Fast Detection of Two Smenamide Family Members Using Molecular Networking

 ,
,  , , and
, , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
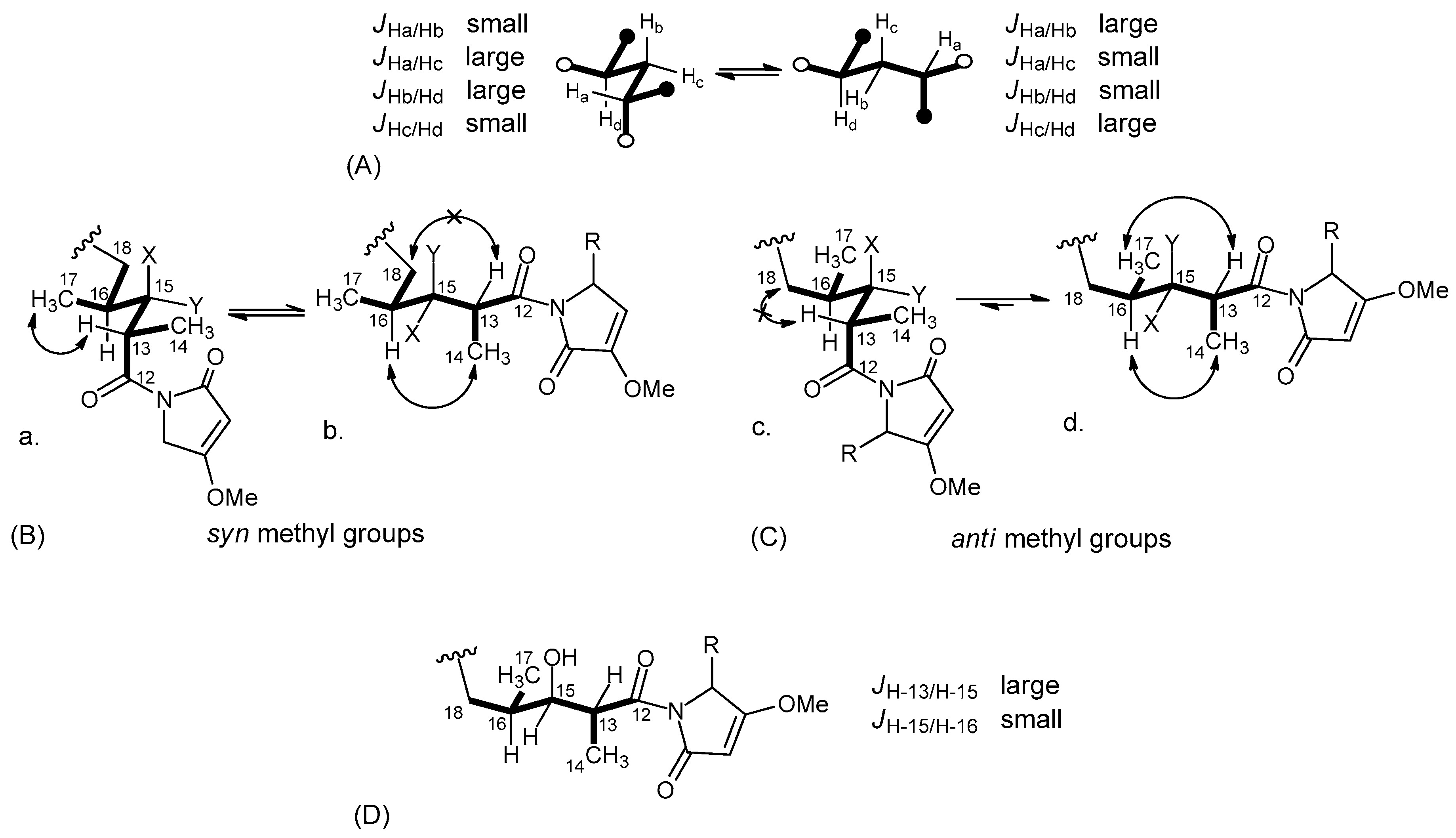{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
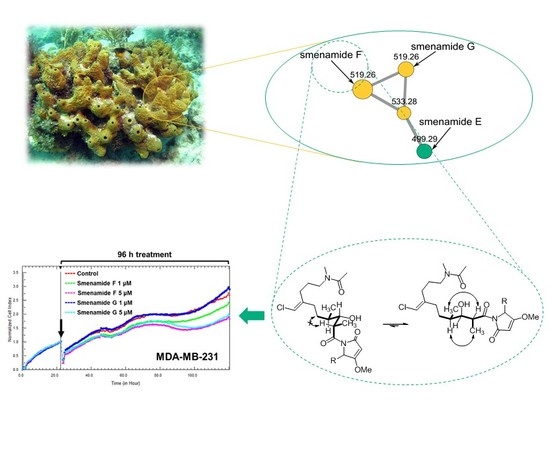
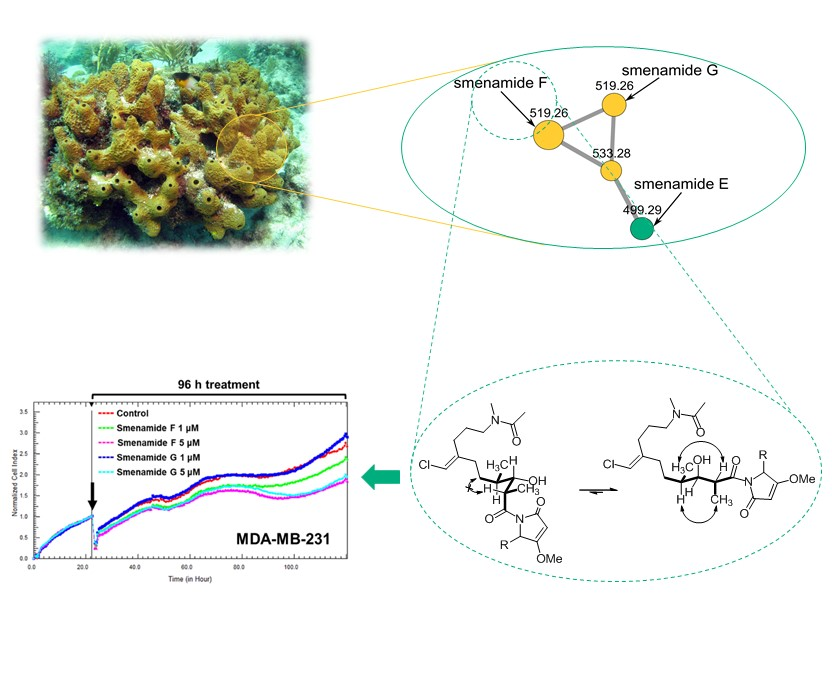
