Discovery of Stealthin Derivatives and Implication of the Amidotransferase FlsN3 in the Biosynthesis of Nitrogen-Containing Fluostatins



Abstract
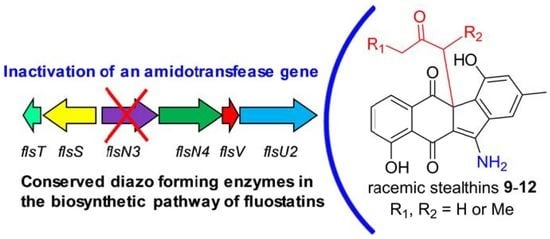
1. Introduction
2. Results and Discussion
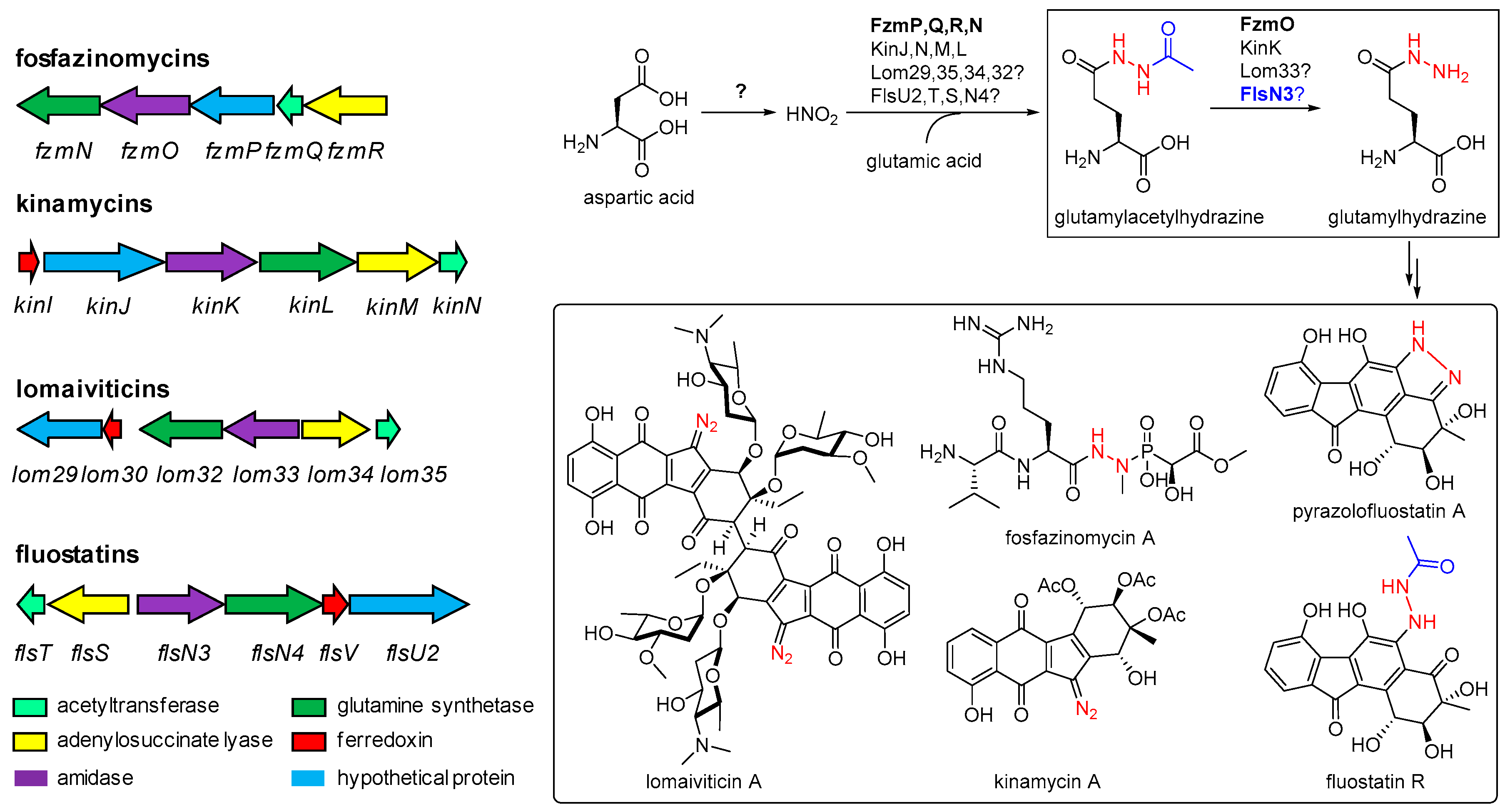
2.1. Gene Inactivation and Compound Isolation
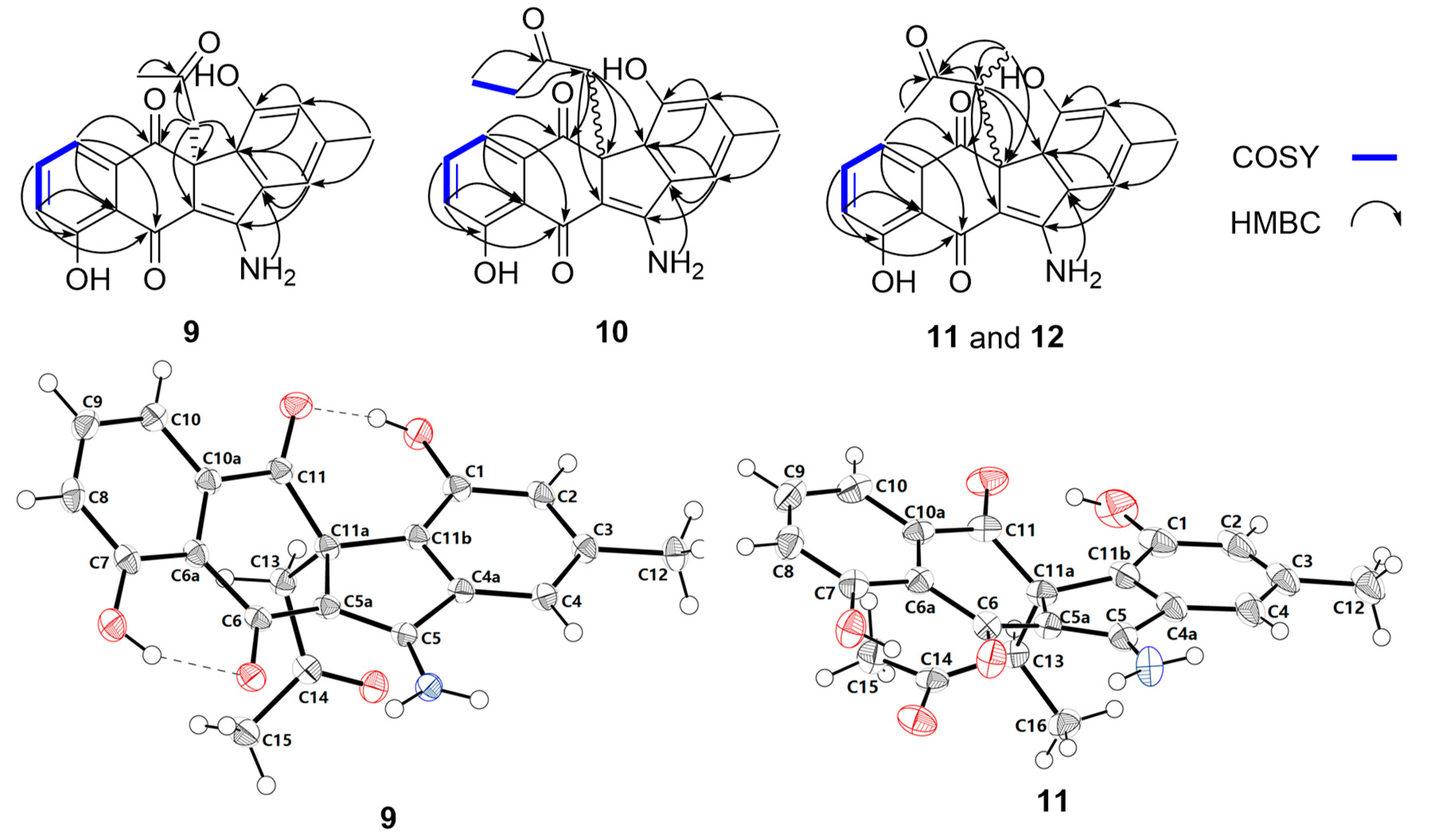
2.2. Structural Elucidation
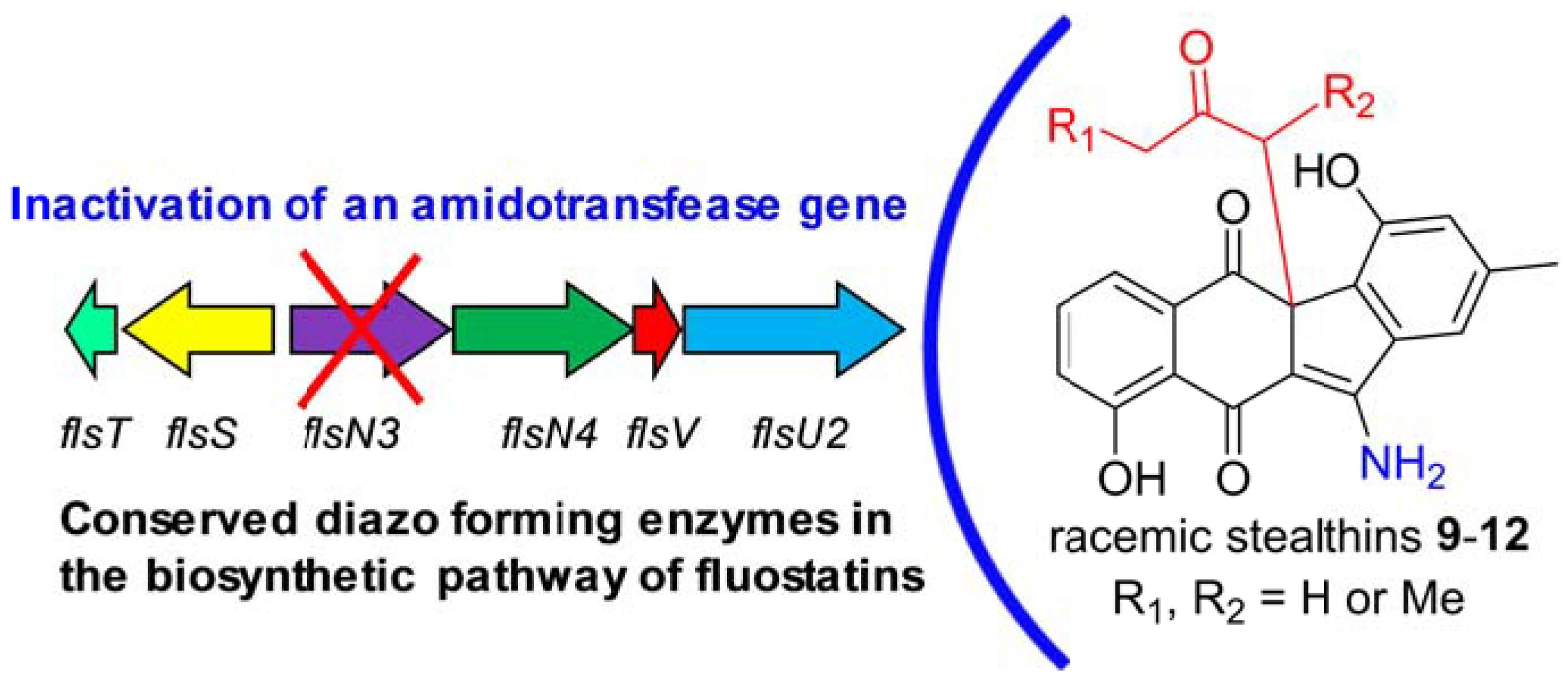
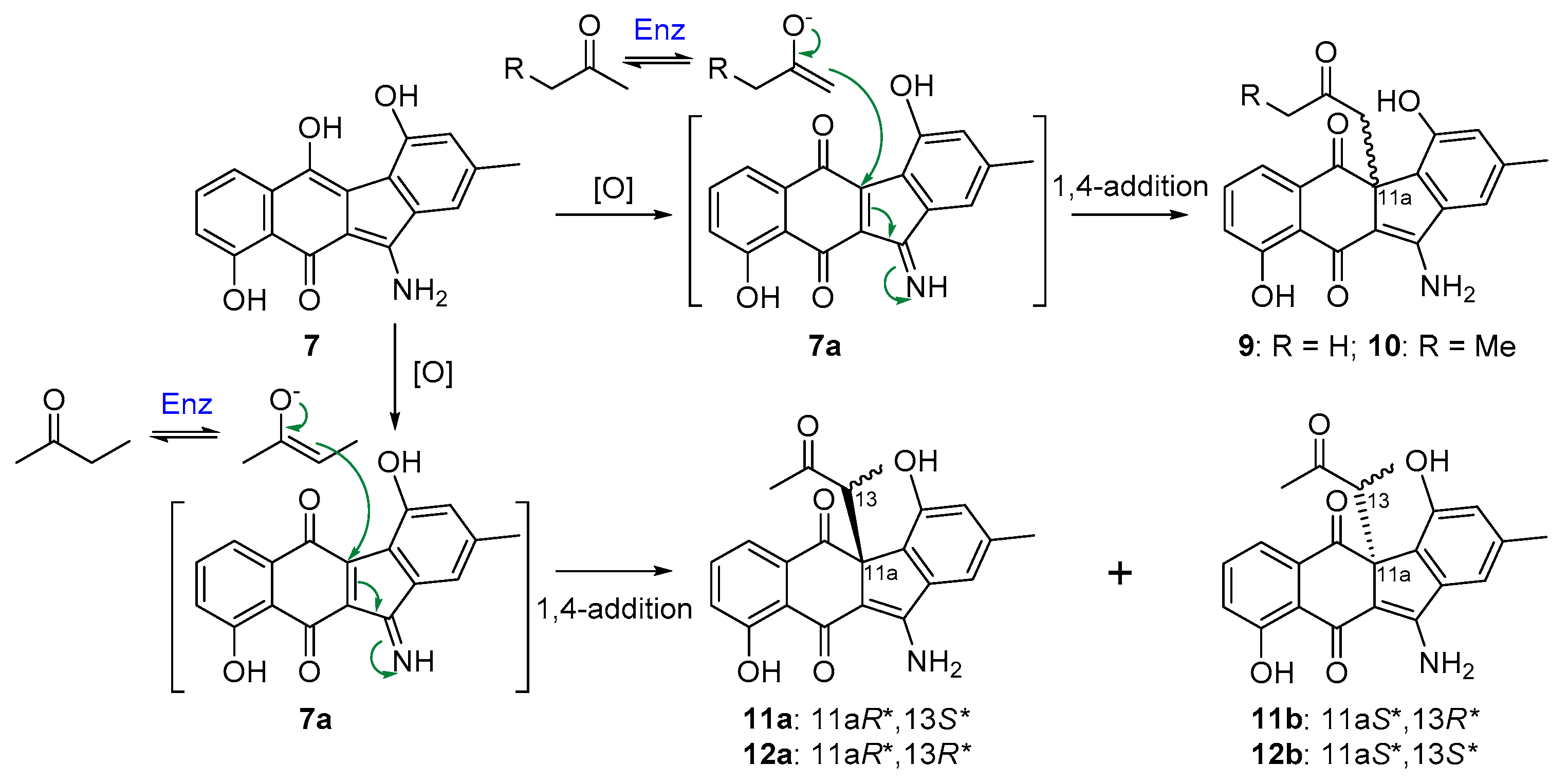
2.3. Biosynthetic Implications
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Construction of the flsN3 Inactivation Mutant
3.3. Fermentation, Extraction, and Isolation
3.4. Synthesis of Trimethylstealthin C (13)
3.5. X-ray Crystallographic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Zhang, Q. The functional differentiation of the post-PKS tailoring oxygenases contributed to the chemical diversities of atypical angucyclines. Synth. Syst. Biotechnol. 2018, 3, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Guo, F.; Ren, J.; Ai, G.; Aigle, B.; Fan, K.; Yang, K. Identification of Alp1U and Lom6 as epoxy hydrolases and implications for kinamycin and lomaiviticin biosynthesis. Nat. Commun. 2015, 6, 7674. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.C.; Woo, C.M.; Hegan, D.C.; Li, Z.; Glazer, P.M.; Herzon, S.B. The cytotoxicity of (-)-lomaiviticin A arises from induction of double-strand breaks in DNA. Nat. Chem. 2014, 6, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.M.; Li, Z.; Paulson, E.K.; Herzon, S.B. Structural basis for DNA cleavage by the potent antiproliferative agent (–)-lomaiviticin A. Proc. Natl. Acad. Sci. USA 2016, 113, 2851–2856. [Google Scholar] [CrossRef] [PubMed]
