Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach †

 ,
,  and
and 
Abstract
1. Introduction
2. Results
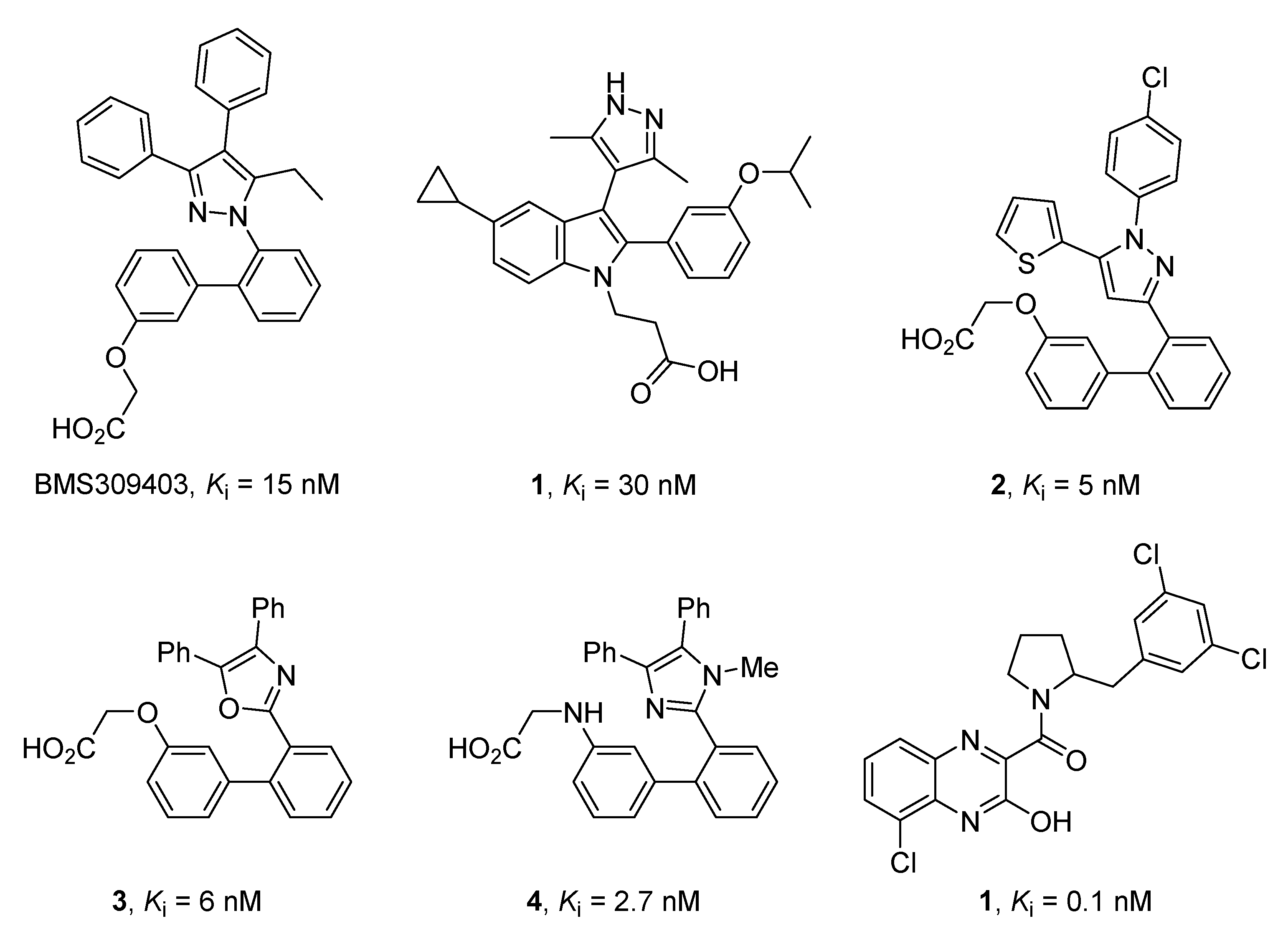
2.1. Design and Application of the Three Filters Used for the MNP Database Screening
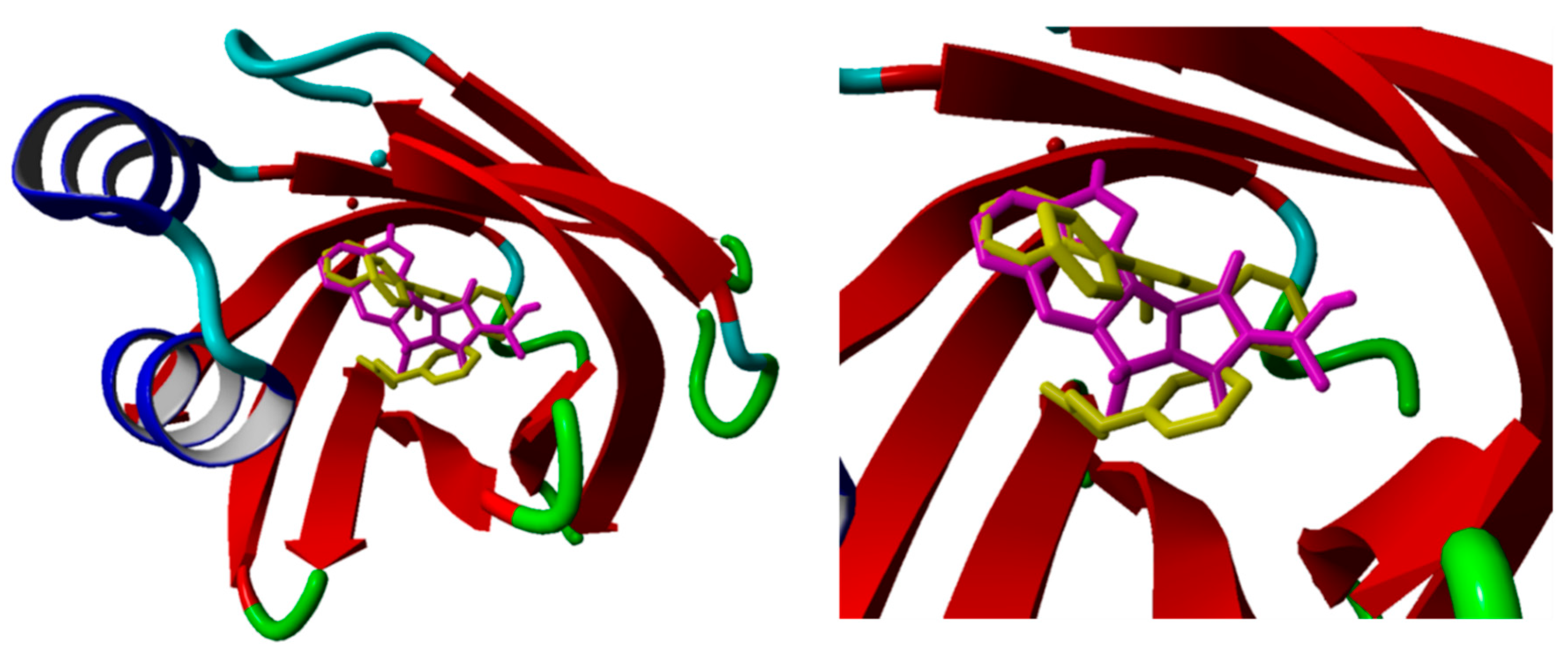
2.2. Merged Ligand- and Structure-Based Filters
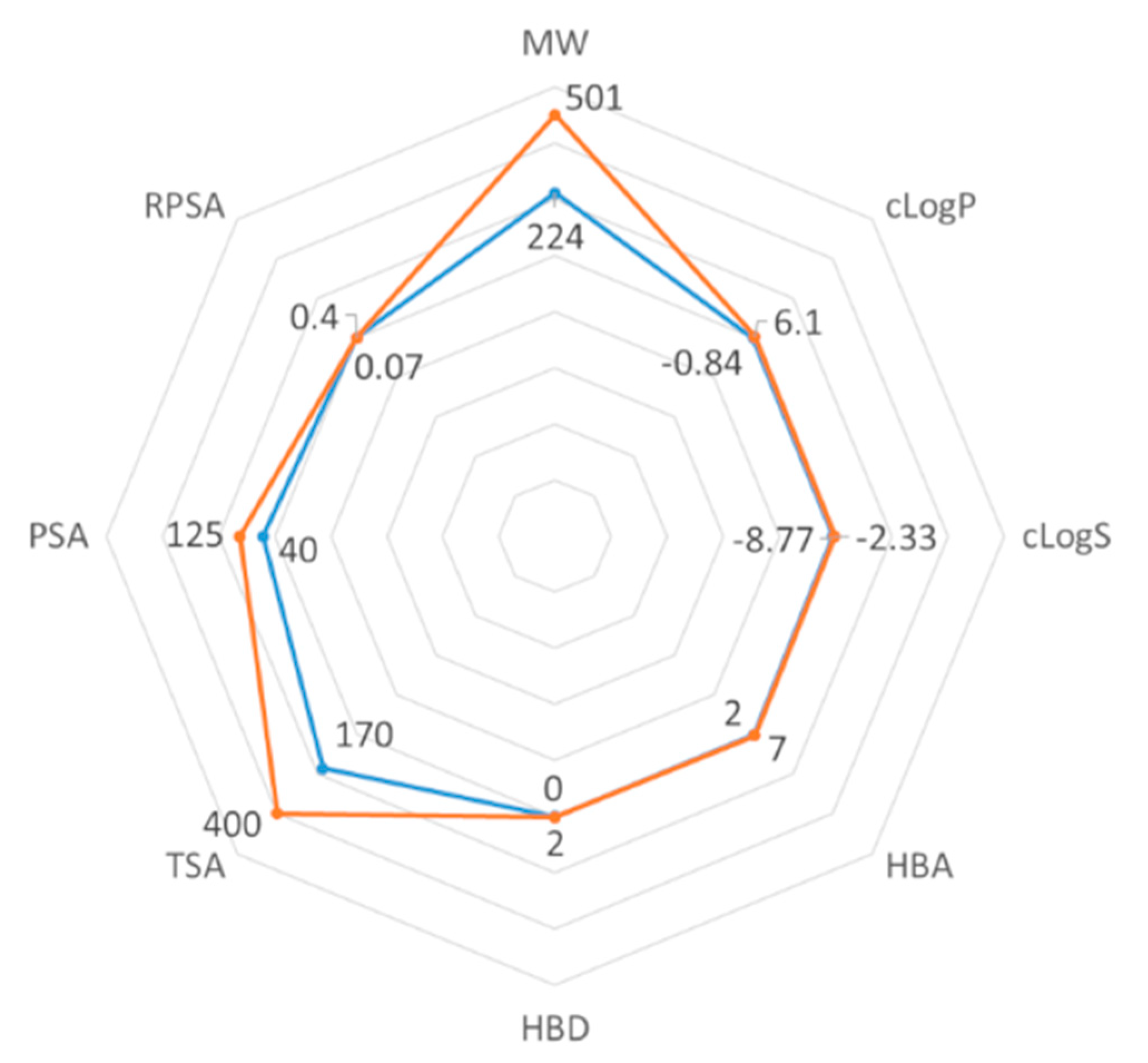
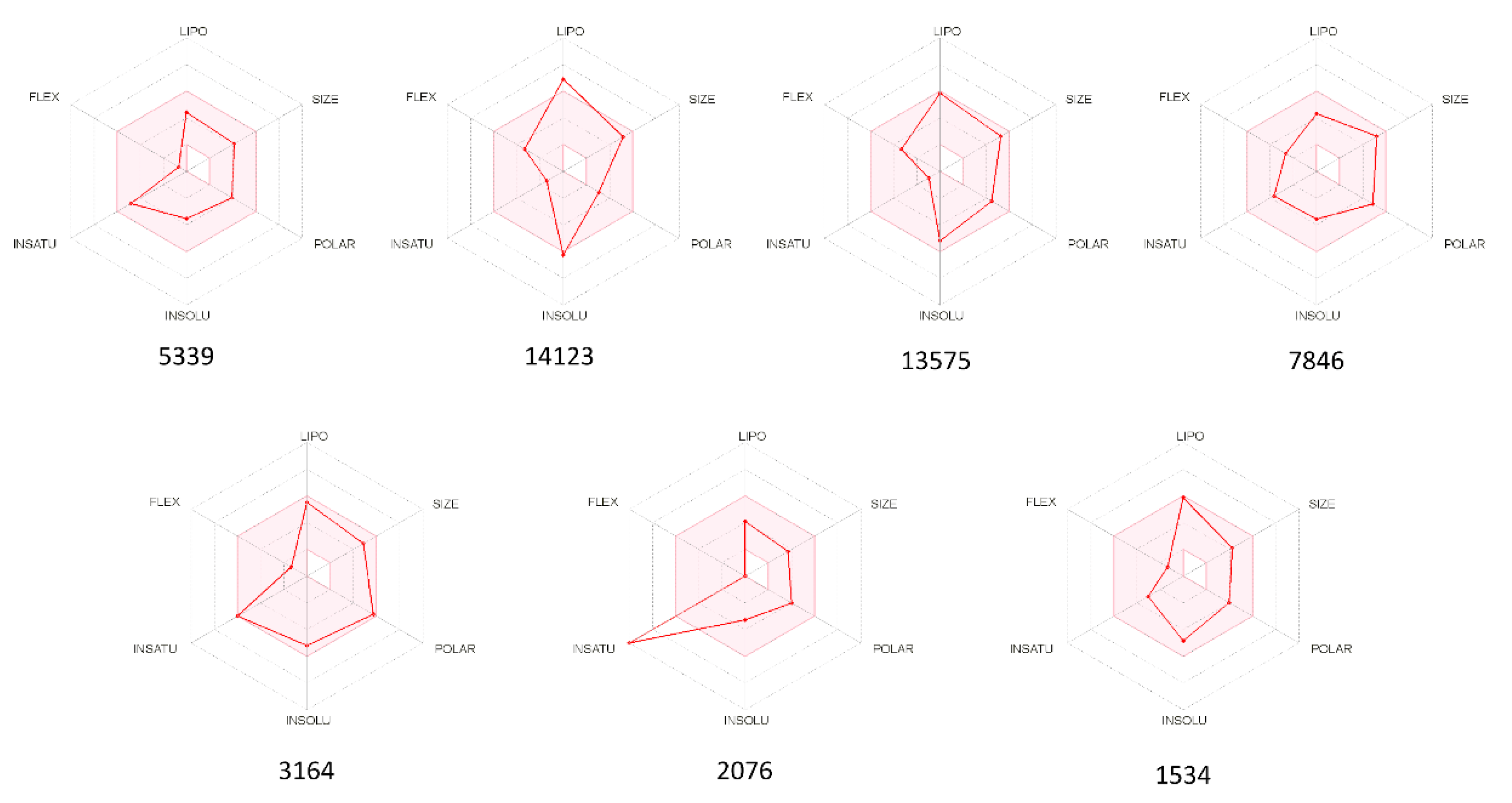
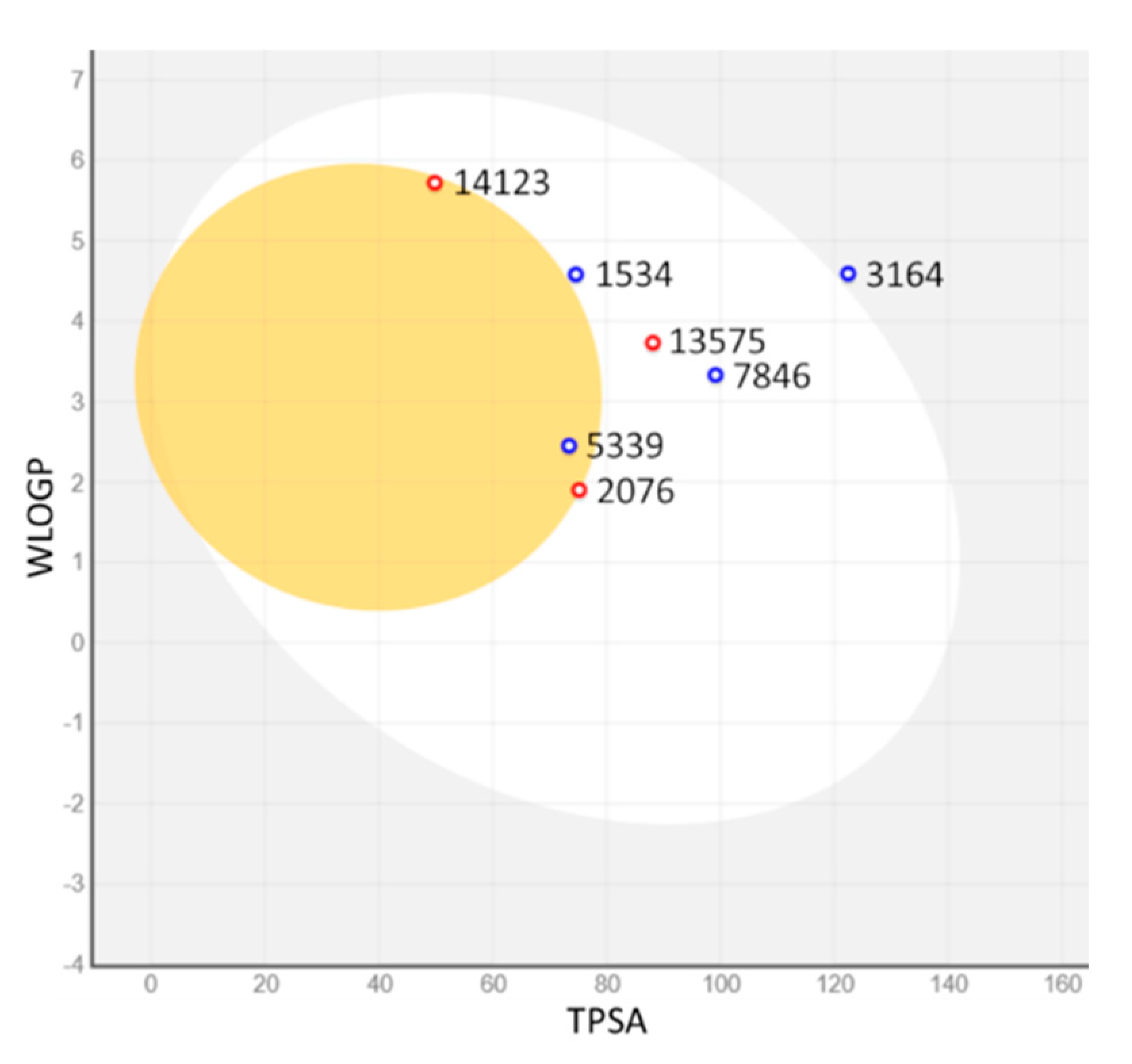
2.3. ADMET Properties
3. Materials and Methods
3.1. Dataset of Compounds
3.2. Structure Preparation and Minimization
3.3. Compound Alignment for the 3D-Ligand Based Filter
3.4. Molecular Docking
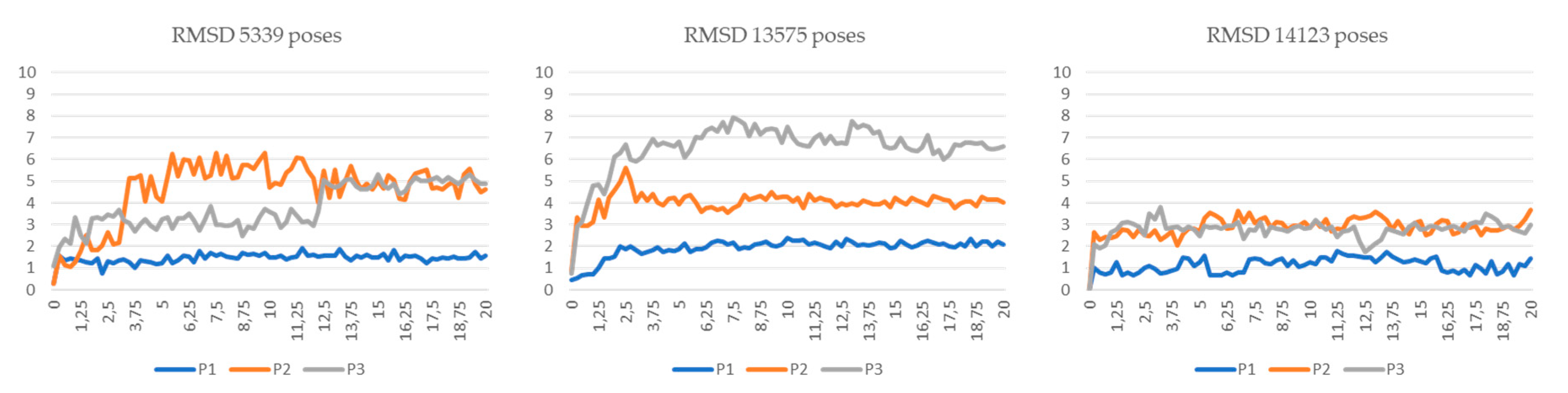
3.5. Molecular Dynamics Simulations
3.6. In Silico ADMET Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Das, U.N. Essential Fatty acids—A review. Curr. Pharm. Biotechnol. 2006, 7, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Free fatty acids (FFA), a link between obesity and insulin resistance. Front. Biosci. 1998, 3, d169–d175. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int. J. Clin. Pract. Suppl. 2004, 143, 9–21. [Google Scholar] [CrossRef]
- Sheth, S.G.; Gordon, F.D.; Chopra, S. Nonalcoholic steatohepatitis. Ann. Int. Med. 1997, 126, 137–145. [Google Scholar] [CrossRef]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef]
- Storch, J.; Thumser, A.E. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta 2000, 1486, 28–44. [Google Scholar] [CrossRef]
