Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes


 ,
,  ,
, 
 and
and 
Abstract
1. Introduction
2. Results
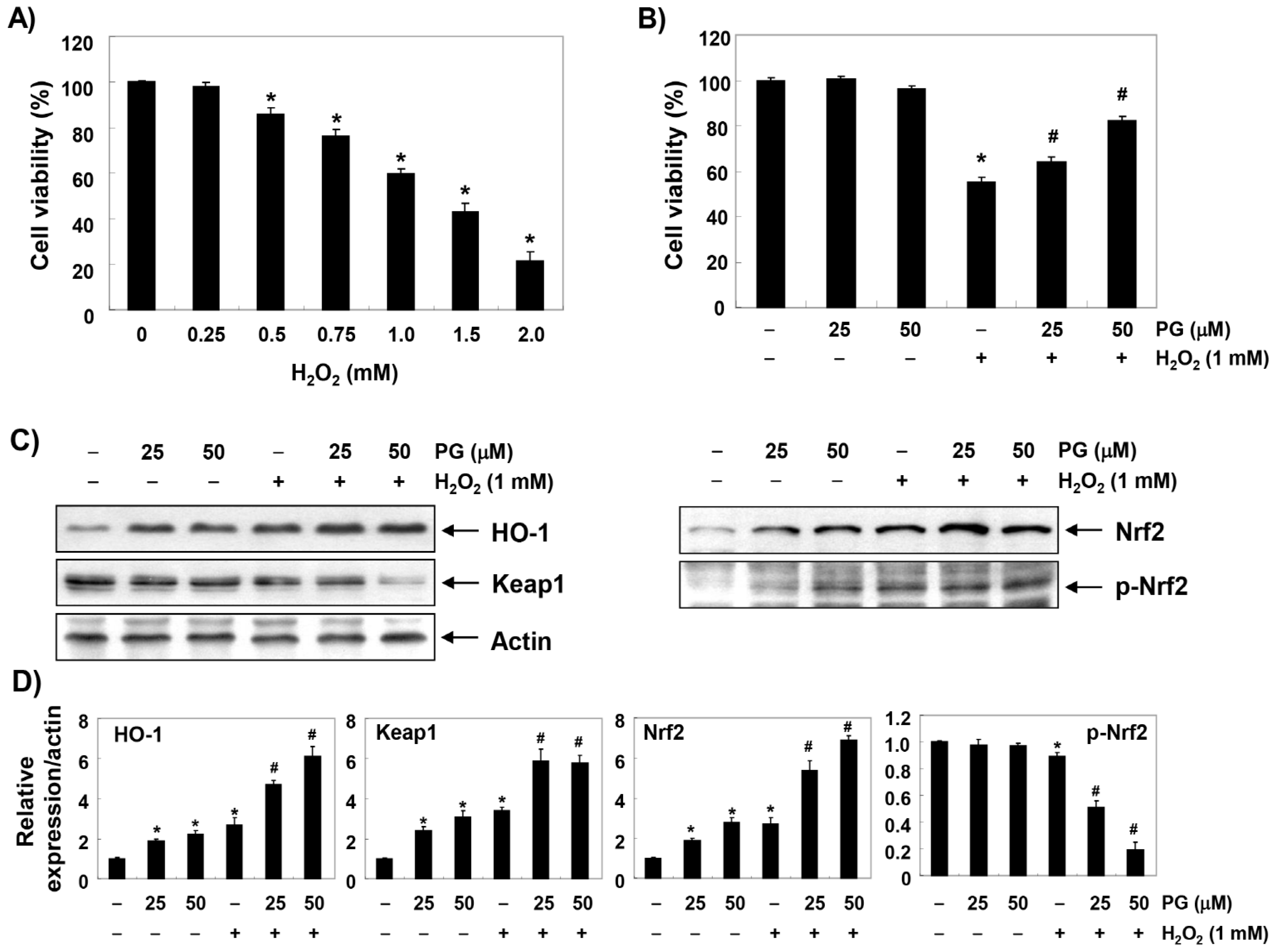
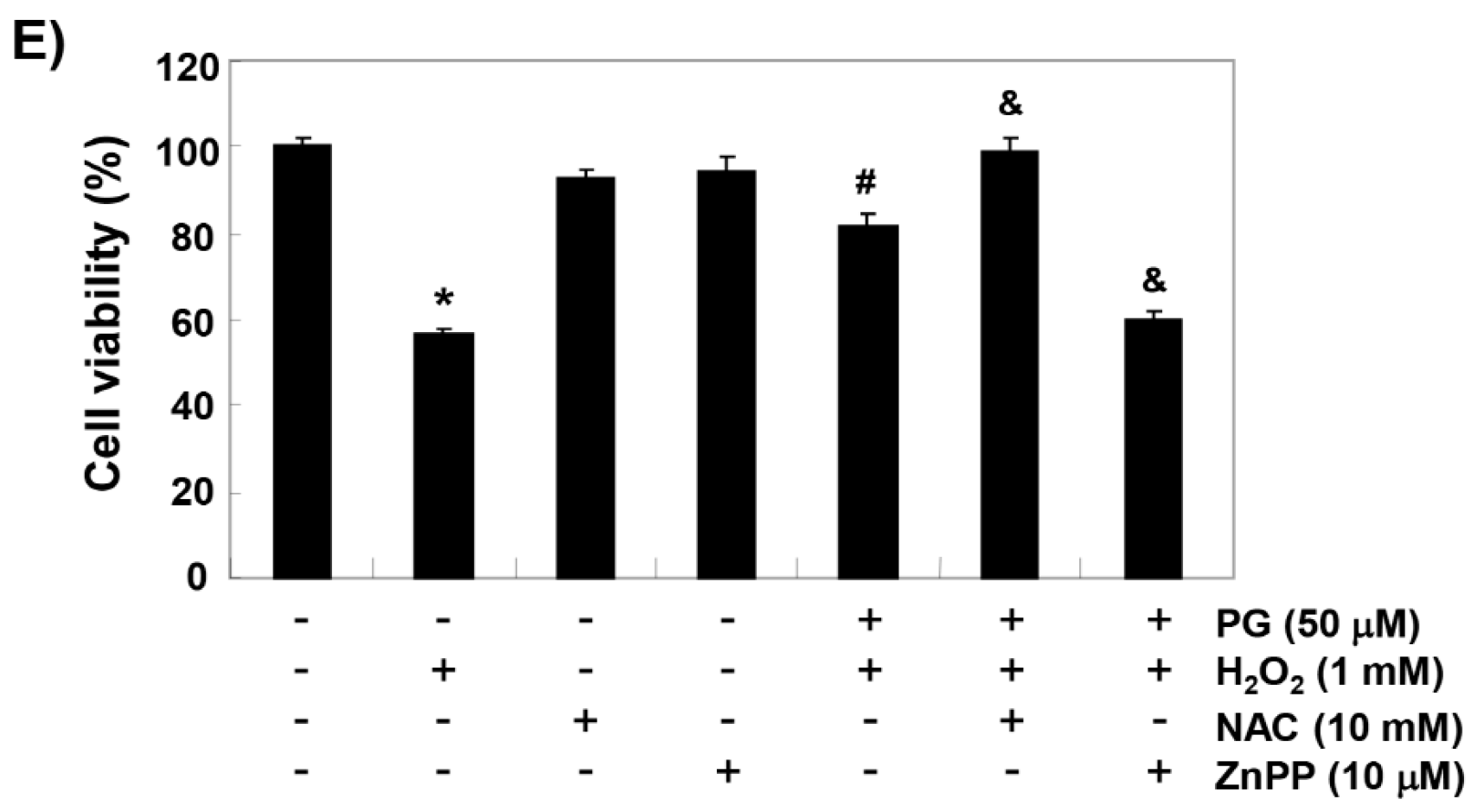
2.1. PG Inhibits H2O2-Induced Cytotoxicity in HaCaT Keratinocytes
2.2. PG Activates the Nrf2/HO-1 Signaling Pathway in HaCaT Keratinocytes
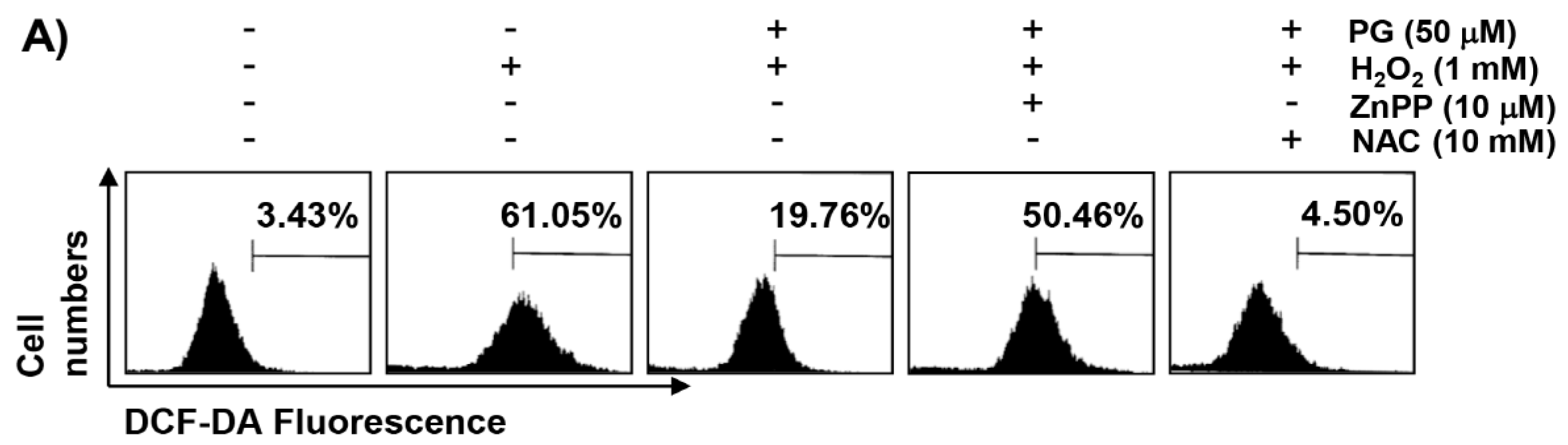
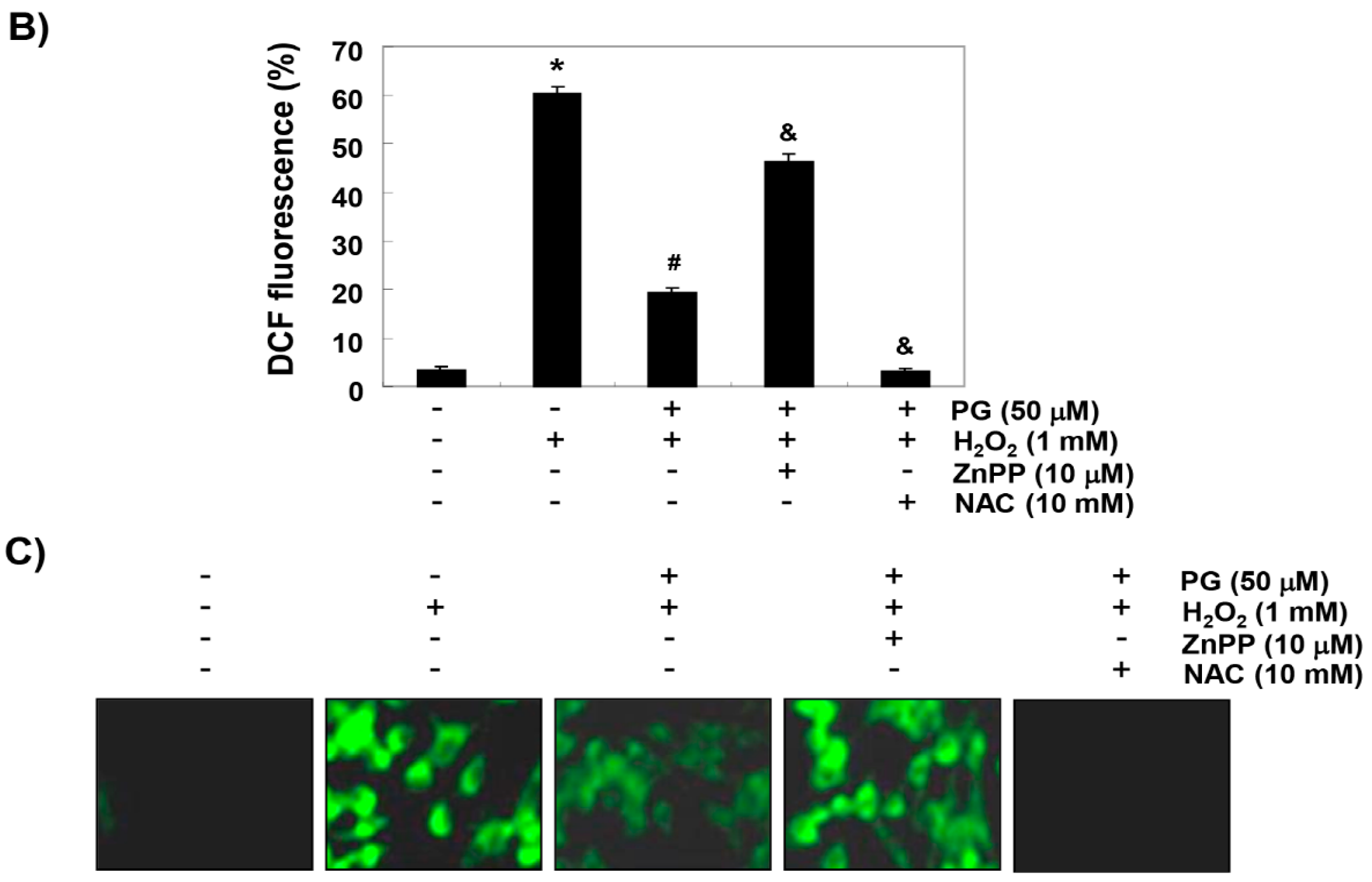
2.3. PG Inhibits H2O2-Induced ROS Generation in HaCaT Keratinocytes
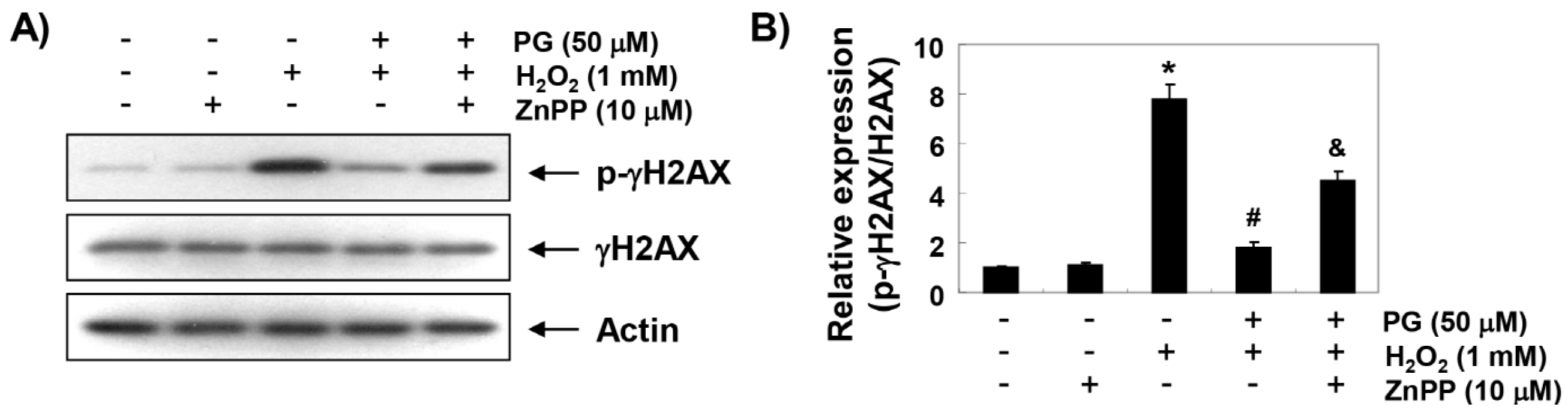
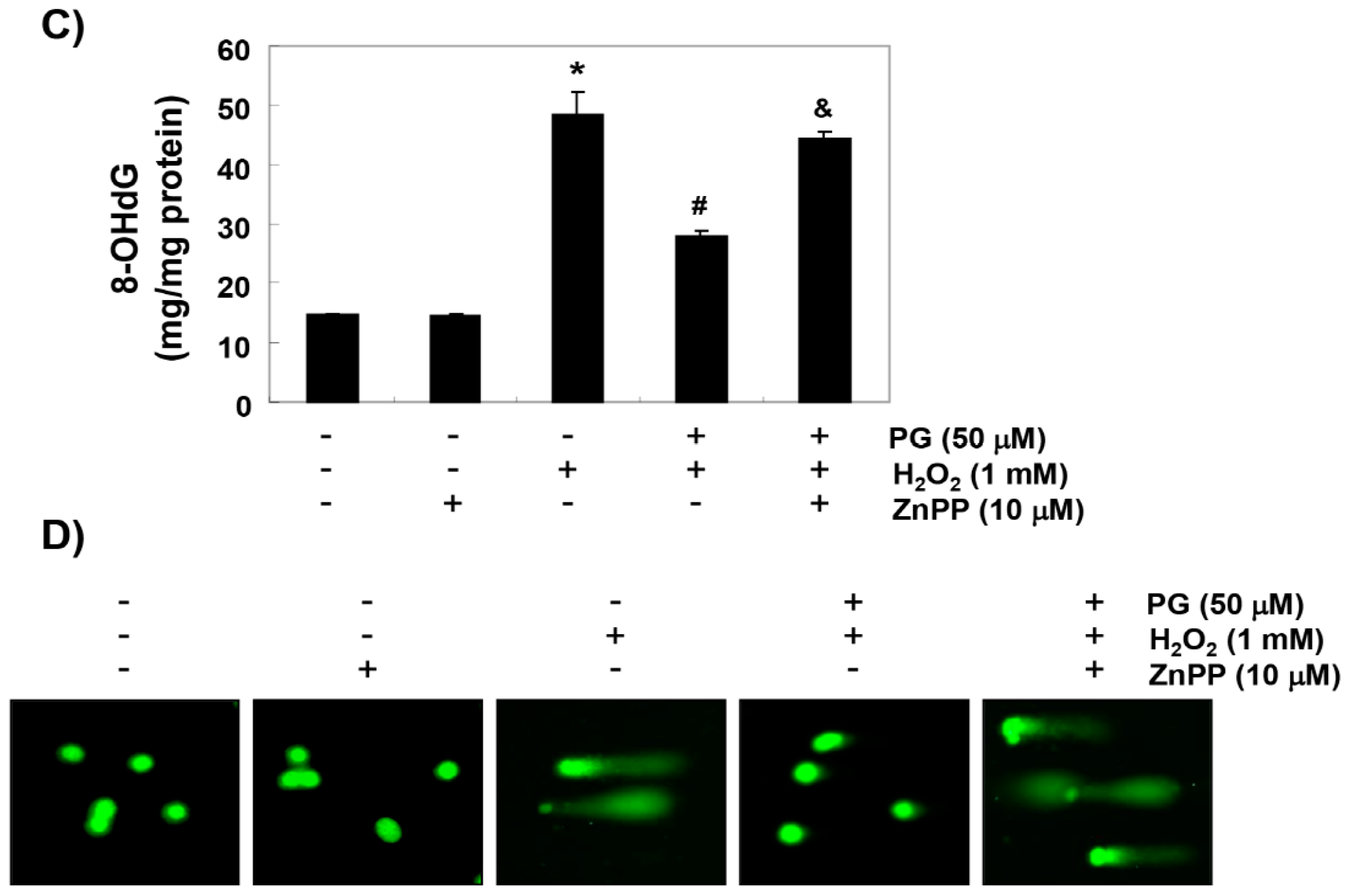
2.4. PG Blocks H2O2-Induced DNA Damage in HaCaT Keratinocytes
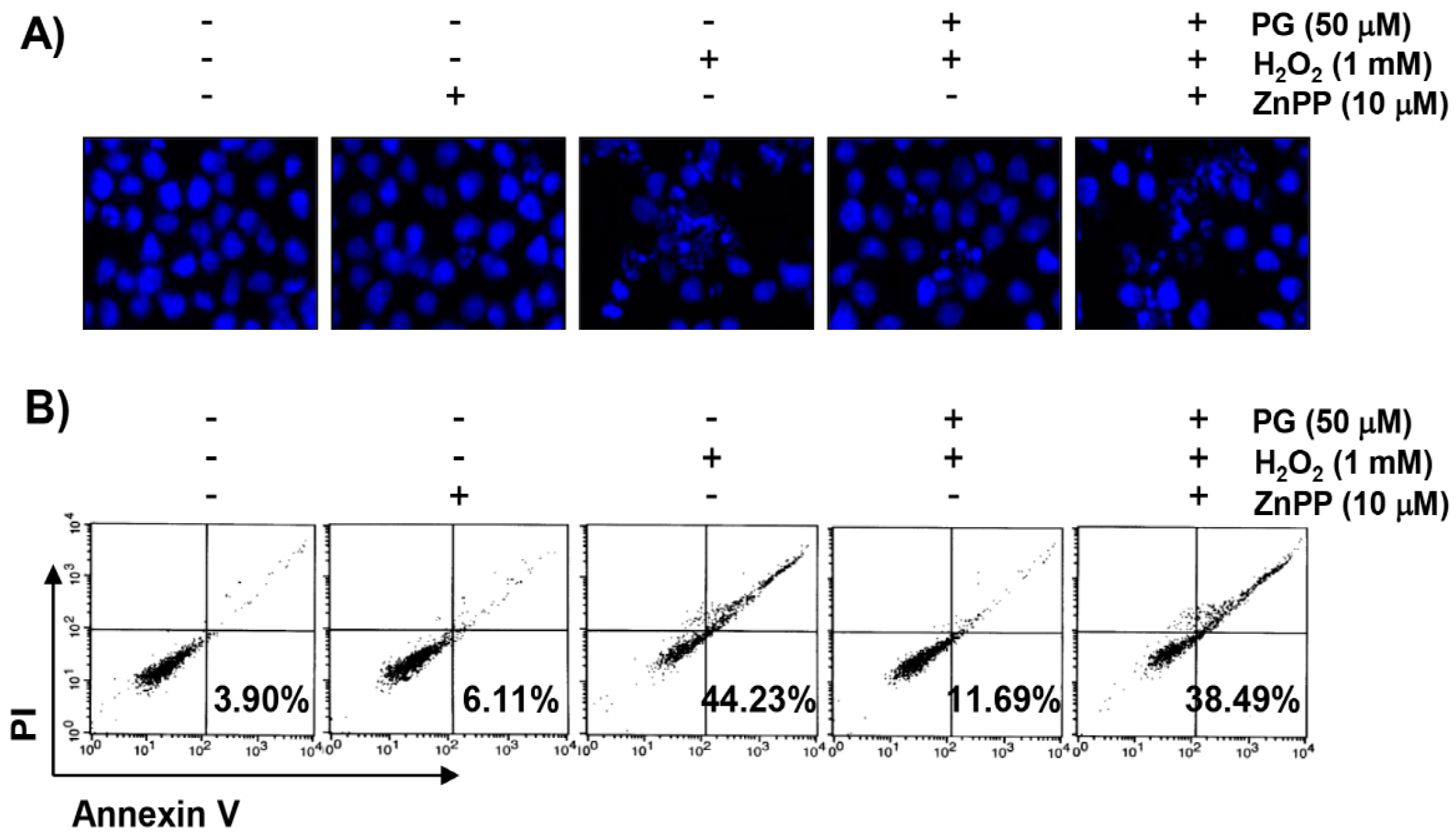
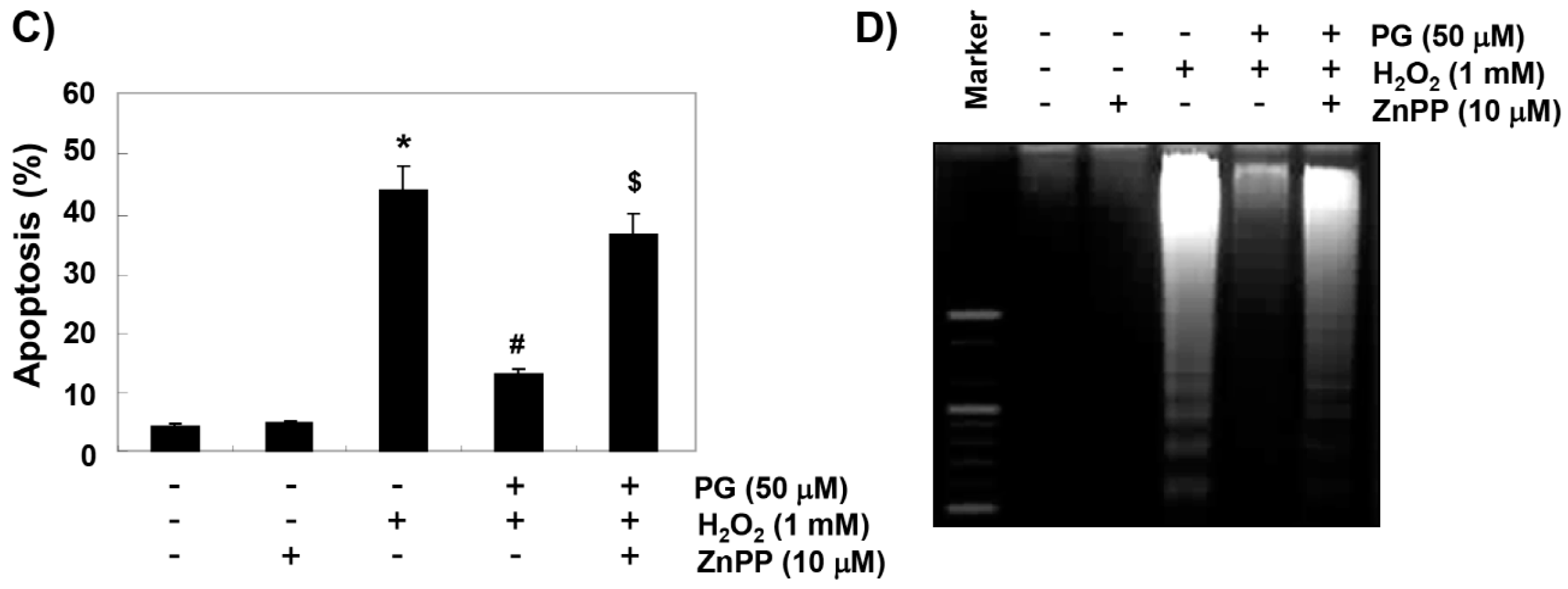
2.5. PG Suppresses H2O2-Induced Apoptosis in HaCaT Keratinocytes
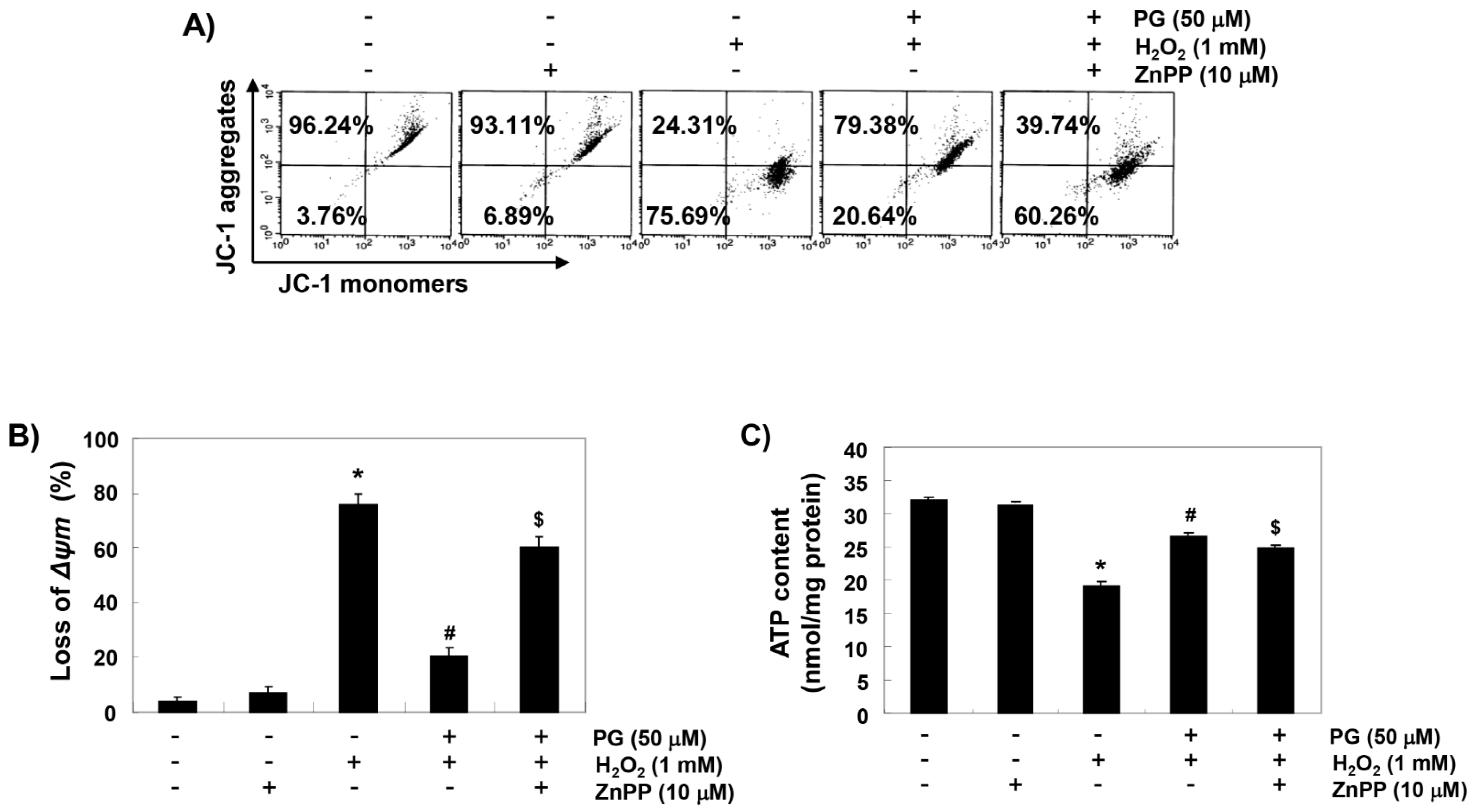
2.6. PG Reduces H2O2-Induced Mitochondrial Dysfunction in HaCaT Keratinocytes
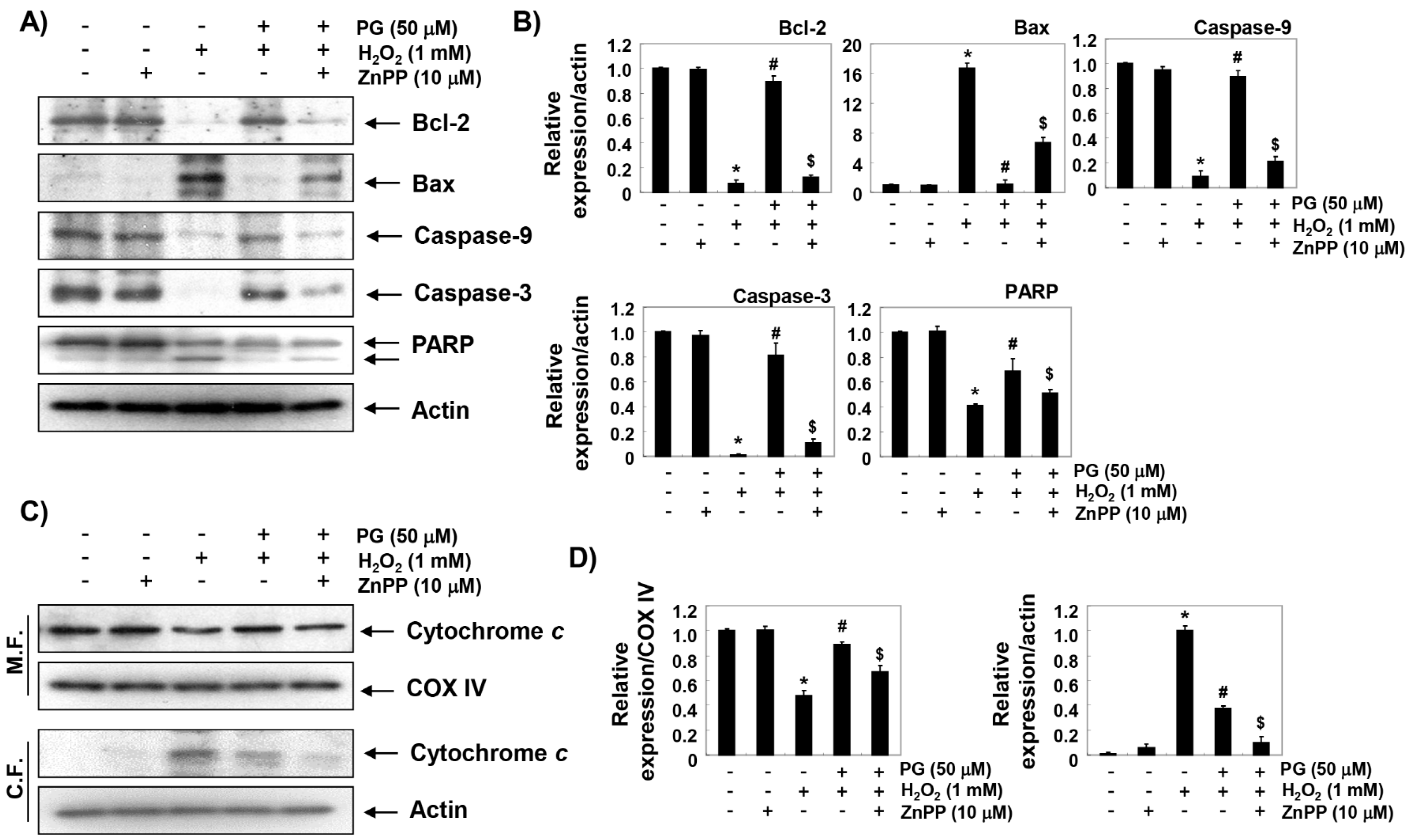
2.7. PG Restores H2O2-Induced Alteration of the Apoptosis Regulatory Genes in HaCaT Keratinocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture and PG Treatment
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. Measurement of ROS Level
4.5. Comet Assay for DNA Damage
4.6. Determination of 8-OHdG
4.7. Apoptosis Assay Using Fluorescence Microscopy
4.8. Apoptosis Analysis Using Flow Cytometry
4.9. Detection of DNA Fragmentation
4.10. Analysis of MMP
4.11. Detection of ATP Levels
4.12. Colorimetric Assay of Caspase-3 Activity
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and nutritional antioxidants in human diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef]
