Cytotoxic Polyketides Isolated from the Deep-Sea-Derived Fungus Penicillium chrysogenum MCCC 3A00292

 , , ,
, , ,
Abstract
1. Introduction
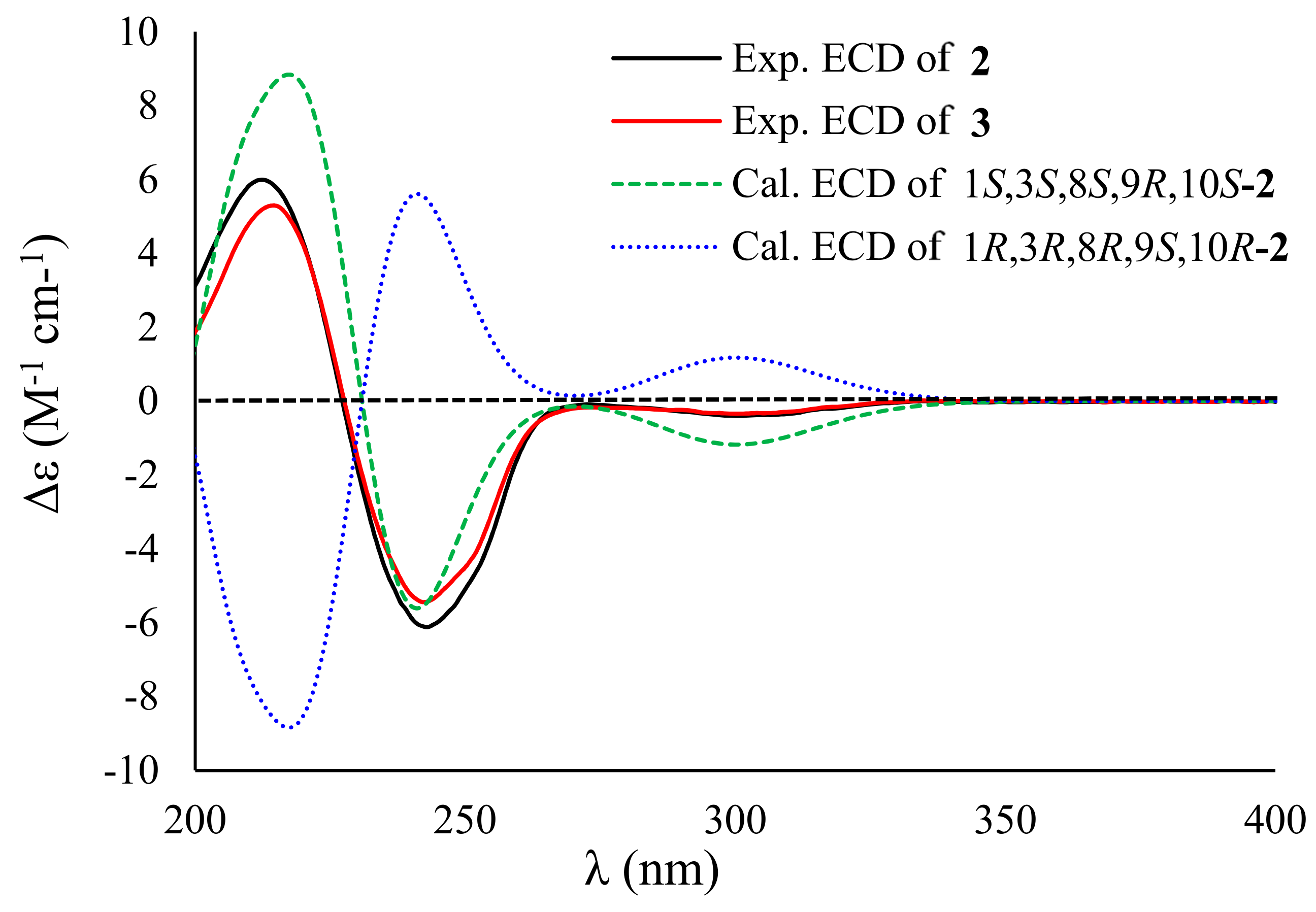
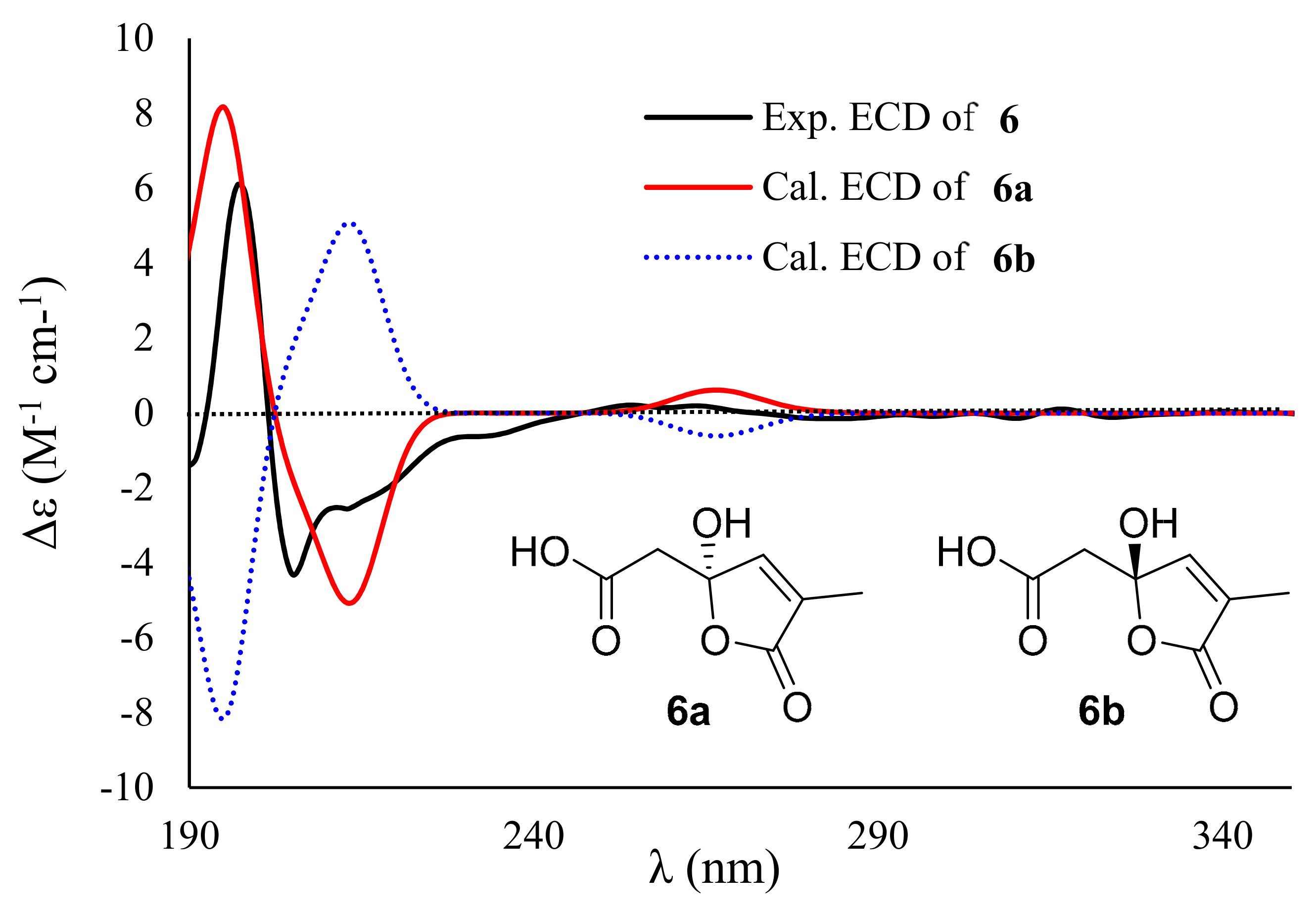
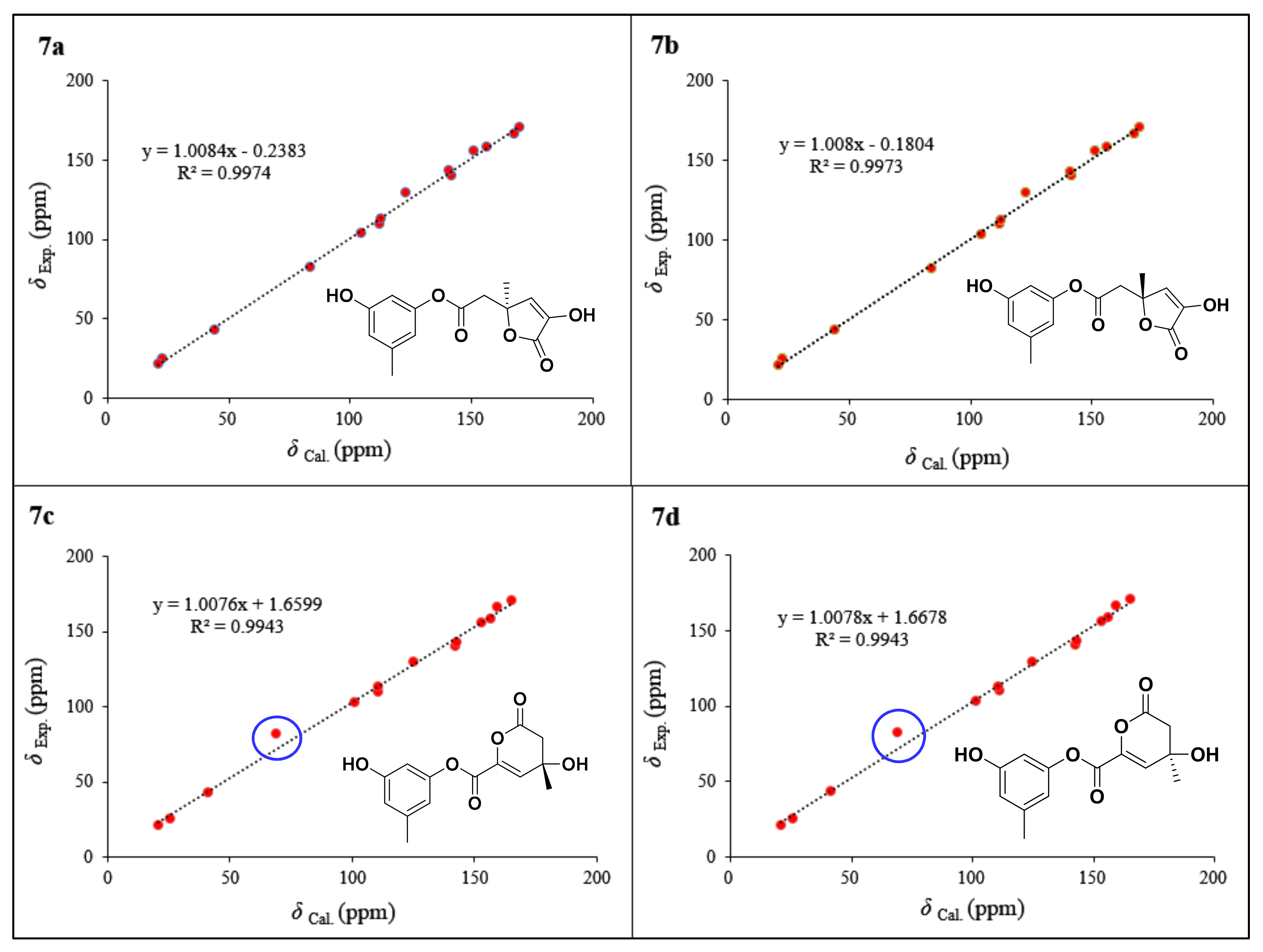
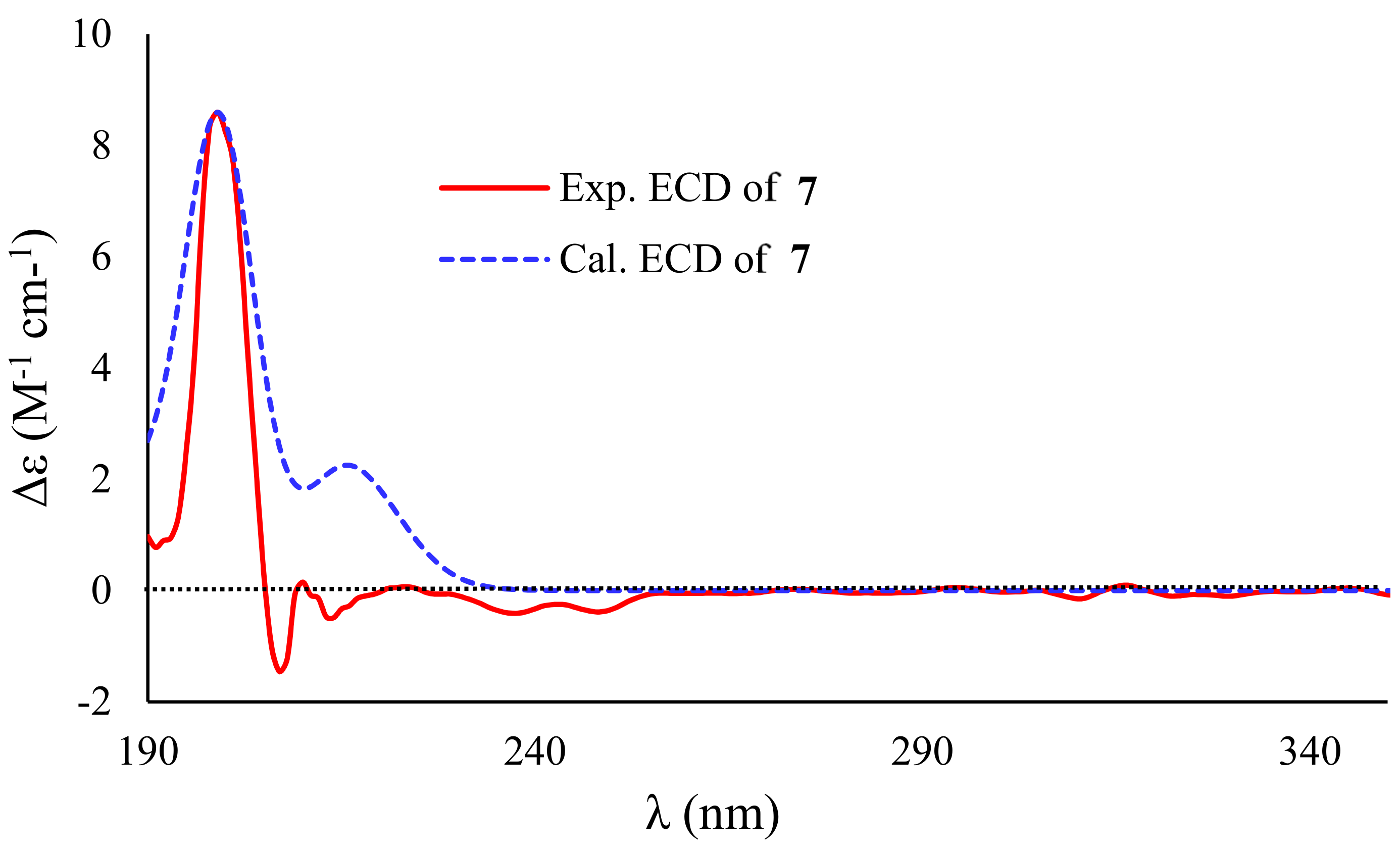
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Cytotoxicity Assay
3.5. Computation Section
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schueffler, A.; Anke, T. Fungal natural products in research and development. Nat. Prod. Rep. 2014, 31, 1425–1448. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, A.M.S.; Elias, N.; Farag, A.M.; Chen, L.; Saeed, A.; Hegazy, F.M.E.; Moustafa, S.M.; Abd El Wahed, A.; Al Mousawi, M.S.; Musharraf, G.S.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 481. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef]
- Daletos, G.; Ebrahim, W.; Ancheeva, E.; El-Neketi, M.; Song, W.; Lin, W.; Proksch, P. Natural products from deep-sea-derived fungi—A new source of novel bioactive compounds? Curr. Med. Chem. 2018, 25, 186–207. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, J.; Wei, X.; Chen, Y.; Fu, T.; Xiang, Y.; Huang, X.; Tian, X.; Xiao, Z.; Zhang, W.; et al. Variecolortins A–C, three pairs of spirocyclic diketopiperazine enantiomers from the marine-derived fungus Eurotium sp. SCSIO F452. Org. Lett. 2018, 20, 4593–4596. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Xu, R.; Li, X.M.; Yang, S.Q.; Meng, L.H.; Wang, B.G. Simpterpenoid A, a meroterpenoid with a highly functionalized cyclohexadiene moiety featuring gem-propane-1,2-dione and methylformate groups, from the mangrove-derived Penicillium simplicissimum MA-332. Org. Lett. 2018, 20, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, Z.; Zhang, G.; Che, Q.; Zhu, T.; Gu, Q.; Li, D. Determination of taichunamide H and structural revision of taichunamide A. Org. Lett. 2018, 20, 1138–1141. [Google Scholar] [CrossRef]
