknown | VI/VII | 3 | |
| CE124 | Ts3-Y01 | P04949 | RCCISPACNDTCYCCQD | M | III | 3 |
| Conomarphin Number | MS/MS Verified Sequence | PTMs Identified by MS | Conomarphin Name in ConoServer Database | Proposed Conomarphin Name a |
|---|---|---|---|---|
| 1 | GWVYHANPEANSWWTc | none | Bt1 | Eb2 |
| 2 | GWVYHANPγANSWWT | γ-carboxylation (E) | Eb2 | Eb2[(Gla)9E] |
| 3 | GWVYHAHPEPNSFWTc | none | Bt2 | Eb1 |
| 4 | GWVYHAHPEONSFWT | Hydroxylation (P) | Eb1 b | Eb1[(Hyp)10P] |
| 5 | GWVYHAHPγONSFWT d | γ-carboxylation (E); Hydroxylation (P) | None | Eb1[(Gla)9E][(Hyp)10P] |
| 6 | GWVYHAHOEONSFWTd | Hydroxylation (P) x 2 | None | Eb1[(Hyp)8E][(Hyp)10P] |
| 7 | GWVYHAHPDANSWWSc | none | Bt3 | Eb3 |
| pH Levels | Conomarphin | Properties | Possible Effect of PTMs | |
|---|---|---|---|---|
| Ave. # of H-Bonds | End-to-End Distance (Å) | |||
| pH 3 | Eb1 | 1.9673 | 20.2473 | There are no significant differences in the properties. |
| Eb1[(Hyp)10P] | 0.1724 | 28.0155 | ||
| Eb1[(Gla)9E][(Hyp)10P] | 0.7245 | 17.2082 | ||
| Eb1[(Hyp)8E][(Hyp)10P] | 0.2952 | 21.3046 | ||
| pH 5 | Eb1 | 0.5768 | 15.8414 | Conomarphins with PTMs are more elongated and polarized. |
| Eb1[(Hyp)10P] | 3.6198 | 19.0677 | ||
| Eb1[(Gla)9E][(Hyp)10P] | 2.0044 | 24.1722 | ||
| Eb1[(Hyp)8E][(Hyp)10P] | 2.586 | 32.0786 | ||
| pH 7 | Eb1 | 2.5545 | 11.1528 | Conomarphins without PTM are more spherical. |
| Eb1[(Hyp)10P] | 4.4091 | 16.0787 | ||
| Eb1[(Gla)9E][(Hyp)10P] | 3.5531 | 13.2177 | ||
| Eb1[(Hyp)8E][(Hyp)10P] | 2.9578 | 16.6192 | ||
| pH 11 | Eb1 | 3.3795 | 8.8539 | The properties are too variable to derive a conclusion. |
| Eb1[(Hyp)10P] | 2.9953 | 14.4752 | ||
| Eb1[(Gla)9E][(Hyp)10P] | 2.6705 | 24.7668 | ||
| Eb1[(Hyp)8E][(Hyp)10P] | 3.1841 | 4.083 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itang, C.E.M.M.; Gaza, J.T.; Masacupan, D.J.M.; Batoctoy, D.C.R.; Chen, Y.-J.; Nellas, R.B.; Yu, E.T. Identification of Conomarphin Variants in the Conus eburneus Venom and the Effect of Sequence and PTM Variations on Conomarphin Conformations. Mar. Drugs 2020, 18, 503. https://doi.org/10.3390/md18100503
Itang CEMM, Gaza JT, Masacupan DJM, Batoctoy DCR, Chen Y-J, Nellas RB, Yu ET. Identification of Conomarphin Variants in the Conus eburneus Venom and the Effect of Sequence and PTM Variations on Conomarphin Conformations. Marine Drugs. 2020; 18(10):503. https://doi.org/10.3390/md18100503
Chicago/Turabian StyleItang, Corazon Ericka Mae M., Jokent T. Gaza, Dan Jethro M. Masacupan, Dessa Camille R. Batoctoy, Yu-Ju Chen, Ricky B. Nellas, and Eizadora T. Yu. 2020. "Identification of Conomarphin Variants in the Conus eburneus Venom and the Effect of Sequence and PTM Variations on Conomarphin Conformations" Marine Drugs 18, no. 10: 503. https://doi.org/10.3390/md18100503
APA StyleItang, C. E. M. M., Gaza, J. T., Masacupan, D. J. M., Batoctoy, D. C. R., Chen, Y.-J., Nellas, R. B., & Yu, E. T. (2020). Identification of Conomarphin Variants in the Conus eburneus Venom and the Effect of Sequence and PTM Variations on Conomarphin Conformations. Marine Drugs, 18(10), 503. https://doi.org/10.3390/md18100503