- Herzon, S.B. The mechanism of action of (-)-lomaiviticin A. Acc. Chem. Res. 2017, 50, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, Q.; Zhu, Y.; Nie, F.; Wu, Z.; Yang, C.; Zhang, L.; Tian, X.; Zhang, C. Isolation, structure elucidation and biosynthesis of benzo[b]fluorene nenestatin A from deep-sea derived Micromonospora echinospora SCSIO 04089. Tetrahedron 2017, 73, 3585–3590. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Z.; Li, S.; Lu, Y.; Chen, Y.; Zhang, H.; Zhang, G.; Zhu, Y.; Zhang, G.; Zhang, W.; et al. Fluostatins I-K from the South China Sea-derived Micromonospora rosaria SCSIO N160. J. Nat. Prod. 2012, 75, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Huang, C.; Zhang, W.; Zhu, Y.; Zhang, C. Heterologous expression of fluostatin gene cluster leads to a bioactive heterodimer. Org. Lett. 2015, 17, 5324–5327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, C.; Huang, C.; Zhang, L.; Zhang, H.; Zhang, Q.; Yuan, C.; Zhu, Y.; Zhang, C. Pyrazolofluostatins A–C, pyrazole-fused benzo[a]fluorenes from South China Sea-derived Micromonospora rosaria SCSIO N160. Org. Lett. 2017, 19, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yang, X.; Liu, T.; Xiao, H.; Wang, G.; Zhou, M.; Liu, F.; Zhang, Y.; Liu, D.; Chen, M.; et al. Fluostatins M-Q featuring a 6-5-6-6 ring skeleton and high oxidized A-rings from marine Streptomyces sp. PKU-MA00045. Mar. Drugs 2018, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, C.; Zhang, W.; Zhang, L.; De, B.C.; Zhu, Y.; Jiang, X.; Fang, C.; Zhang, Q.; Yuan, C.