- Fukuyama, K.; Tsukihara, T.; Katsube, Y.; Hamasaki, T.; Hatsuda, Y. Structure of versiol, a new methabolite from Aspergillus versicolor. Tetrahedron. Lett. 1976, 189–190. [Google Scholar] [CrossRef]
- An, C.L.; Kong, F.D.; Ma, Q.Y.; Xie, Q.Y.; Yuan, J.Z.; Zhou, L.M.; Dai, H.F.; Yu, Z.F.; Zhao, Y.X. Chemical constituents of the marine-derived fungus Aspergillus sp. SCS-KFD66. Mar. Drugs 2018, 16, 468. [Google Scholar] [CrossRef]
- Wijesekera, K.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Metabolite diversification by cultivation of the endophytic fungus Dothideomycete sp. in halogen containing media: Cultivation of terrestrial fungus in seawater. Bioorg. Med. Chem. 2017, 25, 2868–2877. [Google Scholar] [CrossRef]
- Sun, Y.Z.; Kurtan, T.; Mandi, A.; Tang, H.; Chou, Y.; Soong, K.; Su, L.; Sun, P.; Zhuang, C.L.; Zhang, W. Immunomodulatory polyketides from a Phoma-like fungus isolated from a soft coral. J. Nat. Prod. 2017, 80, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kusari, S.; Golz, C.; Laatsch, H.; Strohmann, C.; Spiteller, M. Epigenetic modulation of endophytic Eupenicillium sp. LG41 by a histone deacetylase inhibitor for production of decalin-containing compounds. J. Nat. Prod. 2017, 80, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.; Ransom, T.T.; Sigmund, J.; Tran, T.; Cichewicz, R.H.; Goetz, M.; Beutler, J.A. Growth inhibition of colon cancer and melanoma cells by versiol derivatives from a Paraconiothyrium species. J. Nat. Prod. 