New Napyradiomycin Analogues from Streptomyces sp. Strain CA-271078

 ,
,  , , , and
, , , and 
Abstract
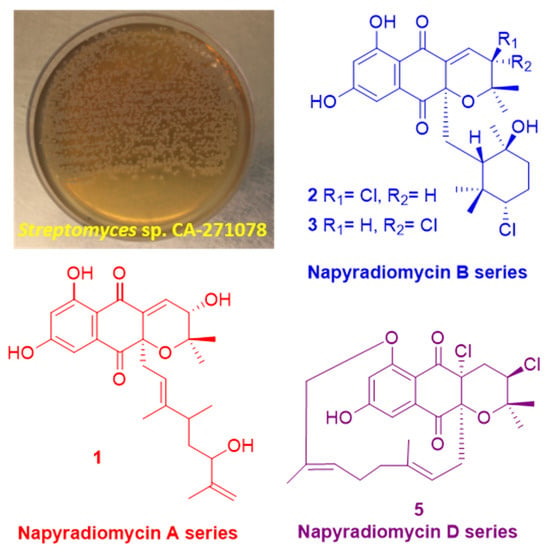
1. Introduction
2. Results
2.1. Isolation and Taxonomy of the Producing Microorganism
2.2. Extraction, Dereplication, and Bioassay-Guided Isolation
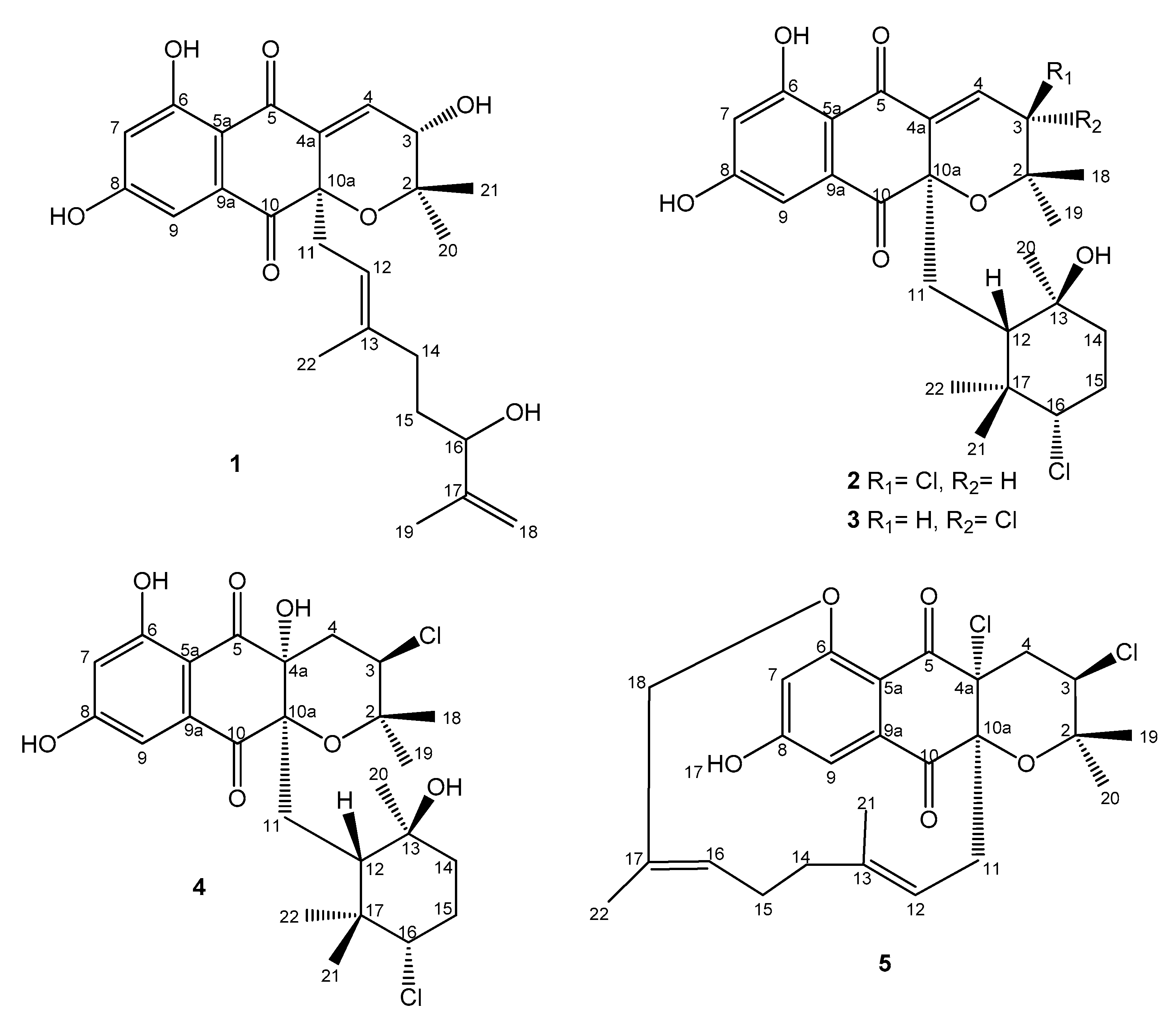
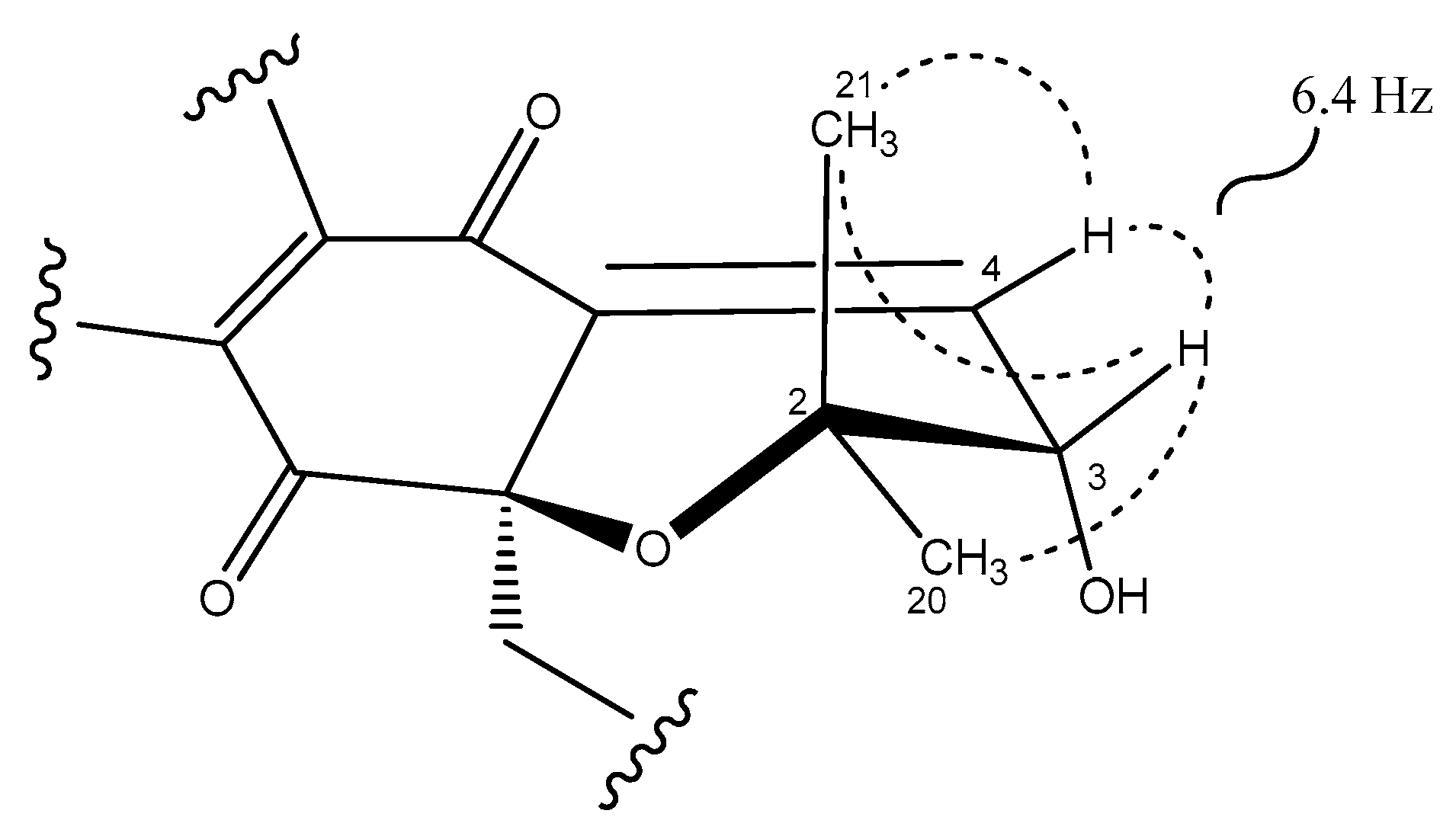
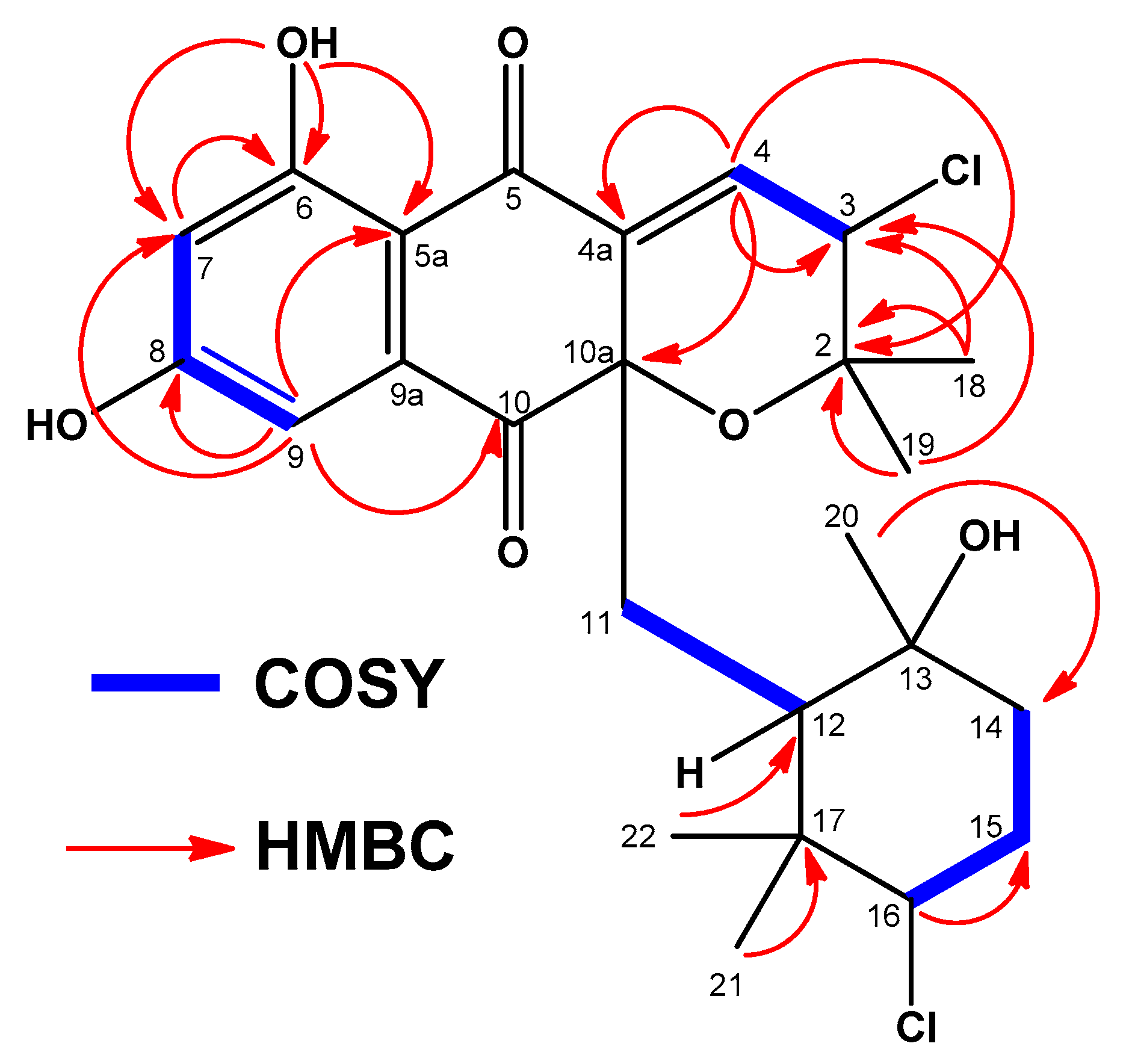
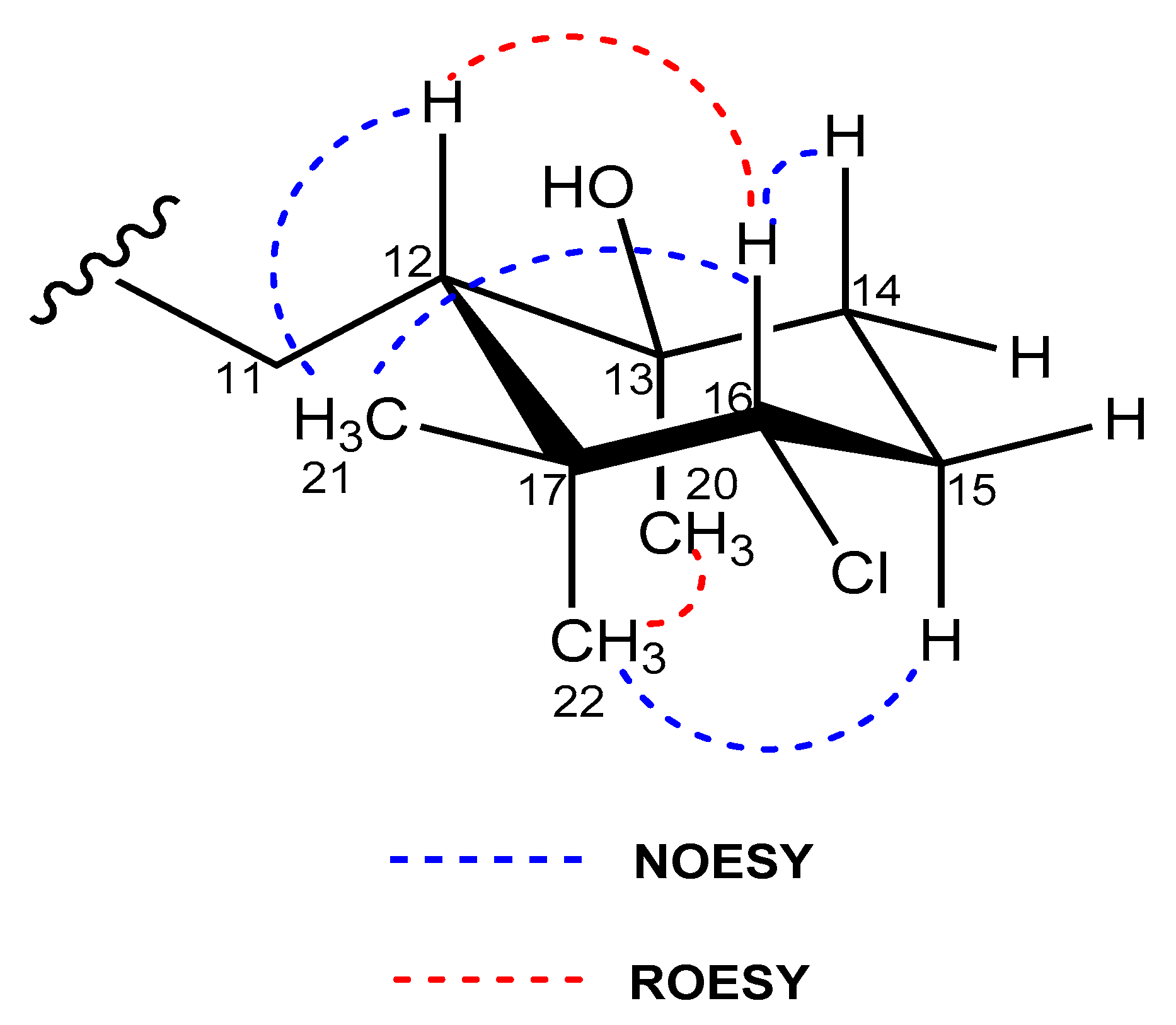
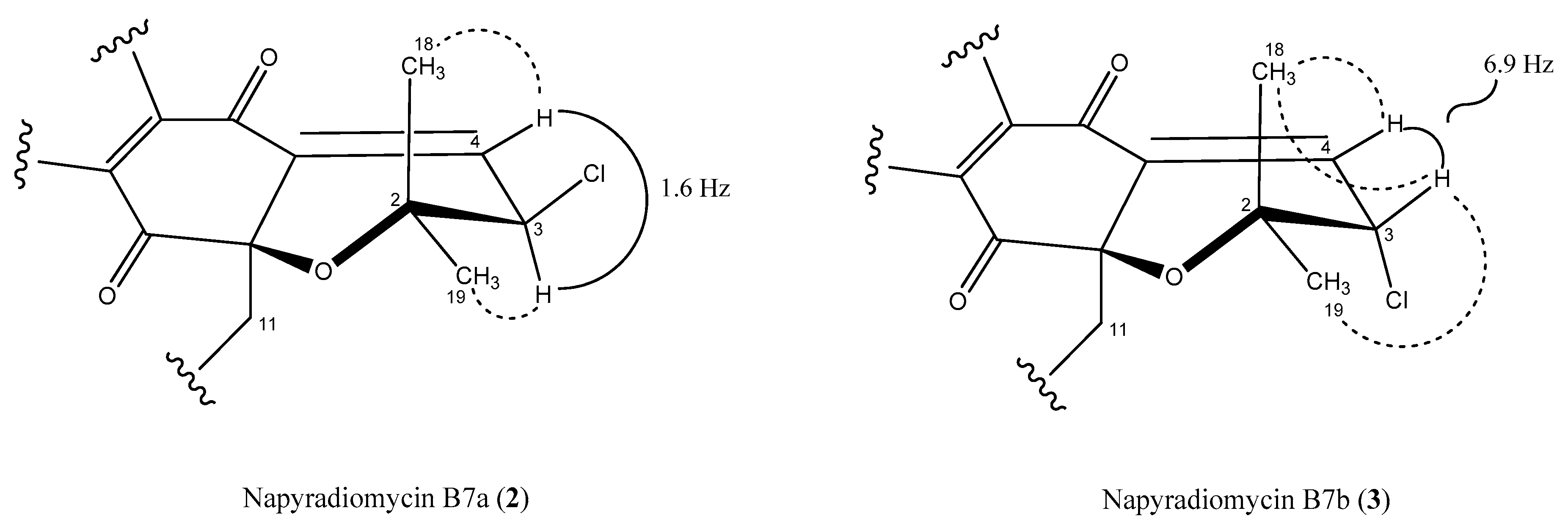
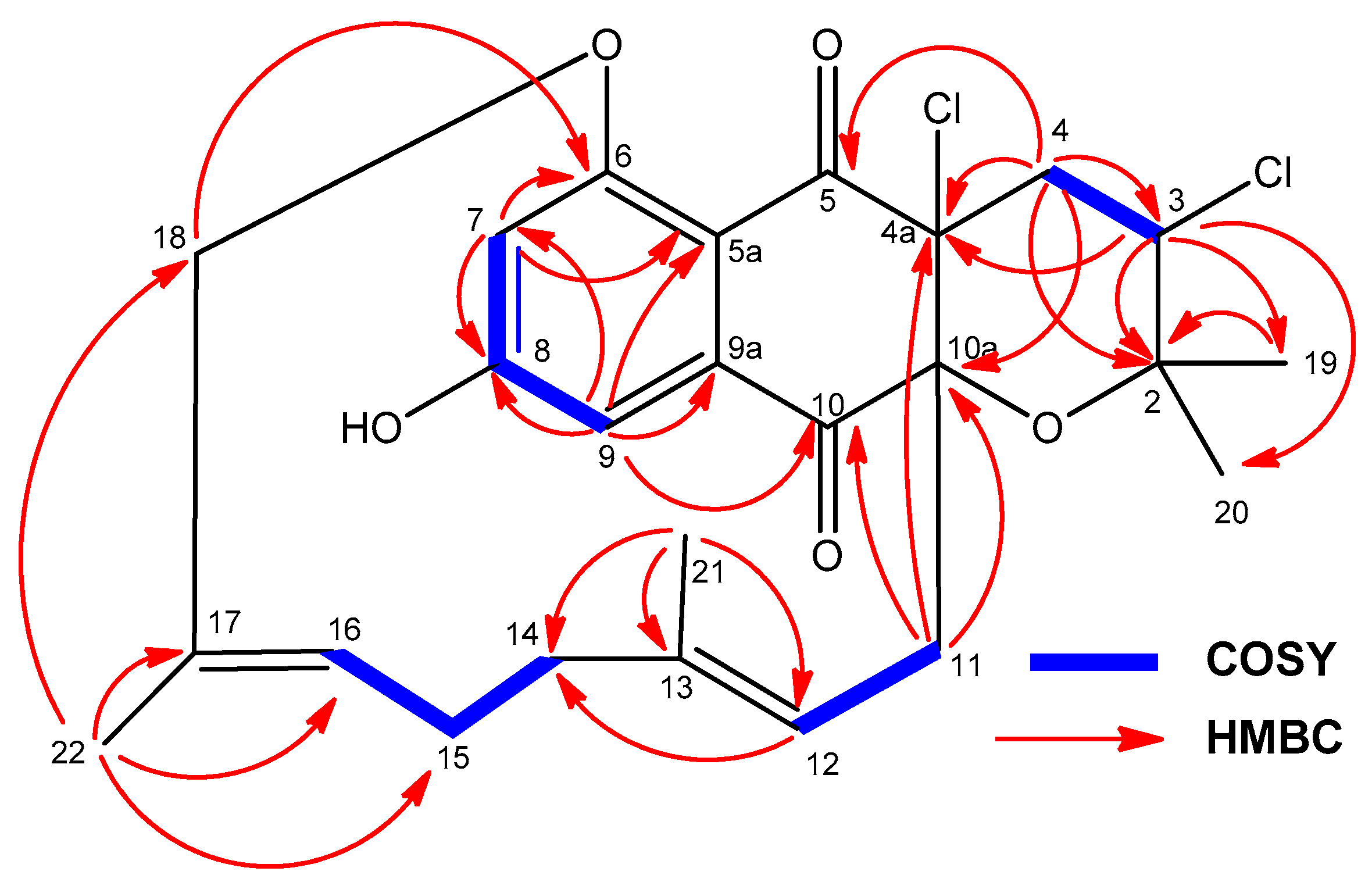
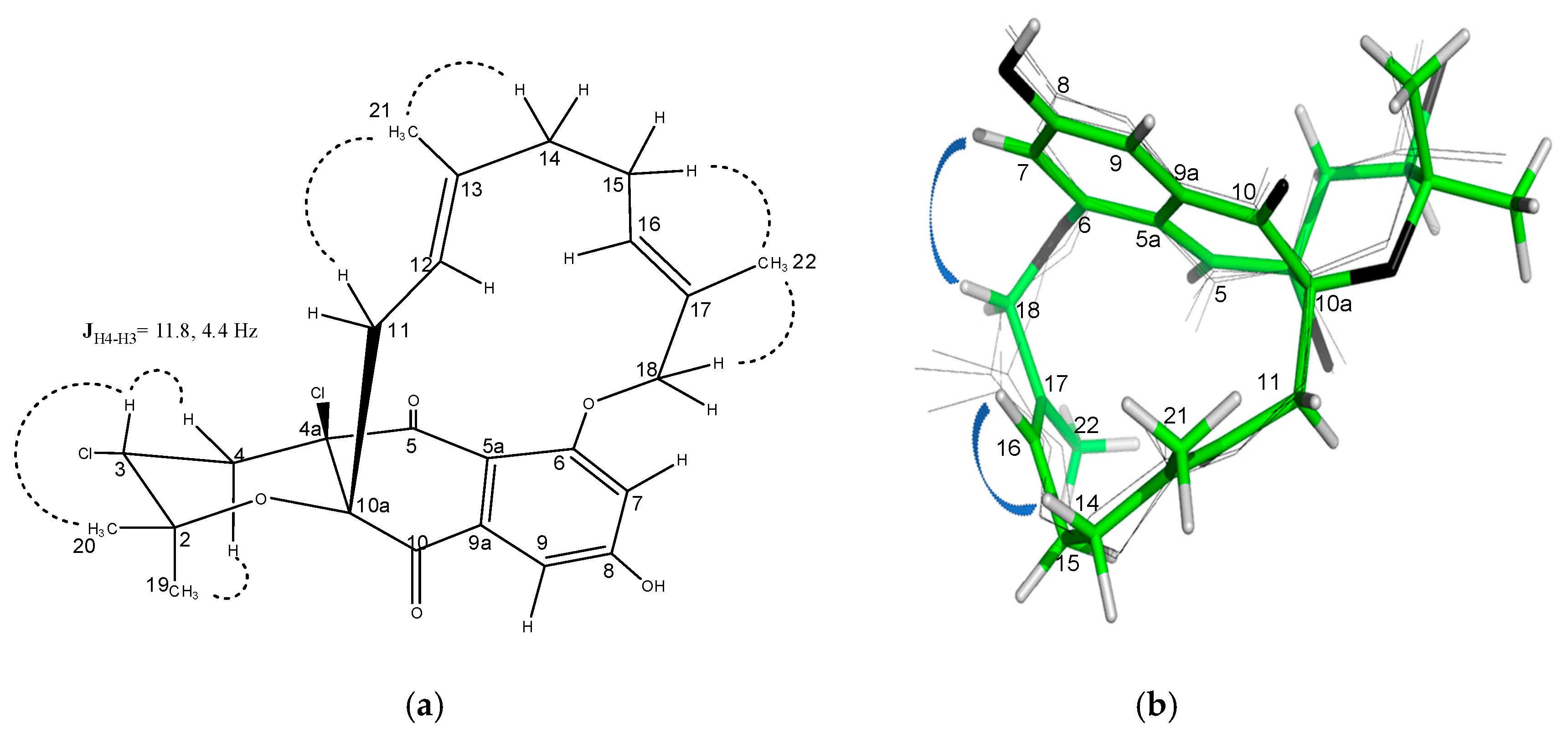
2.3. Structural Elucidation
2.4. Evaluation of Antimicrobial Activity
Antibacterial, Antifungal, and Cytotoxic Activities
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Taxonomic Identification of the Producing Microorganism
4.3. Fermentation of the Producing Microorganism
4.4. Extraction and Bioassay Guided Isolation
4.5. Characterization Data
4.6. Antibacterial and Antifungal Assays Cytotoxic Activities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shiomi, K.; Nakamura, H.; Iinuma, H.; Naganawa, H.; Isshiki, K.; Takeuchi, T.; Umezawa, H.; Iitaka, Y. Structures of new antibiotics napyradiomycins. J. Antibiot. 1986, 39, 494–501. [Google Scholar] [CrossRef]