- Queipo-Ortuno, M.I.; Escote, X.; Ceperuelo-Mallafre, V.; Garrido-Sanchez, L.; Miranda, M.; Clemente-Postigo, M.; Perez-Perez, R.; Peral, B.; Cardona, F.; Fernandez-Real, J.M.; et al. FABP4 dynamics in obesity: Discrepancies in adipose tissue and liver expression regarding circulating plasma levels. PLoS ONE 2012, 7, e48605. [Google Scholar] [CrossRef]
- Thompson, B.R.; Mazurkiewicz-Munoz, A.M.; Suttles, J.; Carter-Su, C.; Bernlohr, D.A. Interaction of Adipocyte Fatty Acid-binding Protein (AFABP) and JAK2 AFABP/aP2 AS A REGULATOR OF JAK2 SIGNALING. J. Biol. Chem. 2009, 284, 13473–13480. [Google Scholar] [CrossRef]
- Adida, A.; Spener, F. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2006, 1761, 172–181. [Google Scholar] [CrossRef]
- Fu, Y.; Luo, N.; Lopes-Virella, M.F.; Garvey, W.T. The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis 2002, 165, 259–269. [Google Scholar] [CrossRef]
- Fu, Y.; Luo, L.; Luo, N.; Garvey, W.T. Lipid metabolism mediated by adipocyte lipid binding protein (ALBP/aP2) gene expression in human THP-1 macrophages. Atherosclerosis 2006, 188, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Tolle, A.; Suhail, S.; Jung, M.; Jung, K.; Stephan, C. Fatty acid binding proteins (FABPs) in prostate, bladder and kidney cancer cell lines and the use of IL-FABP as survival predictor in patients with renal cell carcinoma. BMC Cancer 2011, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Takahashi, T.; Oha, M.; Ogawa, H.; Izumi, K. Exogenous fatty acid binding protein 4 promotes human prostate cancer cell progression. Int. J. Cancer 2014, 135, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, H.; Sun, Y.; Wang, Y.; Yang, X.; Yang, X.; Zhang, H.; Guo, W.; Zhu, G.; Tian, J.; et al. Modulation of FABP4 hypomethylation by DNMT1 and its inverse interaction with miR-148a/152 in the placenta of preeclamptic rats and HTR-8 cells. Placenta 2016, 46, 49–62. [Google Scholar] [CrossRef]