2017, 80, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Afiyatullov, S.S.; Leshchenko, E.V.; Berdyshev, D.V.; Sobolevskaya, M.P.; Antonov, A.S.; Denisenko, V.A.; Popov, R.S.; Pivkin, M.V.; Udovenko, A.A.; Pislyagin, E.A.; et al. Zosteropenillines: Polyketides from the marine-derived fungus Penicillium thomii. Mar. Drugs 2017, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, J.; Huang, M.; Liu, L.; Wang, J.; Lin, Y. Six new polyketide decalin compounds from mangrove endophytic fungus Penicillium aurantiogriseum 328#. Mar. Drugs 2015, 13, 6306–6318. [Google Scholar]
- Fu, Y.; Wu, P.; Xue, J.; Wei, X.; Li, H. Versicorin, a new lovastatin analogue from the fungus Aspergillus versicolor SC0156. Nat. Prod. Res. 2015, 29, 1363–1368. [Google Scholar] [CrossRef]
- Hewage, R.T.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. One strain-many compounds (OSMAC) method for production of polyketides, azaphilones, and an isochromanone using the endophytic fungus Dothideomycete sp. Phytochemistry 2014, 108, 87–94. [Google Scholar] [CrossRef]
- Zhuravleva, O.I.; Afiyatullov, S.S.; Vishchuk, O.S.; Denisenko, V.A.; Slinkina, N.N.; Smetanina, O.F. Decumbenone C, a new cytotoxic decaline derivative from the marine fungus Aspergillus sulphureus KMM 4640. Arch. Pharmacal Res. 2012, 35, 1757–1762. [Google Scholar] [CrossRef]
- Guo, H.; Feng, T.; Li, Z.H.; Liu, J.K. Five new polyketides from the basidiomycete Craterellus odoratus. Nat. Prod. Bioprospect. 2012, 2, 170–173. [Google Scholar] [CrossRef]