- Wölfle, U.; Seelinger, G.; Bauer, G.; Meinke, M.C.; Lademann, J.; Schempp, C.M. Reactive molecule species and antioxidative mechanisms in normal skin and skin aging. Skin Pharmacol. Physiol. 2014, 27, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Mohania, D.; Chandel, S.; Kumar, P.; Verma, V.; Digvijay, K.; Tripathi, D.; Choudhury, K.; Mitten, S.K.; Shah, D. Ultraviolet radiations: Skin defense-damage mechanism. Adv. Exp. Med. Biol. 2017, 996, 71–87. [Google Scholar]
- Karran, P.; Brem, R. Protein oxidation, UVA and human DNA repair. DNA Repair 2016, 44, 178–185. [Google Scholar] [CrossRef]
- D’Errico, M.; Lemma, T.; Calcagnile, A.; Proietti De Santis, L.; Dogliotti, E. Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat. Res. 2007, 614, 37–47. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Berthon, J.Y.; Nachat-Kappes, R.; Bey, M.; Cadoret, J.P.; Renimel, I.; Filaire, E. Marine algae as attractive source to skin care. Free Radic. Res. 2017, 51, 555–567. [Google Scholar] [CrossRef]
- Fernando, I.P.; Kim, M.; Son, K.T.; Jeong, Y.; Jeon, Y.J. Antioxidant activity of marine algal polyphenolic compounds: A mechanistic approach. J. Med. Food. 2016, 19, 615–628. [Google Scholar] [CrossRef]
- Yoon, N.Y.; Eom, T.K.; Kim, M.M.; Kim, S.K. Inhibitory effect of phlorotannins isolated from Ecklonia cava on mushroom tyrosinase activity and melanin formation in mouse B16F10 melanoma cells. J. Agric. Food Chem. 2009, 57, 4124–4129. [Google Scholar] [CrossRef]
- Kang, K.A.; Lee, K.H.; Chae, S.; Zhang, R.; Jung, M.S.; Ham, Y.M.; Baik, J.S.; Lee, N.H.; Hyun, J.W. Cytoprotective effect of phloroglucinol on oxidative stress induced cell damage via catalase activation. J. Cell. Biochem. 2006, 97, 609–620. [Google Scholar] [CrossRef]