- Lee, D.; Wada, K.; Taniguchi, Y.; Al-Shareef, H.; Masuda, T.; Usami, Y.; Aikawa, T.; Okura, M.; Kamisaki, Y.; Kogo, M. Expression of fatty acid binding protein 4 is involved in the cell growth of oral squamous cell carcinoma. Oncol. Rep. 2014, 31, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Pistara, V.; Amata, E.; Dichiara, M.; Marrazzo, A.; Prezzavento, O.; Rescifina, A. Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. A comprehensive systematic review. Eur. J. Med. Chem. 2017, 138, 854–873. [Google Scholar] [CrossRef]
- Wang, Y.; Law, W.K.; Hu, J.S.; Lin, H.Q.; Ip, T.M.; Wan, D.C. Discovery of FDA-approved drugs as inhibitors of fatty acid binding protein 4 using molecular docking screening. J. Chem. Inf. Model. 2014, 54, 3046–3050. [Google Scholar] [CrossRef]
- Zhou, Y.; Nie, T.; Zhang, Y.; Song, M.; Li, K.; Ding, M.; Ding, K.; Wu, D.; Xu, Y. The discovery of novel and selective fatty acid binding protein 4 inhibitors by virtual screening and biological evaluation. Bioorg. Med. Chem. 2016, 24, 4310–4317. [Google Scholar] [CrossRef]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef]
- Floresta, G.; Pittala, V.; Sorrenti, V.; Romeo, G.; Salerno, L.; Rescifina, A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018, 81, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Salerno, L.; Romeo, G.; Prezzavento, O.; Pittalà, V.; Rescifina, A. Identification