- Hao, G.; Zhang, Q.H.; Jiang, M.M.; Tang, J.S.; Miao, C.D.; Hong, K.; Michio, N.; Wang, N.L.; Yao, X.S. Polyketides from a marine sponge-derived fungus Mycelia sterilia and proton-proton long-range coupling. Magn. Reson. Chem. 2008, 46, 1148–1152. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Miura, S.; Yamashita, Y.; Ohta, T. Aspermytin A: A new neurotrophic polyketide isolated from a marine-derived fungus of the genus Aspergillus. Bioorg. Med. Chem. Lett. 2004, 14, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Asahara, M.; Ichinoe, M.; Nakajima, H. Fungal melanin inhibitor and related compounds from Penicillium decumbens. Phytochemistry 2002, 60, 703–708. [Google Scholar] [CrossRef]
- Niu, S.; Xia, J.; Li, Z.; Yang, L.; Yi, Z.; Xie, C.; Peng, G.; Luo, Z.; Shao, Z.; Yang, X. Aphidicolin chemistry of the deep-sea-derived fungus Botryotinia fuckeliana MCCC 3A00494. J. Nat. Prod. 2019, 82, 2307–2331. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Tang, X.; Fan, Z.; Xia, J.; Xie, C.; Yang, X. Fusarisolins A–E, polyketides from the marine-derived fungus Fusarium solani H918. Mar. Drugs 2019, 17, 125. [Google Scholar] [CrossRef]
- Niu, S.; Liu, Q.; Xia, J.; Xie, C.; Luo, Z.; Shao, Z.; Liu, G.; Yang, X. Polyketides from the deep-sea-derived fungus Graphostroma sp. MCCC 3A00421 showed potent antifood allergic activities. J. Agric. Food Chem. 2018, 66, 1369–1376. [Google Scholar] [CrossRef]
- Niu, S.; Liu, D.; Shao, Z.; Proksch, P.; Lin, W. Eremophilane-type sesquiterpenoids in a deep-sea fungus Eutypella sp. activated by chemical epigenetic manipulation. Tetrahedron 2018, 74, 7310–7325. [Google Scholar] [CrossRef]
- Niu, S.; Fan, Z.; Tang, X.; Liu, Q.; Shao, Z.; Liu, G.; Yang, X. Cyclopiane-type diterpenes from the deep-sea-derived fungus Penicillium commune MCCC 3A00940. Tetrahedron Lett. 2018, 59, 375–378. [Google Scholar] [CrossRef]