- Kang, K.A.; Zhang, R.; Chae, S.; Lee, S.J.; Kim, J.; Kim, J.; Jeong, J.; Lee, J.; Shin, T.; Lee, N.H.; et al. Phloroglucinol (1,3,5-trihydroxybenzene) protects against ionizing radiation-induced cell damage through inhibition of oxidative stress in vitro and in vivo. Chem. Biol. Interact. 2010, 185, 215–226. [Google Scholar] [CrossRef]
- Park, S.J.; Ahn, G.; Lee, N.H.; Park, J.W.; Jeon, Y.J.; Jee, Y. Phloroglucinol (PG) purified from Ecklonia cava attenuates radiation-induced apoptosis in blood lymphocytes and splenocytes. Food Chem. Toxicol. 2011, 49, 2236–2242. [Google Scholar] [CrossRef]
- Kim, K.C.; Piao, M.J.; Cho, S.J.; Lee, N.H.; Hyun, J.W. Phloroglucinol protects human keratinocytes from ultraviolet B radiation by attenuating oxidative stress. Photodermatol. Photoimmunol. Photomed. 2012, 28, 322–331. [Google Scholar] [CrossRef]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Hyun, C.L.; Kang, H.K.; Lee, N.H.; et al. Phloroglucinol inhibits ultraviolet B radiation-induced oxidative stress in the mouse skin. Int. J. Radiat. Biol. 2014, 90, 928–935. [Google Scholar] [CrossRef]
- Park, M.H.; Han, J.S. Phloroglucinol protects INS-1 pancreatic β-cells against glucotoxicity-induced apoptosis. Phytother. Res. 2015, 29, 1700–1706. [Google Scholar] [CrossRef]
- So, M.J.; Cho, E.J. Phloroglucinol attenuates free radical-induced oxidative stress. Prev. Nutr. Food Sci. 2014, 19, 129–135. [Google Scholar] [CrossRef]