-S.; et al. Molecular basis of dimer formation during the biosynthesis of benzofluorene-containing atypical angucyclines. Nat. Commun. 2018, 9, 2088. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, C.; Zhu, Y.; Zhang, W.; Yuan, C.; Zhang, C. Marine bacterial aromatic polyketides from host-dependent heterologous expression and fungal mode of cyclization. Front. Chem. 2018, 6, 528. [Google Scholar] [CrossRef] [PubMed]
- Sugai, Y.; Katsuyama, Y.; Ohnishi, Y. A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat. Chem. Biol. 2016, 12, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Molina, M.T. Naturally occurring diazo compounds: The kinamycins. Curr. Org. Chem. 2003, 7, 1433–1442. [Google Scholar] [CrossRef]
- Neumann, C.S.; Jiang, W.; Heemstra, J.R.; Gontang, E.A.; Kolter, R.; Walsh, C.T. Biosynthesis of piperazic acid via N5-hydroxy-ornithine in Kutzneria spp. 744. ChemBioChem 2012, 13, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.J.; Balskus, E.P. Discovery of a diazo-forming enzyme in cremeomycin biosynthesis. J. Org. Chem. 2018, 83, 7539–7546. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, K.-K.A.; van der Donk, W.A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 2016, 7, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-K.A.; Ng, T.L.; Wang, P.; Huang, Z.; Balskus, E.P.; van der Donk, W.A. Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kinamycin. Nat. Commun. 2018, 9, 3687. [Google Scholar] [CrossRef] [PubMed]