- Niu, S.; Fan, Z.; Xie, C.; Liu, Q.; Luo, Z.; Liu, G.; Yang, X. Spirograterpene A, a tetracyclic spiro-diterpene with a fused 5/5/5/5 ring system from the deep-sea-derived fungus Penicillium granulatum MCCC 3A00475. J. Nat. Prod. 2017, 80, 2174–2177. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Itabashi, T.; Nozawa, K.; Nakajima, S.; Kawai, K. A new azaphilone, falconensin H, from Emericella falconensis. Chem. Pharm. Bull. 1993, 41, 2040–2041. [Google Scholar] [CrossRef]
- Ballantine, J.A.; Hassall, C.H.; Jones, B.D. Biosynthesis of phenols. XVII. Some phenolic metabolites of mutant strains of Aspergillus rugulosus. Phytochemistry 1968, 7, 1529–1534. [Google Scholar] [CrossRef]
- Taniguchi, M.; Kaneda, N.; Shibata, K.; Kamikawa, T. Isolation and biological activity of aspermutarubrol, a self-growth inhibitor from Aspergillus sydowi. Agric. Biol. Chem. 1978, 42, 1629–1630. [Google Scholar]
- He, P.; Tian, S.; Xu, Y.; Yu, H.; Ji, Y.; Zhu, H.; Li, J. Three new phenyl ether derivatives from Aspergillus carneus HQ889708. Helv. Chim. Acta 2015, 98, 819–822. [Google Scholar] [CrossRef]
- Yamazaki, M.; Maebayashi, Y. Structure determination of violaceol-I and -II, new fungal metabolites from a strain of Emericella violacea. Chem. Pharm. Bull. 1982, 30, 514–518. [Google Scholar] [CrossRef]
- Takenaka, Y.; Tanahashi, T.; Nagakura, N.; Hamada, N. Phenyl ethers from cultured lichen mycobionts of Graphis scripta var. serpentina and G. rikuzensis. Chem. Pharm. Bull. 2003, 51, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Maes, C.M.; Steyn, P.S. Polyketide-derived fungal metabolites from Bipolaris sorokiniana and their significance in the biosynthesis of sterigmatocystin and aflatoxin B1. J. Chem. Soc. Perkin Trans. 1 1984, 1137–1140. [Google Scholar] [CrossRef]