- Fukuda, D.S.; Mynderse, J.S.; Baker, P.J.; Berry, D.M.; Boeck, L.D.; Yao, R.C.; Mertz, F.P.; Nakatsukasa, W.M.; Mabe, J.; Ott, J.; et al. A80915, a new antibiotic complex produced by Streptomyces aculeolatus. Discovery, taxonomy, fermentation, isolation, characterization, and antibacterial evaluation. J. Antibiot. 1990, 43, 623–633. [Google Scholar] [CrossRef]
- Soria-Mercado, I.E.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. Antibiotic terpenoid chloro-dihydroquinones from a new marine actinomycete. J. Nat. Prod. 2005, 68, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, S.; Li, J.; Chen, Y.; Saurav, K.; Zhang, Q.; Zhang, H.; Zhang, W.; Zhang, W.; Zhang, S.; et al. Antibacterial and cytotoxic new napyradiomycins from the marine-derived Streptomyces sp. SCSIO 10428. Mar. Drugs 2013, 11, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Haste, N.M.; Farnaes, L.; Perera, V.R.; Fenical, W.; Nizet, V.; Hensler, M.E. Bactericidal kinetics of marine-derived napyradiomycins against contemporary methicillin-resistant Staphylococcus aureus. Mar. Drugs 2011, 9, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, K.; Sue, M.; Furihata, K.; Ito, S.; Seto, H. Terpenoids produced by actinomycetes: Napyradiomycins from Streptomyces antimycoticus NT17. J. Nat. Prod. 2008, 71, 595–601. [Google Scholar] [CrossRef]
- Henkel, T.; Zeeck, A. Secondary metabolites by chemical screening. 15. Structure and absolute configuration of naphthomevalin, a new dihydro-naphthoquinone antibiotic from Streptomyces sp. J. Antibiot. 1991, 44, 665–669. [Google Scholar] [CrossRef]
- Shiomi, K.; Nakamura, H.; Iinuma, H.; Naganawa, H.; Takeuchi, T.; Umezawa, H.; Iitaka, Y. New antibiotic napyradiomycins A2 and B4 and stereochemistry of napyradiomycins. J. Antibiot. 1987, 40, 1213–1219. [Google Scholar] [CrossRef]
- Soria-Mercado, I.E.; Jensen, P.R.; Fenical, W.; Kassel, S.; Golen, J. 3,4a-Dichloro-10a-(3-chloro-6-hydroxy-2,2,6-trimethylcyclohexylmethyl)-6,8-dihydroxy-2,2,7-trimethyl-3,4,4a,10a-tetrahydro-2H-benzo[g]chromene-5,10-dione. Acta Crystallogr. Sect. E 2004, 60, o1627–o1629. [Google Scholar] [CrossRef]