of Potentially Potent Heme Oxygenase 1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem 2018, 13, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittala, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistara, V.; Pittala, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Sigma-2 receptor ligands QSAR model dataset. Data Brief. 2017, 13, 514–535. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef]
- Floresta, G.; Rescifina, A.; Abbate, V. Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. Int. J. Mol. Sci. 2019, 20, E2311. [Google Scholar] [CrossRef]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief. 2019, 22, 471–483. [Google Scholar] [CrossRef]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. 3D-QSAR assisted identification of FABP4 inhibitors: An effective scaffold hopping analysis/QSAR evaluation. Bioorg. Chem. 2019, 84, 276–284. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liu, Q.; Gao, D.; Wang, T.; Chen, T.; Yan, G.; Chen, K.; Xu, Y.; Wang, H.; Li, Y.; et al. Novel fatty acid binding protein 4 (FABP4) inhibitors: Virtual screening, synthesis and crystal structure determination. Eur. J. Med. Chem. 2015, 90, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yan, G.; Zhang, X.; Gorbenko, O.; Wang, H.; Zhu, W. Discovery of highly selective inhibitors of human fatty acid binding protein 4 (FABP4) by virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 3675–3679. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Sulsky, R.; Magnin, D.R.; Huang, Y.; Simpkins, L.; Taunk, P.; Patel, M.; Zhu, Y.; Stouch, T.R.; Bassolino-Klimas, D.; Parker, R.; et al. Potent and selective biphenyl azole inhibitors of adipocyte fatty acid binding protein (aFABP). Bioorg. Med. Chem. Lett. 2007, 17, 3511–3515. [Google Scholar] [CrossRef]