- Yurchenko, A.N.; Smetanina, O.F.; Kalinovsky, A.I.; Pivkin, M.V.; Dmitrenok, P.S.; Kuznetsova, T.A. A new meroterpenoid from the marine fungus Aspergillus versicolor (Vuill.) Tirab. Russ. Chem. Bull. 2010, 59, 852–856. [Google Scholar] [CrossRef]
- Martin, P.K.; Rapoport, H.; Smith, H.W.; Wong, J.L. Synthesis of cyclopenin and isocyclopenin. J. Org. Chem. 1969, 34, 1359–1363. [Google Scholar] [CrossRef]
- Hodge, R.P.; Harris, C.M.; Harris, T.M. Verrucofortine, a major metabolite of Penicillium verrucosum var. cyclopium, the fungus that produces the mycotoxin verrucosidin. J. Nat. Prod. 1988, 51, 66–67. [Google Scholar]
- Sassa, T.; Takemura, T.; Ikeda, M.; Miura, Y. Structure of radiclonic acid, a new plant growth-regulator produced by a fungus. Tetrahedron Lett. 1973, 2333–2334. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision. D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]

 ) and key HMBC (
) and key HMBC ( ) correlations of compounds 1−3.
) correlations of compounds 1−3.








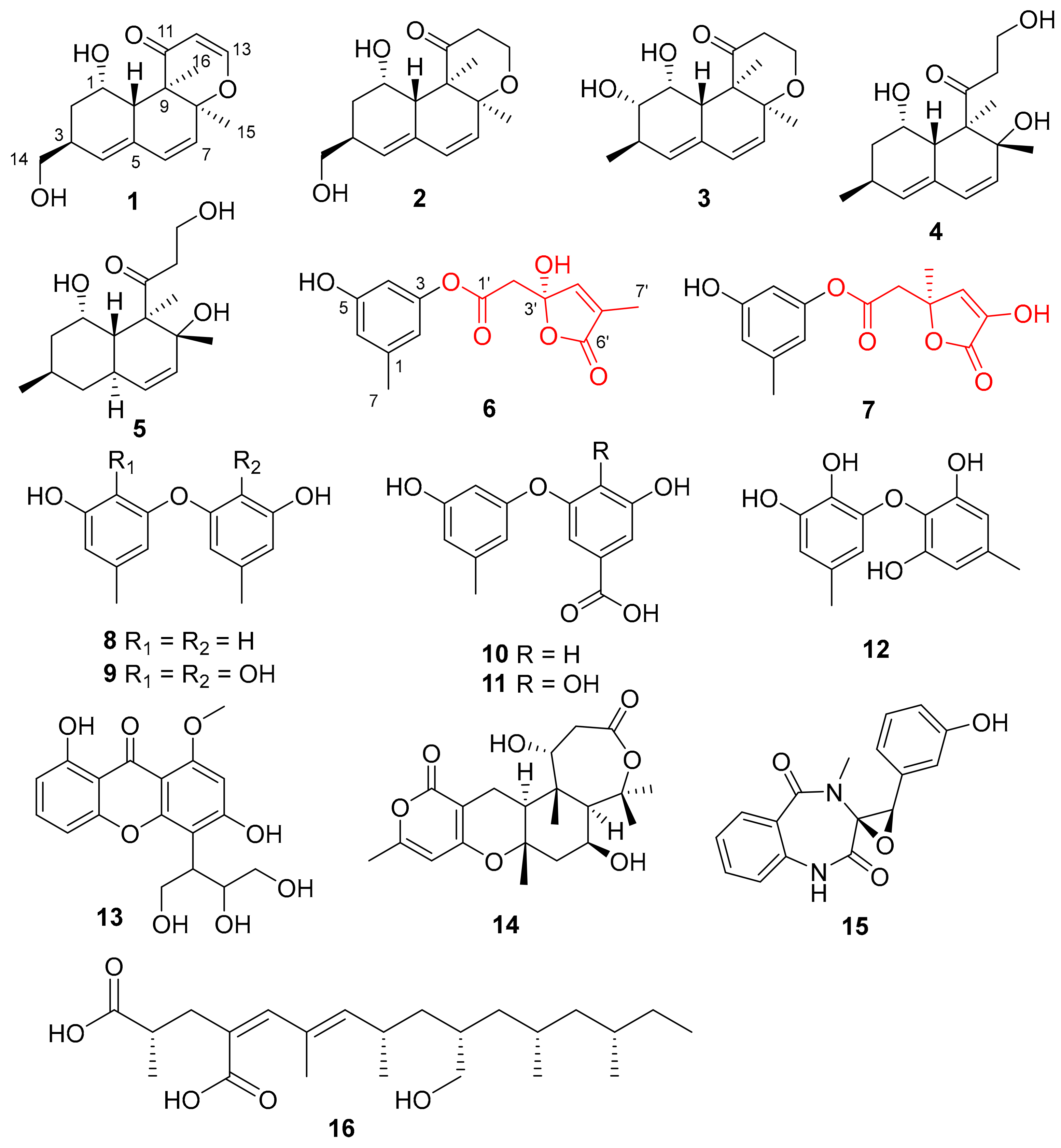{kind=link}
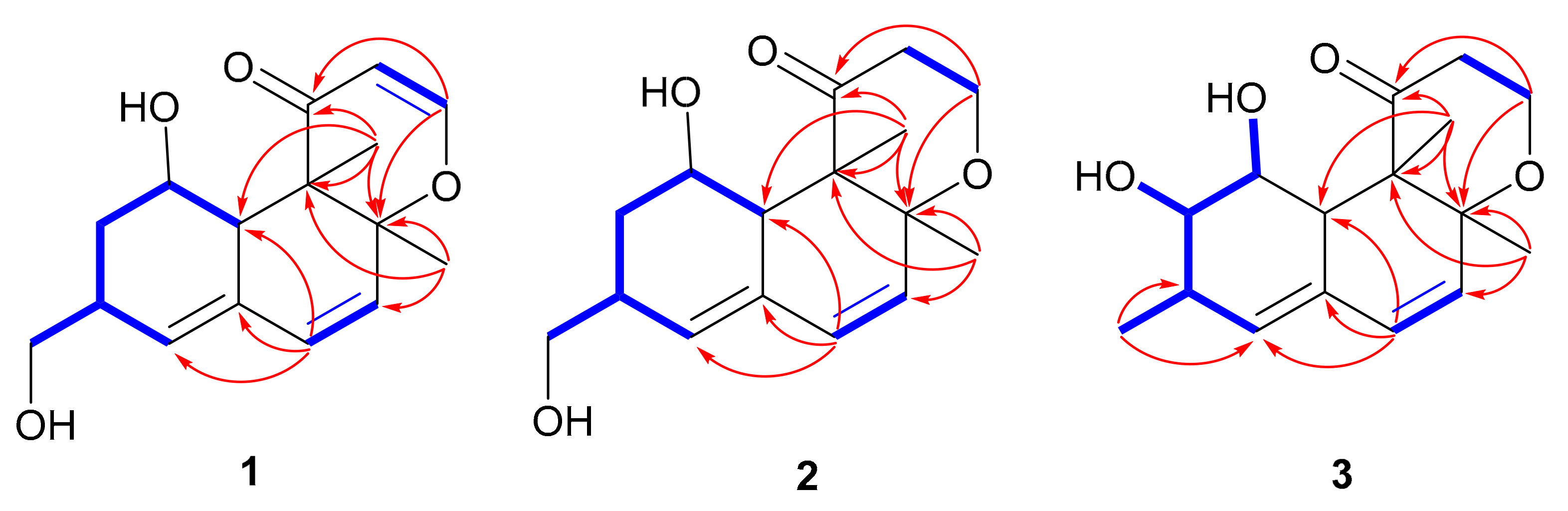{kind=link}
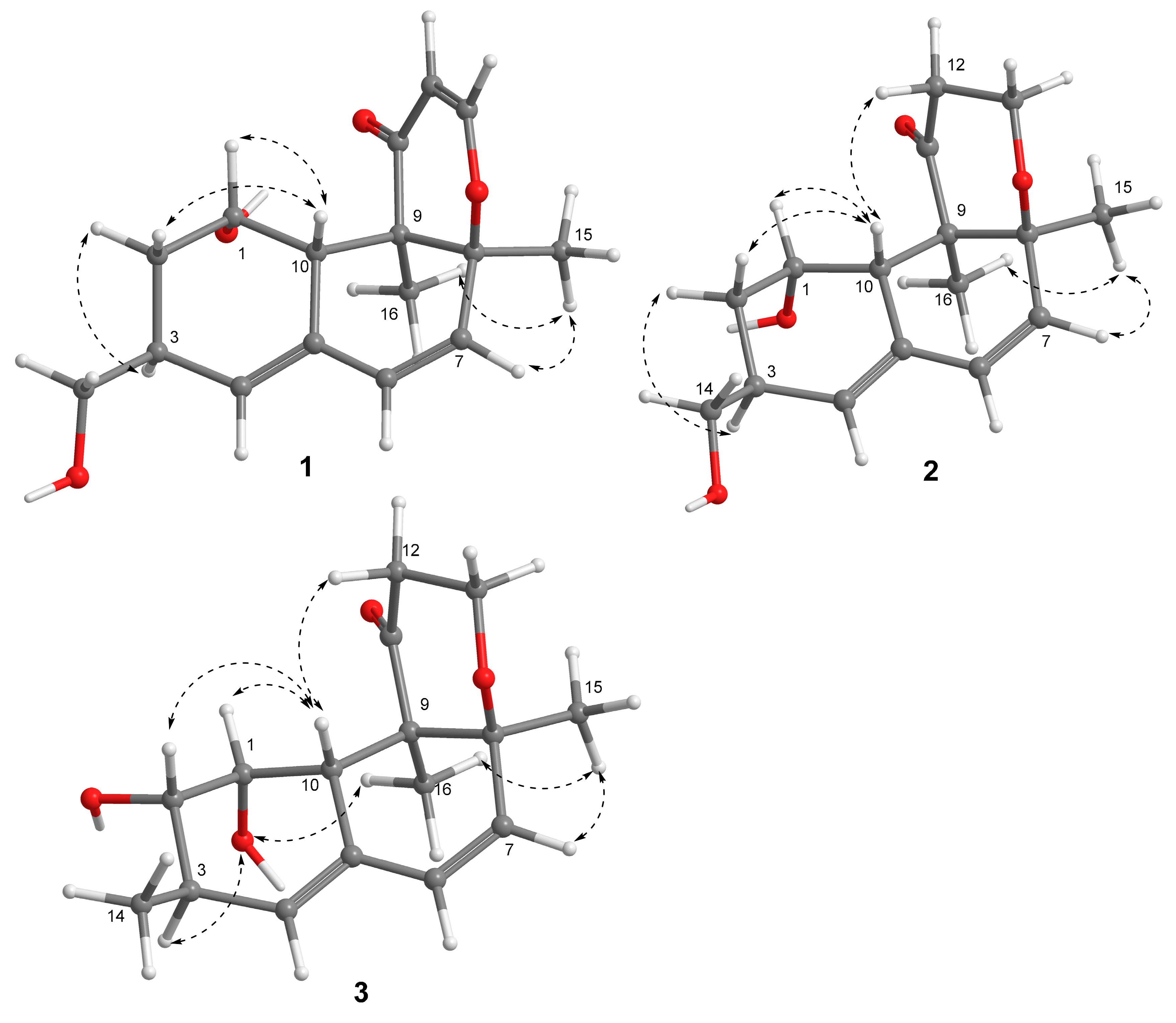{kind=link}
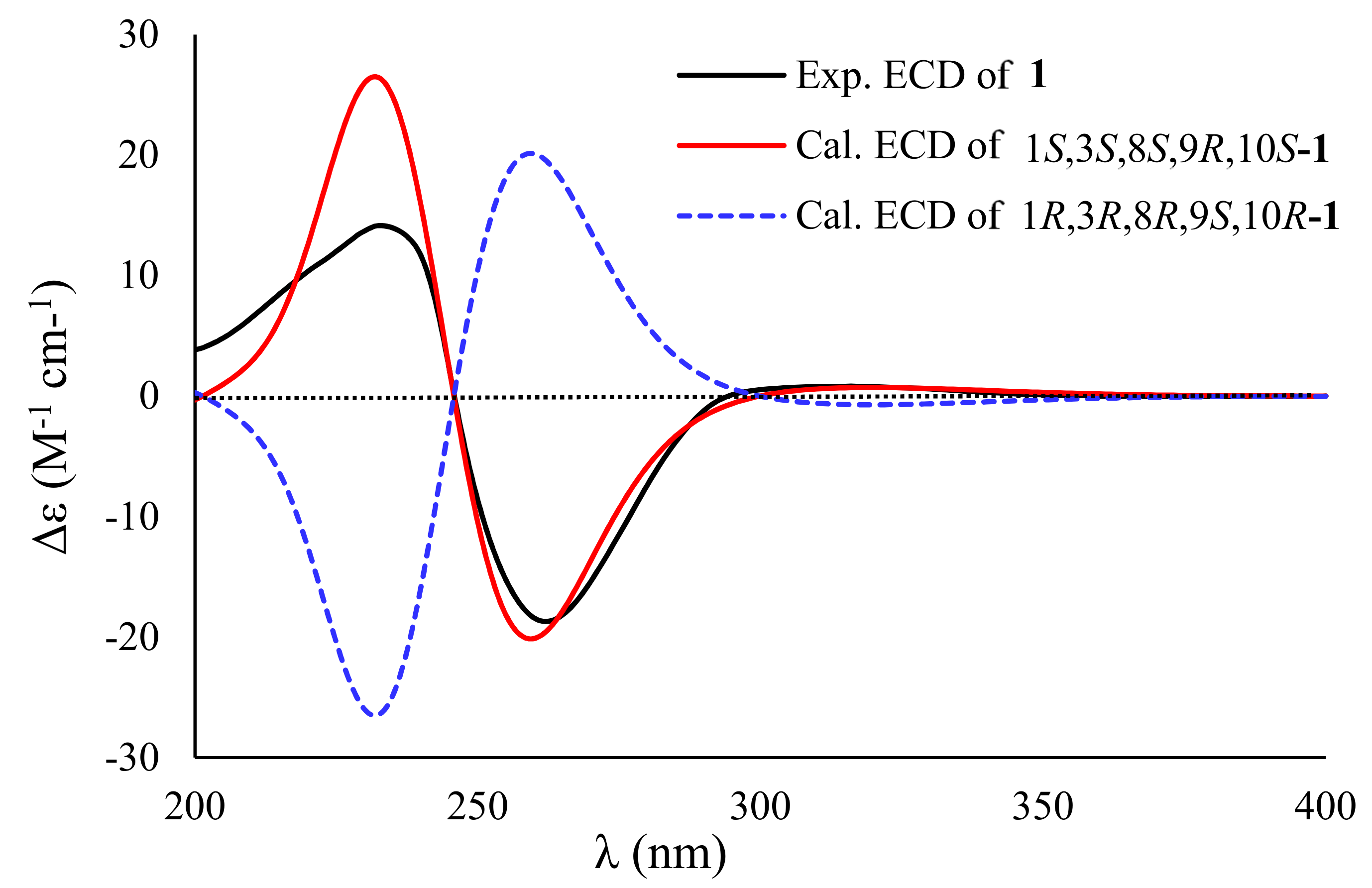{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| no. | 1 a | 2 a | 3 b | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 3.93, t (3.5) | 67.3, CH | 3.95, t (3.2) | 67.2, CH | 3.52, q (2.4) | 70.1, CH |
| 2 | 1.89, dtd (12.5, 4.8, 1.3); 1.33, td (12.5, 1.6) | 35.1, CH2 | 1.88, dtd (12.6, 5.0, 1.3); 1.44, td (12.6, 1.8) | 35.6, CH2 | 3.18, ddd (8.6, 5.9, 1.7) | 75.9, CH |
| 3 | 2.68, m | 35.6, CH | 2.67, m | 35.5, CH | 2.36, m | 33.0, CH |