- Gomi, S.; Ohuchi, S.; Sasaki, T.; Itoh, J.; Sezaki, M. Studies on new antibiotics SF2415. 2. The structural elucidation. J. Antibiot. 1987, 40, 740–749. [Google Scholar] [CrossRef]
- Umezawa, K.; Masuoka, S.; Ohse, T.; Naganawa, H.; Kondo, S.; Ikeda, Y.; Kinoshita, N.; Hamada, M.; Sawa, T.; Takeuchi, T. Isolation from Streptomyces of a novel naphthoquinone compound, naphthablin, that inhibits Abl oncogene functions. J. Antibiot. 1995, 48, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Kwon, H.C.; Williams, P.G.; Jensen, P.R.; Fenical, W. Azamerone, a terpenoid phthalazinone from a marine-derived bacterium related to the genus Streptomyces (actinomycetales). Org. Lett. 2006, 8, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, K.; Irie, K.; Toda, T.; Matsuo, Y.; Kasai, H.; Sue, M.; Furihata, K.; Seto, H. Studies on terpenoids produced by actinomycetes. 5-Dimethylallylindole-3-carboxylic acid and A80915G-8”-acid produced by marine-derived Streptomyces sp. MS239. J. Antibiot. 2008, 61, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Farnaes, L.; Coufal, N.G.; Kauffman, C.A.; Rheingold, A.L.; DiPasquale, A.G.; Jensen, P.R.; Fenical, W. Napyradiomycin derivatives, produced by a marine-derived actinomycete, illustrate cytotoxicity by induction of apoptosis. J. Nat. Prod. 2014, 77, 15–21. [Google Scholar] [CrossRef]