- Janso, J.E.; Haltli, B.A.; Eustaquio, A.S.; Kulowski, K.; Waldman, A.J.; Zha, L.; Nakamura, H.; Bernan, V.S.; He, H.; Carter, G.T.; et al. Discovery of the lomaiviticin biosynthetic gene cluster in Salinispora pacifica. Tetrahedron 2014, 70, 4156–4164. [Google Scholar] [CrossRef] [PubMed]
- Echavarren, A.M.; Tamayo, N.; Cardenas, D.J. Synthesis of antibiotics WS 5995 A and C and related compounds by palladium-catalyzed coupling of 2-bromonaphthoquinones with organostannanes. J. Org. Chem. 1994, 59, 6075–6083. [Google Scholar] [CrossRef]
- Wang, P.; Hong, G.J.; Wilson, M.R.; Balskus, E.P. Production of stealthin C involves an S-N-type Smiles rearrangement. J. Am. Chem. Soc. 2017, 139, 2864–2867. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.C.; Gontang, E.A.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinipyrones and pacificanones, mixed-precursor polyketides from the marine actinomycete Salinispora pacifica. J. Nat. Prod. 2008, 71, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Awakawa, T.; Crusemann, M.; Munguia, J.; Ziemert, N.; Nizet, V.; Fenical, W.; Moore, B.S. Salinipyrone and pacificanone are biosynthetic by-products of the rosamicin polyketide synthase. ChemBioChem 2015, 16, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, S.; Zhang, H.; Zhang, Q.; Zhang, G.; Zhang, G.; Zhu, Y.; Zhang, C. Isolation and structural elucidation of macrolide antibiotics from marine-derived Micromonospora rosaria SCSIO N160. Nat. Prod. Res. Dev. 2013, 25, 466–469. [Google Scholar]