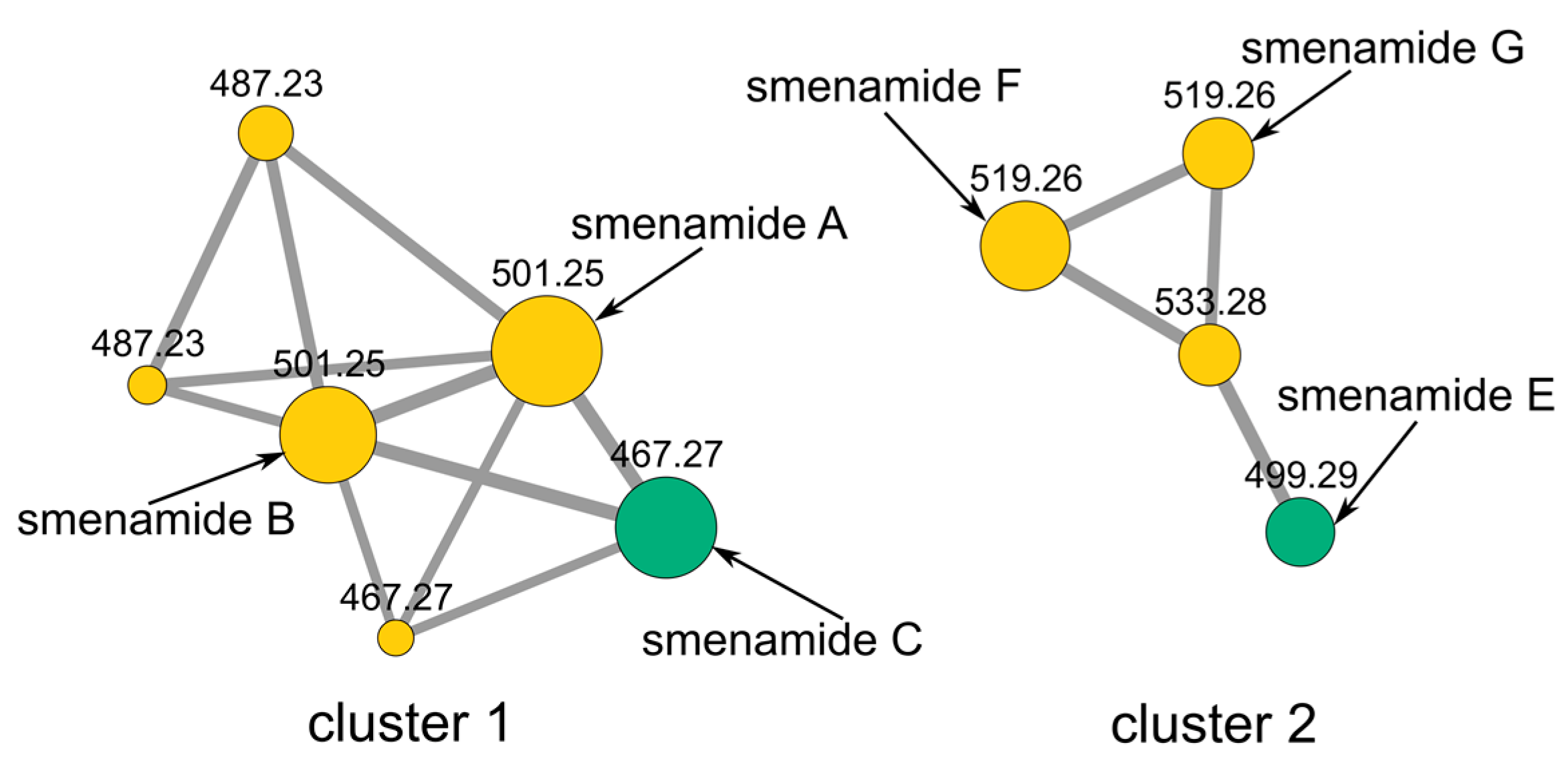
2.1. Fast Detection of Two New Analogues of the Smenamide Family: Construction of the Molecular Network and Isolation