- Farnaes, L.; La Clair, J.J.; Fenical, W. Napyradiomycins CNQ525.510B and A80915C target the Hsp90 paralogue Grp94. Org. Biomol. Chem. 2014, 12, 418–423. [Google Scholar] [CrossRef]
- Hori, Y.; Abe, Y.; Shigematsu, N.; Goto, T.; Okuhara, M.; Kohsaka, M. Napyradiomycin A and B1: Non-steroidal estrogen-receptor antagonists produced by a Streptomyces. J. Antibiot. 1993, 46, 1890–1893. [Google Scholar] [CrossRef]
- Dantzig, A.H.; Minor, P.L.; Garrigus, J.L.; Fukuda, D.S.; Mynderse, J.S. Studies on the mechanism of action of A80915A, a semi-naphthoquinone natural product, as an inhibitor of gastric (H+-K+)-ATPase. Biochem. Pharmacol. 1991, 42, 2019–2026. [Google Scholar] [CrossRef]
- Shiomi, K.; Iinuma, H.; Hamada, M.; Naganawa, H.; Manabe, M.; Matsuki, C.; Takeuchi, T.; Umezawa, H. Novel antibiotics napyradiomycins. Production, isolation, physicochemical properties and biological activity. J. Antibiot. 1986, 39, 487–493. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, A.R.; Davis, R.A.; Keyzersd, R.A.; Prinsepe, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef]
- Hassan, S.S.; Shaikh, A.L. Marine actinobacteria as a drug treasure house. Biomed. Pharmacother. 2017, 87, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Lacret, R.; Oves-Costales, D.; Gómez, C.; Díaz, C.; de la Cruz, M.; Pérez-Victoria, I.; Vicente, F.; Genilloud, O.; Reyes, F. New ikarugamycin derivatives with antifungal and antibacterial properties from Streptomyces zhaozhouensis. Mar. Drugs 2015, 13, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Lacret, R.; Pérez-Victoria, I.; Oves-Costales, D.; de la Cruz, M.; Domingo, E.; Martín, J.; Díaz, C.; Vicente, F.; Genilloud, O.; Reyes, F. MDN-0170, a new napyradiomycin from Streptomyces sp. strain CA-271078. Mar. Drugs 2016, 14, 188. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Victoria, I.; Martín, J.; Reyes, F. Combined LC/UV/MS and NMR strategies for the dereplication of marine natural products. Planta Med. 2016, 82, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Dictionary of Natural Products (DNP 2019). Chapman & Hall/CRC. Available online: http://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml (accessed on 26 November 2019).