- Kang, S.M.; Cha, S.H.; Ko, J.Y.; Kang, M.C.; Kim, D.; Heo, S.J.; Kim, J.S.; Heu, M.S.; Kim, Y.T.; Jung, W.K.; et al. Neuroprotective effects of phlorotannins isolated from a brown alga, Ecklonia cava, against H2O2-induced oxidative stress in murine hippocampal HT22 cells. Environ. Toxicol. Pharmacol. 2012, 34, 96–105. [Google Scholar] [CrossRef]
- Kang, K.A.; Lee, K.H.; Chae, S.; Koh, Y.S.; Yoo, B.S.; Kim, J.H.; Ham, Y.M.; Baik, J.S.; Lee, N.H.; Hyun, J.W. Triphlorethol-A from Ecklonia cava protects V79-4 lung fibroblast against hydrogen peroxide induced cell damage. Free Radic. Res. 2005, 39, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Zhang, R.; Hong, B.H.; Yang, E.J.; Kang, K.A.; Choi, M.; Kim, K.C.; Noh, S.J.; Kim, H.S.; Lee, N.H.; et al. Phloroglucinol attenuates motor functional deficits in an animal model of Parkinson’s disease by enhancing Nrf2 activity. PLoS ONE 2013, 8, e71178. [Google Scholar] [CrossRef] [PubMed]
- Rahim, A.H.; Setiawan, B.; Dewi, F.R.; Noor, Z. Regulation by phloroglucinol of Nrf2/Maf-mediated expression of antioxidant enzymes and inhibition of osteoclastogenesis via the RANKL/RANK signaling pathway: In Silico study. Acta. Inform. Med. 2015, 23, 228–232. [Google Scholar] [CrossRef]
- Lee, S.E.; Jeong, S.I.; Kim, G.D.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Upregulation of heme oxygenase-1 as an adaptive mechanism for protection against crotonaldehyde in human umbilical vein endothelial cells. Toxicol. Lett. 2011, 201, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Sun Jang, J.; Piao, S.; Cha, Y.N.; Kim, C. Taurine chloramine activates Nrf2, increases HO-1 expression and protects cells from death caused by hydrogen peroxide. J. Clin. Biochem. Nutr. 2009, 45, 37–43. [Google Scholar] [CrossRef]