- Gould, S.J.; Melville, C.R.; Cone, M.C.; Chen, J.; Carney, J.R. Kinamycin biosynthesis. synthesis, isolation, and incorporation of stealthin C, an aminobenzo[b]fluorene. J. Org. Chem. 1997, 62, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Melville, C.R. NMR silent, naturally-occurring quinones: A case of radicals. Tetrahedron Lett. 1997, 38, 1473–1476. [Google Scholar] [CrossRef]
- Heider, J.; Schuhle, K.; Frey, J.; Schink, B. Activation of acetone and other simple ketones in anaerobic bacteria. J. Mol. Microbiol. Biotechnol. 2016, 26, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Dong, C.L.; Guan, Z.; He, Y.H. Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: Direct enantioselective synthesis of 2,2-disubstituted indol-3-ones from 2-arylindoles. Angew. Chem. Int. Ed. 2019, 58, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Gust, B.; Challis, G.L.; Fowler, K.; Kieser, T.; Chater, K.F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 2003, 100, 1541–1546. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 9 | 10 | 11 | 12 |
|---|---|---|---|---|
| 2 | 6.85, s | 6.84, s | 6.88, s | 6.83, s |
| 4 | 7.35, s | 7.34, s | 7.38, s | 7.37, s |
| 8 | 7.17, d (8.2) | 7.16, dd (0.8, 8.2) | 7.19, d (8.2) | 7.12, d (8.2) |