- Tagami, U.; Takahashi, K.; Igarashi, S.; Ejima, C.; Yoshida, T.; Takeshita, S.; Miyanaga, W.; Sugiki, M.; Tokumasu, M.; Hatanaka, T.; et al. Interaction Analysis of FABP4 Inhibitors by X-ray Crystallography and Fragment Molecular Orbital Analysis. ACS Med. Chem. Lett. 2016, 7, 435–439. [Google Scholar] [CrossRef][Green Version]
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef]
- Yao, S.; Gallenkamp, D.; Wolfel, K.; Luke, B.; Schindler, M.; Scherkenbeck, J. Synthesis and SERCA activities of structurally simplified cyclopiazonic acid analogues. Bioorg. Med. Chem. 2011, 19, 4669–4678. [Google Scholar] [CrossRef]
- Kobayashi, M.; Lee, N.K.; Son, B.W.; Yanag, K.; Kyogoku, Y.; Kitagawa, I. Stoloniferone-a, -b, -c, and -d, four new cytotoxic steroids from the okinawan soft coral clavularia viridis. Tetrahedron Lett. 1984, 25, 5925–5928. [Google Scholar] [CrossRef]
- Rudi, A.; Erez, Y.; Benayahu, Y.; Kashman, Y. Omriolide A and B; two new rearranged spongian diterpenes from the marine sponge Dictyodendrilla aff. retiara. Tetrahedron Lett. 2005, 46, 8613–8616. [Google Scholar] [CrossRef]
- Iwashima, M.; Matsumoto, Y.; Takenaka, Y.; Iguchi, K.; Yamori, T. New marine diterpenoids from the Okinawan soft coral Clavularia koellikeri. J. Nat. Prod. 2002, 65, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.I.; Hirase, T.; Shigemori, H.; Ishibashi, M.; Bae, M.-A.; Tsuji, T.; Sasaki, T. New Pentacyclic Compounds from the Okinawan Marine Sponge Xestospongia sapra. J. Nat. Prod. 1992, 55, 994–998. [Google Scholar] [CrossRef]