- Cheng, Y.-B.; Jensen, P.R.; Fenical, W. Cytotoxic and antimicrobial napyradiomycins from two marine-derived Streptomyces strains. Eur. J. Org. Chem. 2013, 18, 3751–3757. [Google Scholar] [CrossRef]
- Kamimura, D.; Yamada, K.; Tsuji, T. Agent for Inhibiting Production of Venous Cell-Adhering Molecule-1 and Napyradiomycin SC. Japanese Patent 09110689, 28 April 1997. [Google Scholar]
- Winter, J.M.; Moffitt, M.C.; Zazopoulos, E.; McAlpine, J.B.; Dorrestein, P.C.; Moore, B.S. Molecular basis for chloronium-mediated meroterpene cyclization: Cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J. Biol. Chem. 2007, 282, 16362–16368. [Google Scholar] [CrossRef]
- McKinnie, S.M.K.; Miles, Z.D.; Jordan, P.A.; Awakawa, T.; Pepper, H.P.; Murray, L.A.M.; George, J.H.; Moore, B.S. Total enzyme syntheses of napyradiomycins A1 and B1. J. Am. Chem. Soc. 2018, 140, 17840–17845. [Google Scholar] [CrossRef]
- Barbeau, X.; Vincent, A.T.; Lagüe, P. ConfBuster: Open-source tools for macrocycle conformational search and analysis. J. Open Res. Softw. 2018, 6, 1. [Google Scholar] [CrossRef]
- Bernhardt, P.; Okino, T.; Winter, J.M.; Miyanaga, A.; Moore, B.S. A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J. Am. Chem. Soc. 2011, 133, 4268–4270. [Google Scholar] [CrossRef]
- Wever, R.; Krenn, B.E.; Renirie, R. Marine vanadium-dependent haloperoxidases, their isolation, characterization, and application. Methods Enzymol. 2018, 605, 141–201. [Google Scholar] [CrossRef]
- Diethelm, S.; Teufel, R.; Kaysser, L.; Moore, B.S. A multitasking vanadium-dependent chloroperoxidase as an inspiration for the chemical synthesis of the merochlorins. Angew. Chem. Int. Ed. 2014, 53, 11023–11026. [Google Scholar] [CrossRef] [PubMed]
- McKinnie, S.M.K.; Miles, Z.D.; Moore, B.S. Characterization and biochemical assays of Streptomyces vanadium-dependent chloroperoxidases. Methods Enzymol. 2018, 604, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kaysser, L.; Bernhardt, P.; Nam, S.J.; Loesgen, S.; Ruby, J.G.; Skewes-Cox, P.; Jensen, P.R.; Fenical, W.; Moore, B.S. Merochlorins A-D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc. 2012, 134, 11988–119991. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; da S Sousa, T.; Crespo, G.; Palomo, S.; González, I.; Tormo, J.R.; de la Cruz, M.; Anderson, M.; Hill, R.T.; Vicente, F.; et al. Kocurin, the true structure of PM181104, an anti-methicillin-resistant Staphylococcus aureus (MRSA) thiazolyl peptide from the marine-derived bacterium Kocuria palustris. Mar. Drugs 2013, 11, 387–398. [Google Scholar] [CrossRef]
- Monteiro, M.C.; de la Cruz, M.; Cantizani, J.; Moreno, C.; Tormo, J.R.; Mellado, E.; De Lucas, J.R.; Asensio, F.; Valiante, V.; Brakhage, A.A.; et al. A new approach to drug discovery: High-throughput screening of microbial natural extracts against Aspergillus fumigatus using resazurin. J. Biomol. Screen. 2012, 17, 542–549. [Google Scholar] [CrossRef]
- Zhang, L.; Ravipati, A.S.; Koyyalamudi, S.R.; Jeong, S.C.; Reddy, N.; Bartlett, J.; Smith, P.T.; de la Cruz, M.; Monteiro, M.C.; Melguizo, A.; et al. Anti-fungal and anti-bacterial activities of ethanol extracts of selected traditional chinese medicinal herbs. Asian Pac. J. Trop. Med. 2013, 6, 673–681. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation andvalidation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | δ 1H (mult, J, Hz) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| 2 | |||||
| 3 | 3.72, d (6.4) | 4.98, d (1.6) | 4.70, d (6.9) | 4.38, dd (11.8, 4.4) | 4.40, dd (11.8, 4.4) |
| 4 | 6.89, d (6.4) | 6.73, d (1.6) | 7.01, d (6.9) | 2.12, dd (13.4, 4.4) | 2.25 a, dd (14.4, 4.4) |
| 2.19, dd, (13.4, 11.8) | 2.52, dd (14.4, 11.8) | ||||
| 4a | 6.74, s | ||||
| 5 | |||||
| 5a | |||||
| 6 | 12.52, s | 12.66, s | 12.56, s | 11.94, s | |
| 7 | 6.57, d (2.1) | 6.59, d (2.2) | 6.60, d (1.9) | 6.66, d (2.0) | 7.04, d (2.4) |
| 8 | -- | -- | -- | -- | -- |
| 9 | 6.84, d (2.1) | 6.88, d (2.2) | 6.88, d (1.9) | 6.99, d (2.0) | 7.18, d (2.4) |
| 9a | |||||
| 10 | |||||
| 10a | |||||
| 11 | 2.43, m | 1.52 a, dd (13.3, 3.4) | 1.68, dd (15.2, 2.4) | 1.27, br d (16.4, 2.3) | 2.26 a, m |
| 1.85 b, dd (13.3, 3.20) | 1.93, dd (15.2, 6.0) | 2.32, dd (16.4, 6.0) | 2.77, dd (13.5,8.9) | ||
| 12 | 4.98, br t (7.25) | 1.31, br t (3.3) | 1.60 a, dd (6.0, 2.4) | 1.55, dd (6.0, 2.3) | 4.26, br t (8.9) |
| 13 | 4.75, s | 4.40, s | 5.04, s | ||