| 4 | 5.92, br s | 133.4, CH | 5.84, br s | 131.4, CH | 5.50, t (2.3) | 132.6, CH |
| 5 | 132.5, C | 133.4, C | 130.2, C | |||
| 6 | 6.34, d (9.6) | 135.4, CH | 6.21, d (9.6) | 133.2, CH | 6.15, d (9.6) | 130.7, CH |
| 7 | 5.54, d (9.6) | 126.2, CH | 5.42, d (9.6) | 129.6, CH | 5.39, d (9.6) | 129.0, CH |
| 8 | 86.6, C | 80.4, C | 78.1, C | |||
| 9 | 52.0, C | 58.6, C | 56.5, C | |||
| 10 | 2.79, m | 42.5, CH | 3.44, t (3.2) | 43.1, CH | 3.38, overlap | 41.1, CH |
| 11 | 200.2, C | 213.7, C | 210.2, C | |||
| 12 | 5.38, d (5.9) | 104.8, CH | 3.03, ddd (14.6, 12.2, 8.8); 2.22, dd (14.6, 3.0) | 39.9, CH2 | 2.98, ddd (14.2, 12.0, 8.9); 2.11, dd (14.2, 3.0) | 38.4, CH2 |
| 13 | 7.31, d (5.9) | 162.6, CH | 4.06, dd (11.9, 8.3); 3.91, dd (11.9, 3.5) | 61.6, CH2 | 3.98, dd (11.8, 8.8); 3.79, td (11.8, 3.2) | 60.0, CH2 |
| 14 | 3.55, dd (10.6, 5.9); 3.47, dd (10.6, 6.5) | 66.9, CH2 | 3.54, dd (10.5, 6.2); 3.47, dd (10.5, 6.6) | 67.0, CH2 | 1.04, d (7.2) | 17.8, CH3 |
| 15 | 1.43, s | 19.5, CH3 | 1.22, s | 20.9, CH3 | 1.11, s | 20.1, CH3 |
| 16 | 1.17, s | 13.4, CH3 | 1.08, s | 13.6, CH3 | 0.99, s | 13.2, CH3 |
| 1-OH | 4.15, d (3.3) | |||||
| 2-OH | 4.54, d (5.9) |
| Position | 6 | 7 | ||
|---|---|---|---|---|
| δH, Mult (J in Hz) | δC, Type | δH, Mult (J in Hz) | δC, Type | |
| 1 | 139.4, C | 140.4, C | ||
| 2 | 6.25, br s | 113.9, CH | 6.45, br s | 112.7, CH |
| 3 | 153.2, C | 155.7, C | ||
| 4 | 6.23, br s | 107.0, CH | 6.38, br s | 103.0, CH |
| 5 | 157.8, C | 158.5, C | ||
| 6 | 6.38, br s | 113.1, CH | 6.45, br s | 109.7, CH |
| 7 | 2.16, s | 21.1, CH3 | 2.22, s | 21.1, CH3 |
| 1′ | 169.2, C | 170.5, C | ||
| 2′ | 3.15, d (15.5); 3.01, d (15.5) | 41.9, CH2 | 2.80, d (15.5); 2.74, d (15.5) | 43.1, CH2 |