- Tomczyk, M.; Kraszewska, I.; Dulak, J.; Jazwa-Kusior, A. Modulation of the monocyte/macrophage system in heart failure by targeting heme oxygenase-1. Vascul. Pharmacol. 2019, 112, 79–90. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp. Neurobiol. 2014, 23, 93–103. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, A.; Speit, G. Overexpression of heme oxygenase-1 (HO-1) in V79 cells results in increased resistance to hyperbaric oxygen (HBO)-induced DNA damage. Environ. Mol. Mutagen. 2002, 40, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Li, W.; Ma, J.Y.; Shao, N.; Zhang, Y.J.; Liu, R.M.; Wu, W.B.; Lin, Y.; Wang, S.M. Knockdown of heme oxygenase-1 promotes apoptosis and autophagy and enhances the cytotoxicity of doxorubicin in breast cancer cells. Oncol. Lett. 2015, 10, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Zhang, X.T.; Sun, X.Q.; Wu, C.; Chen, J.L.; Yuan, J.J.; Pang, Q.F.; Wang, Z.P. Heme oxygnease-1 induction by methylene blue protects RAW264.7 cells from hydrogen peroxide-induced injury. Biochem. Pharmacol. 2018, 148, 265–277. [Google Scholar] [CrossRef]
- Luo, T.; Shen, X.Y.; Li, S.; Ouyang, T.; Mai, Q.A.; Wang, H.Q. The protective effect of jatrorrhizine against oxidative stress in primary rat cortical neurons. CNS Neurol. Disord. Drug Targets 2017, 16, 617–623. [Google Scholar] [CrossRef]
- Guo, X.M.; Chen, B.; Lv, J.M.; Lei, Q.; Pan, Y.J.; Yang, Q. Knockdown of IRF6 attenuates hydrogen dioxide-induced oxidative stress via inhibiting mitochondrial dysfunction in HT22 cells. Cell Mol. Neurobiol. 2016, 36, 1077–1086. [Google Scholar] [CrossRef]