| 14 | 1.74, m | 1.46, m | 1.37, m | 1.47, m | 1.42, m |
| 1.83, m | 1.51 a, m | 1.60 a, m | 1.67, m | 1.93 b, m | |
| 15 | 1.31, m | 1.70, dd (13.5, 12.1) | 1.70, dd (13.2, 12.1) | 1.72, m | 1.95 b, m |
| 1.81 b, dd (13.5, 3.8) | 1.83, dd (13.2, 3.1) | 1.82, m | |||
| 16 | 3.78, t (6.2, 6.2) | 4.02, dd (12.1, 3.8) | 3.77, dd (12.1, 3.1) | 3.81, dd (11.9, 3.9) | 4.91, br t (11.5, 6.5) |
| 17 | |||||
| 18 | 4.71, br s | 0.94 c, s | 1.02, s | 1.15, s | 4.67, s |
| 4.85, br s | 4.73, s | ||||
| 19 | 1.61, s | 1.40, s | 1.42, s | 1.35, s | 1.37, s |
| 20 | 1.27, s | 0.79, s | 0.91, s | 1.02, s | 1.11, s |
| 21 | 0.88, s | 0.94 c, s | 0.77, s | 0.38, s | 1.32, s |
| 22 | 1.23, s | 0.64, s | 0.63, s | 0.59, s | 1.52, s |
| No. | δ 13C | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| 2 | 75.4, C | 76.1, C | 74.8, C | 79.1, C | 78.2, C |
| 3 | 65.8, CH | 59.9, CH | 57.3, CH | 58.9, CH | 59.8, CH |
| 4 | 134.7, CH | 136,2 CH | 132.1, CH | 40.4, CH2 | 41.6 CH2 |
| 4a | 137.6, C | 136.3, C | 137.8, C | 79.2, C | 81.7, C |
| 5 | 189.7, C | 187.7, C | 188.3, C | 194.4, C | 183.4, C |
| 5a | 110.8, C | 110.5, C | 110.4, C | 107.9, C | 116.7, C |
| 6 | 164.7, C | 164.8, C | 164.8, C | 163.9, C | 162.7, C |
| 7 | 107.8, CH | 108.1, CH | 108.1, CH | 108.5, CH | 113.5, CH |
| 8 | 165.7, C | 165.3, C | 165.6, C | 165.7, C | 163.8, C |
| 9 | 107.7, CH | 107.7, CH | 107.9, CH | 107.8, CH | 108.3, CH |
| 9a | 136.5, C | 135.9, C | 135.9, C | 135.1, C | 136.4, C |
| 10 | 195.5, C | 195.3, C | 194.9, C | 199.5, C | 196.0, C |
| 10a | 82.2, C | 82.9, C | 82.6, C | 83.7, C | 82.9, C |
| 11 | 40.4, CH2 | 37.0, CH2 | 37.8, CH2 | 33.7, CH2 | 41.5, CH2 |
| 12 | 117.7, CH | 50.5, CH | 50.1, CH | 48.6, CH | 116.9, CH |
| 13 | 138.9, C | 70.5, C | 70.1, C | 70.1, C | 140.6, C |
| 14 | 35.5, CH2 | 41.2, CH2 | 41.3, CH2 | 41.2, CH2 | 39.7, CH2 |
| 15 | 32.9, CH2 | 31.1, CH2 | 30.8, CH2 | 30.5, CH2 | 23.7, CH2 |
| 16 | 73.4, CH | 72.2, CH | 72.4, CH | 71.8, CH | 126.5, CH |
| 17 | 148.3, C | 40.4, C | 40.4, C | 40.2, C | 129.2, C |
| 18 | 110.3, CH2 | 20.5, CH3 | 25.8, CH3 | 21.6, CH3 | 76.3, CH2 |
| 19 | 17.8, CH3 | 26.5, CH3 | 26.5, CH3 | 28.7, CH3 | 28.9, CH3 |
| 20 | 24.9, CH3 | 23.6, CH3 | 24.2, CH3 | 24.4, CH3 | 22.5, CH3 |
| 21 | 25.7, CH3 | 30.1, CH3 | 29.0, CH3 | 28.4, CH3 | 15.2, CH3 |
| 22 | 16.1, CH3 | 16.6, CH3 | 16.7, CH3 | 16.0, CH3 | 15.1, CH3 |
| MIC (μg/mL) | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| MRSA | Mt | Ec | Ab | Af | HepG-2 | |
| 1 | >96 | NT a | >96 | >96 | NT a | >67.8 |
| 2 | 48 | 12–24 | >96 | >96 | >96 | 41.7 |
| 3 | >64 | >64 | >64 | >64 | >64 | 109.5 |
| 4 | >96 | 24–48 | >96 | >96 | >96 | 263.5 |
| 5 | 12–24 | 24–48 | >96 | >96 | NT a | 14.9 |
| 6 | >96 | >96 | >96 | >96 | >96 | 277.2 |
| 7 | 48–96 | 12–24 | >96 | >96 | >96 | 186.9 |
| 8 | >64 | >64 | >64 | >64 | >64 | 30.2 |
| 9 | 48–96 | 12–24 | >96 | >96 | >96 | 71.2 |
| 10 | 48–96 | >96 | >96 | >96 | >96 | 64.4 |
| 11 | 12–24 | 48–96 | >96 | >96 | >96 | 30.4 |
| 12 | 12–24 | 48–96 | >96 | >96 | >96 | 28.6 |
| 13 | 12–24 | 12–24 | >96 | >96 | >96 | 15.6 |
| 14 | 3–6 | 24–48 | >96 | >96 | >96 | 27.1 |
| 15 | 12–24 | 24–48 | >96 | >96 | >96 | 40.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carretero-Molina, D.; Ortiz-López, F.J.; Martín, J.; Oves-Costales, D.; Díaz, C.; de la Cruz, M.; Cautain, B.; Vicente, F.; Genilloud, O.; Reyes, F. New Napyradiomycin Analogues from Streptomyces sp. Strain CA-271078. Mar. Drugs 2020, 18, 22. https://doi.org/10.3390/md18010022
Carretero-Molina D, Ortiz-López FJ, Martín J, Oves-Costales D, Díaz C, de la Cruz M, Cautain B, Vicente F, Genilloud O, Reyes F. New Napyradiomycin Analogues from Streptomyces sp. Strain CA-271078. Marine Drugs. 2020; 18(1):22. https://doi.org/10.3390/md18010022
Chicago/Turabian StyleCarretero-Molina, Daniel, Francisco Javier Ortiz-López, Jesús Martín, Daniel Oves-Costales, Caridad Díaz, Mercedes de la Cruz, Bastien Cautain, Francisca Vicente, Olga Genilloud, and Fernando Reyes. 2020. "New Napyradiomycin Analogues from Streptomyces sp. Strain CA-271078" Marine Drugs 18, no. 1: 22. https://doi.org/10.3390/md18010022
APA StyleCarretero-Molina, D., Ortiz-López, F. J., Martín, J., Oves-Costales, D., Díaz, C., de la Cruz, M., Cautain, B., Vicente, F., Genilloud, O., & Reyes, F. (2020). New Napyradiomycin Analogues from Streptomyces sp. Strain CA-271078. Marine Drugs, 18(1), 22. https://doi.org/10.3390/md18010022