- Delfourne, E.; Bastide, J. Marine pyridoacridine alkaloids and synthetic analogues as antitumor agents. Med. Res. Rev. 2003, 23, 234–252. [Google Scholar] [CrossRef]
- Motti, A.C.; Bourguet-Kondracki, M.-L.; Longeon, A.; Doyle, R.J.; Llewellyn, E.L.; Tapiolas, M.D.; Yin, P. Comparison of the Biological Properties of Several Marine Sponge-Derived Sesquiterpenoid Quinones. Molecules 2007, 12, 1376–1388. [Google Scholar] [CrossRef]
- Khanaki, K.; Sadeghi, M.R.; Akhondi, M.M.; Darabi, M.; Mehdizadeh, A.; Shabani, M.; Rahimipour, A.; Nouri, M. High ω-3:ω-6 fatty acids ratio increases fatty acid binding protein 4 and extracellular secretory phospholipase A2IIa in human ectopic endometrial cells. IJRM 2014, 12, 755–764. [Google Scholar]
- Wang, Y.; Lin, H.-Q.; Xiao, C.-Y.; Law, W.-K.; Hu, J.-S.; Ip, T.-M.; Wan, D.C.-C. Using molecular docking screening for identifying hyperoside as an inhibitor of fatty acid binding protein 4 from a natural product database. J. Funct. Foods 2016, 20, 159–170. [Google Scholar] [CrossRef]
- Matlock, M.K.; Hughes, T.B.; Dahlin, J.L.; Swamidass, S.J. Modeling Small-Molecule Reactivity Identifies Promiscuous Bioactive Compounds. J. Chem. Inf. Model. 2018, 58, 1483–1500. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Barf, T.; Lehmann, F.; Hammer, K.; Haile, S.; Axen, E.; Medina, C.; Uppenberg, J.; Svensson, S.; Rondahl, L.; Lundback, T. N-Benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1745–1748. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Qiao, F.; Luo, L.; Peng, H.; Luo, S.; Huang, W.; Cui, J.; Li, X.; Kong, L.; Jiang, D.; Chitwood, D.J.; et al. Characterization of Three Novel Fatty Acid- and Retinoid-Binding Protein Genes (Ha-far-1, Ha-far-2 and Hf-far-1) from the Cereal Cyst Nematodes Heterodera avenae and H. filipjevi. PLoS ONE 2016, 11, e0160003. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MNP ID | Structure | pIC50 (QSAR) | pKi (Docking) | Mean |
|---|---|---|---|---|
| 5339 a |  | 6.30 | 7.66 | 6.98 |
| 14123 b |  | 6.30 | 7.41 | 6.85 |
| 13575 b |  | 6.10 | 7.95 | 7.02 |
| 7846 b |  | 6.40 | 7.35 | 6.87 |
| 3164 b |  | 6.30 | 7.17 | 6.73 |
| 2076 b |  | 6.10 | 7.68 | 6.89 |
| 1534 b |  | 6.10 | 6.87 | 6.48 |
| MNP ID | 5339 | 14123 | 13575 | 7846 | 3164 | 2076 | 1534 | |
|---|---|---|---|---|---|---|---|---|
| Drug-likeness | Lipinski violations | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Ghose violations | 0 | 2 | 0 | 0 | 0 | 0 | 0 | |
| Veber violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Egan violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Muegge violations | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Lead-likeness violations | 0 | 2 | 2 | 1 | 2 | 0 | 1 | |
| PAINS alerts | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| MNP ID | 5339 | 14123 | 13575 | 7846 | 3164 | 2076 | 1534 | |
|---|---|---|---|---|---|---|---|---|
| Absorption | Caco-2 permeability | 0.967 | 1.318 | 1.700 | 0.916 | −0.363 | 1.236 | 0.596 |
| Human intestinal absorption | 92.279 | 95.061 | 97.33 | 88.869 | 68.223 | 98.368 | 91.388 | |