- Tian, X.; He, W.; Yang, R.; Liu, Y. Dl-3-n-butylphthalide protects the heart against ischemic injury and H9c2 cardiomyoblasts against oxidative stress: Involvement of mitochondrial function and biogenesis. J. Biomed. Sci. 2017, 24, 38. [Google Scholar] [CrossRef]
- Mao, C.Y.; Lu, H.B.; Kong, N.; Li, J.Y.; Liu, M.; Yang, C.Y.; Yang, P. Levocarnitine protects H9c2 rat cardiomyocytes from H2O2-induced mitochondrial dysfunction and apoptosis. Int. J. Med. Sci. 2014, 11, 1107–1115. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Schultz, D.R.; Harrington, W.J., Jr. Apoptosis: Programmed cell death at a molecular level. Semin. Arthritis Rheum. 2003, 32, 345–369. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Gottlieb, R.A. Bcl-2 family members and apoptosis, taken to heart. Am. J. Physiol. Cell. Physiol. 2007, 292, C45–C51. [Google Scholar] [CrossRef]
- Kulikov, A.V.; Shilov, E.S.; Mufazalov, I.A.; Gogvadze, V.; Nedospasov, S.A.; Zhivotovsky, B. Cytochrome c: The Achilles’ heel in apoptosis. Cell Mol. Life Sci. 2012, 69, 1787–1797. [Google Scholar] [CrossRef]
- Imahashi, K.; Schneider, M.D.; Steenbergen, C.; Murphy, E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ. Res. 2004, 95, 734–741. [Google Scholar] [CrossRef]
- Yin, Y.; Lu, L.; Wang, D.; Shi, Y.; Wang, M.; Huang, Y.; Chen, D.; Deng, C.; Chen, J.; Lv, P.; et al. Astragalus polysaccharide inhibits autophagy and apoptosis from peroxide-induced injury in C2C12 myoblasts. Cell. Biochem. Biophys. 2015, 73, 433–439. [Google Scholar] [CrossRef]
- Siu, P.M.; Wang, Y.; Always, S.E. Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci. 2009, 84, 468–481. [Google Scholar] [CrossRef]