| 9 | 7.55, dd (7.3, 8.2) | 7.55, dd (7.4, 8.2) | 7.54, dd (7.4, 8.2) | 7.54, dd (7.2, 8.2) |
| 10 | 7.23, d (7.3) | 7.24, dd (0.8, 7.4) | 7.33, d, (7.4) | 7.22, d (7.2) |
| 12 | 2.33, s | 2.32, s | 2.32, s | 2.32, s |
| 13 | 2.87, d (13.7) | 2.87, d (13.8) | 3.52, q (7.0) | 3.35, q (7.2) |
| 3.40, d (13.7) | 3.31, d (13.8) | |||
| 15 | 1.71, s | 2.02, dq (18.2, 7.2) | 1.75, s | 1.91, s |
| 2.12, dq (18.2, 7.2) | ||||
| 16 | 0.62, t (7.2) | 0.73, d (7.0) | 0.65, d (7.2) | |
| 1-OH | 9.69, s | 9.66, s | 9.69, s | 9.61, s |
| 5-NH2 | 8.12, s | 8.11, s | 8.23, s | 8.19, s |
| 8.55, s | 8.54, s | 8.58, s | 8.59, s | |
| 7-OH | 13.32, s | 13.32, s | 13.45, s | 13.17, s |
| No. | 9 | 10 | 11 | 12 |
|---|---|---|---|---|
| 1 | 154.3, C | 154.2, C | 153.7, C | 154.0, C |
| 2 | 119.6, CH | 119.6, CH | 120.3, CH | 119.8, CH |
| 3 | 139.7, C | 139.7, C | 139.9, C | 140.0, C |
| 4 | 114.1, CH | 114.1, CH | 114.4, CH | 114.3, CH |
| 4a | 138.0, C | 137.9, C | 138.0, C | 138.5, C |
| 5 | 159.1, C | 159.1, C | 159.1, C | 159.4, C |
| 5a | 105.0, C | 105.2, C | 103.8, C | 105.6, C |
| 6 | 182.7, C | 182.6, C | 182.6, C | 182.7, C |
| 6a | 118.8, C | 118.9, C | 119.8, C | 118.8, C |
| 7 | 160.4, C | 160.4, C | 160.4, C | 160.1, C |
| 8 | 122.2, CH | 122.1, CH | 123.2, CH | 121.6, CH |
| 9 | 134.3, CH | 134.2, CH | 134.0, CH | 134.2, CH |
| 10 | 118.0, CH | 118.0, CH | 118.3, CH | 118.3, CH |
| 10a | 136.6, C | 136.7, C | 135.8, C | 138.2, C |
| 11 | 196.6, C | 196.7, C | 198.3, C | 198.2, C |
| 11a | 60.9, C | 60.9, C | 65.6, C | 65.2, C |