| Skin permeability | −3.198 | −2.864 | −2.829 | −3.486 | −2.735 | −2.895 | −3.482 | |
| Distribution | VDss (human) | 0.458 | −0.177 | 0.031 | −0.276 | −1.88 | 0.145 | −0.014 |
| Fraction unbound (human) | 0.157 | 0.000 | 0.055 | 0.338 | 0.021 | 0.029 | 0.067 | |
| BBB permeability | 0.536 | 0.016 | −0.707 | −0.616 | −0.802 | 0.021 | −0.053 | |
| CNS permeability | −2.124 | −1.628 | −2.183 | −2.859 | −3.041 | −2.176 | −1.869 | |
| Excretion | Total clearance | 0.553 | 0.501 | 0.228 | 1.374 | 0.181 | 0.444 | 0.925 |
| Renal OCT2 substrate b | No | No | Yes | No | No | No | No | |
| Toxicity | AMES toxicity | No | No | No | No | No | Yes | No |
| Oral rat acute toxicity (LD50) | 2.564 | 2.175 | 2.119 | 2.823 | 2.664 | 2.305 | 2.341 | |
| Minnow toxicity | −0.338 | −0.467 | 0.169 | 2.126 | −0.189 | 0.260 | 0.038 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs 2019, 17, 624. https://doi.org/10.3390/md17110624
Floresta G, Gentile D, Perrini G, Patamia V, Rescifina A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Marine Drugs. 2019; 17(11):624. https://doi.org/10.3390/md17110624
Chicago/Turabian StyleFloresta, Giuseppe, Davide Gentile, Giancarlo Perrini, Vincenzo Patamia, and Antonio Rescifina. 2019. "Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach" Marine Drugs 17, no. 11: 624. https://doi.org/10.3390/md17110624
APA StyleFloresta, G., Gentile, D., Perrini, G., Patamia, V., & Rescifina, A. (2019). Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Marine Drugs, 17(11), 624. https://doi.org/10.3390/md17110624