- Haramizu, S.; Asano, S.; Butler, D.C.; Stanton, D.A.; Hajira, A.; Mohamed, J.S.; Always, S.E. Dietary resveratrol confers apoptotic resistance to oxidative stress in myoblasts. J. Nutr. Biochem. 2017, 50, 103–115. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, W.J.; Lee, M.H.; Kim, S.Y.; Seo, D.H.; Kim, H.S.; Gelinsky, M.; Kim, T.J. Anti-skeletal muscle atrophy effect of Oenothera odorata root extract via reactive oxygen species-dependent signaling pathways in cellular and mouse model. Biosci. Biotechnol. Biochem. 2016, 80, 80–88. [Google Scholar] [CrossRef]
- Ha, D.; Bing, S.J.; Cho, J.; Ahn, G.; Kim, D.S.; Al-Amin, M.; Park, S.J.; Jee, Y. Phloroglucinol protects small intestines of mice from ionizing radiation by regulating apoptosis-related molecules: A comparative immunohistochemical study. J. Histochem. Cytochem. 2013, 61, 63–74. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, X.; Qi, X.; Che, J.; Cao, B. Paeoniflorin protects human EA.hy926 endothelial cells against gamma-radiation induced oxidative injury by activating the NF-E2-related factor 2/heme oxygenase-1 pathway. Toxicol. Lett. 2013, 218, 224–234. [Google Scholar] [CrossRef]
- Chen, H.; Tang, X.; Zhou, B.; Zhou, Z.; Xu, N.; Wang, Y. A ROS-mediated mitochondrial pathway and Nrf2 pathway activation are involved in BDE-47 induced apoptosis in Neuro-2a cells. Chemosphere 2017, 184, 679–686. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer | Item No. | Dilution |
|---|---|---|---|
| HO-1 | Merck Millipore | 374090 | 1:1000 |
| Nrf2 | Santa Cruz Biotechnology, Inc. | sc-13032 | 1:1000 |