| 11b | 127.6, C | 127.8, C | 127.0, C | 126.2, C |
| 12 | 21.1, CH3 | 21.1, CH3 | 20.9, CH3 | 21.0, CH3 |
| 13 | 46.8, CH2 | 46.3, CH2 | 52.7, CH | 52.3, CH |
| 14 | 204.1, C | 206.5, C | 208.1, C | 209.5, C |
| 15 | 30.9, CH3 | 36.3, CH2 | 30.7, CH3 | 29.1, CH3 |
| 16 | 7.2, CH3 | 13.3, CH3 | 12.5, CH3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Yang, C.; Fang, Z.; Zhang, L.; Zhang, W.; Zhu, Y.; Zhang, C. Discovery of Stealthin Derivatives and Implication of the Amidotransferase FlsN3 in the Biosynthesis of Nitrogen-Containing Fluostatins. Mar. Drugs 2019, 17, 150. https://doi.org/10.3390/md17030150
Huang C, Yang C, Fang Z, Zhang L, Zhang W, Zhu Y, Zhang C. Discovery of Stealthin Derivatives and Implication of the Amidotransferase FlsN3 in the Biosynthesis of Nitrogen-Containing Fluostatins. Marine Drugs. 2019; 17(3):150. https://doi.org/10.3390/md17030150
Chicago/Turabian StyleHuang, Chunshuai, Chunfang Yang, Zhuangjie Fang, Liping Zhang, Wenjun Zhang, Yiguang Zhu, and Changsheng Zhang. 2019. "Discovery of Stealthin Derivatives and Implication of the Amidotransferase FlsN3 in the Biosynthesis of Nitrogen-Containing Fluostatins" Marine Drugs 17, no. 3: 150. https://doi.org/10.3390/md17030150
APA StyleHuang, C., Yang, C., Fang, Z., Zhang, L., Zhang, W., Zhu, Y., & Zhang, C. (2019). Discovery of Stealthin Derivatives and Implication of the Amidotransferase FlsN3 in the Biosynthesis of Nitrogen-Containing Fluostatins. Marine Drugs, 17(3), 150. https://doi.org/10.3390/md17030150