| 3′ | 106.3, C | 82.0, C | ||
| 4′ | 7.33, br s | 146.0, CH | 6.69, s | 129.5, CH |
| 5′ | 132.3, C | 142.9, C | ||
| 6′ | 170.5, C | 166.3, C | ||
| 7′ | 1.71, s | 10.0, CH3 | 1.49, s | 25.0, CH3 |
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Compounds | BIU-87 | ECA109 | BEL-7402 | PANC-1 | Hela-S3 |
| 1 | 10.21 | >20 | >20 | >20 | >20 |
| 4 | >20 | 12.41 | >20 | >20 | >20 |
| 5 | >20 | 15.60 | >20 | >20 | >20 |
| 8 | 16.41 | >20 | >20 | >20 | >20 |
| 12 | >20 | 8.95 | >20 | >20 | >20 |
| 13 | >20 | >20 | 15.94 | >20 | >20 |
| 14 | >20 | >20 | 12.75 | >20 | >20 |
| 15 | 8.34 | >20 | 7.81 | >20 | >20 |
| 16 | 12.47 | 7.70 | 13.75 | >20 | >20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, S.; Xia, M.; Chen, M.; Liu, X.; Li, Z.; Xie, Y.; Shao, Z.; Zhang, G. Cytotoxic Polyketides Isolated from the Deep-Sea-Derived Fungus Penicillium chrysogenum MCCC 3A00292. Mar. Drugs 2019, 17, 686. https://doi.org/10.3390/md17120686
Niu S, Xia M, Chen M, Liu X, Li Z, Xie Y, Shao Z, Zhang G. Cytotoxic Polyketides Isolated from the Deep-Sea-Derived Fungus Penicillium chrysogenum MCCC 3A00292. Marine Drugs. 2019; 17(12):686. https://doi.org/10.3390/md17120686
Chicago/Turabian StyleNiu, Siwen, Manli Xia, Mingliang Chen, Xiupian Liu, Zengpeng Li, Yunchang Xie, Zongze Shao, and Gaiyun Zhang. 2019. "Cytotoxic Polyketides Isolated from the Deep-Sea-Derived Fungus Penicillium chrysogenum MCCC 3A00292" Marine Drugs 17, no. 12: 686. https://doi.org/10.3390/md17120686
APA StyleNiu, S., Xia, M., Chen, M., Liu, X., Li, Z., Xie, Y., Shao, Z., & Zhang, G. (2019). Cytotoxic Polyketides Isolated from the Deep-Sea-Derived Fungus Penicillium chrysogenum MCCC 3A00292. Marine Drugs, 17(12), 686. https://doi.org/10.3390/md17120686