| p-Nrf2 | Abcam, Inc. | ab76026 | 1:1000 |
| Keap1 | Santa Cruz Biotechnology, Inc. | sc-15246 | 1:1000 |
| p-γH2AX | Cell Signaling Technology, Inc. | 9718 | 1:500 |
| γH2AX | Cell Signaling Technology, Inc. | 7631 | 1:500 |
| Bcl-2 | Cell Signaling Technology, Inc. | sc-509 | 1:1000 |
| Bax | Cell Signaling Technology, Inc. | sc-493 | 1:1000 |
| Caspase-9 | Santa Cruz Biotechnology, Inc. | sc-7885 | 1:1000 |
| Caspase-3 | Santa Cruz Biotechnology, Inc. | sc-7272 | 1:1000 |
| Cytochrome c | Santa Cruz Biotechnology, Inc. | sc-7159 | 1:500 |
| COX IV | Santa Cruz Biotechnology, Inc. | sc-376731 | 1:1000 |
| Actin | Santa Cruz Biotechnology, Inc. | sc-47778 | 1:1000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.; Cha, H.-J.; Hong, S.H.; Kim, G.-Y.; Kim, S.; Kim, H.-S.; Kim, B.W.; Jeon, Y.-J.; Choi, Y.H. Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes. Mar. Drugs 2019, 17, 225. https://doi.org/10.3390/md17040225
Park C, Cha H-J, Hong SH, Kim G-Y, Kim S, Kim H-S, Kim BW, Jeon Y-J, Choi YH. Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes. Marine Drugs. 2019; 17(4):225. https://doi.org/10.3390/md17040225
Chicago/Turabian StylePark, Cheol, Hee-Jae Cha, Su Hyun Hong, Gi-Young Kim, Suhkmann Kim, Heui-Soo Kim, Byung Woo Kim, You-Jin Jeon, and Yung Hyun Choi. 2019. "Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes" Marine Drugs 17, no. 4: 225. https://doi.org/10.3390/md17040225
APA StylePark, C., Cha, H.-J., Hong, S. H., Kim, G.-Y., Kim, S., Kim, H.-S., Kim, B. W., Jeon, Y.-J., & Choi, Y. H. (2019). Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes. Marine Drugs, 17(4), 225. https://doi.org/10.3390/md17040225