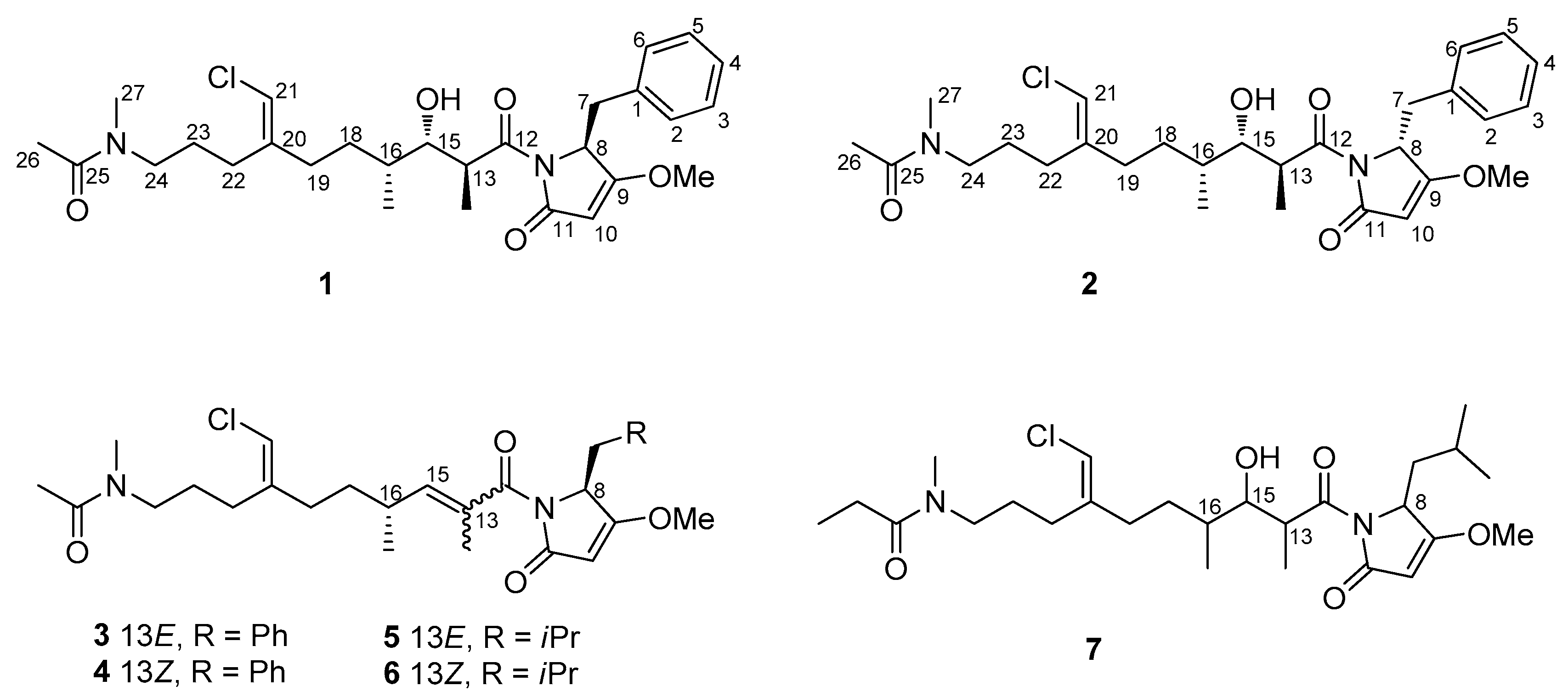
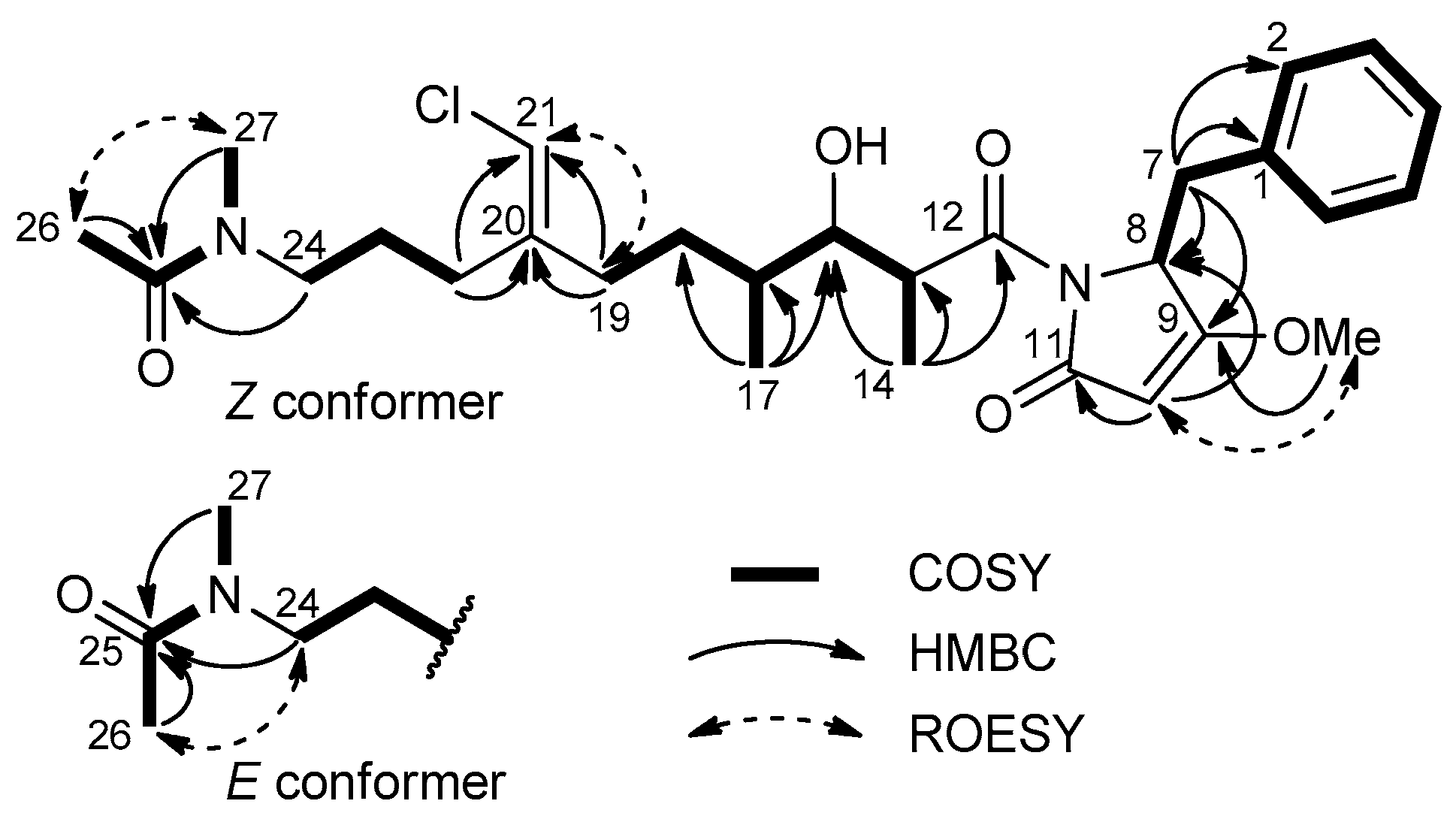
2.2. Structure Elucidation of Smenamides F and G
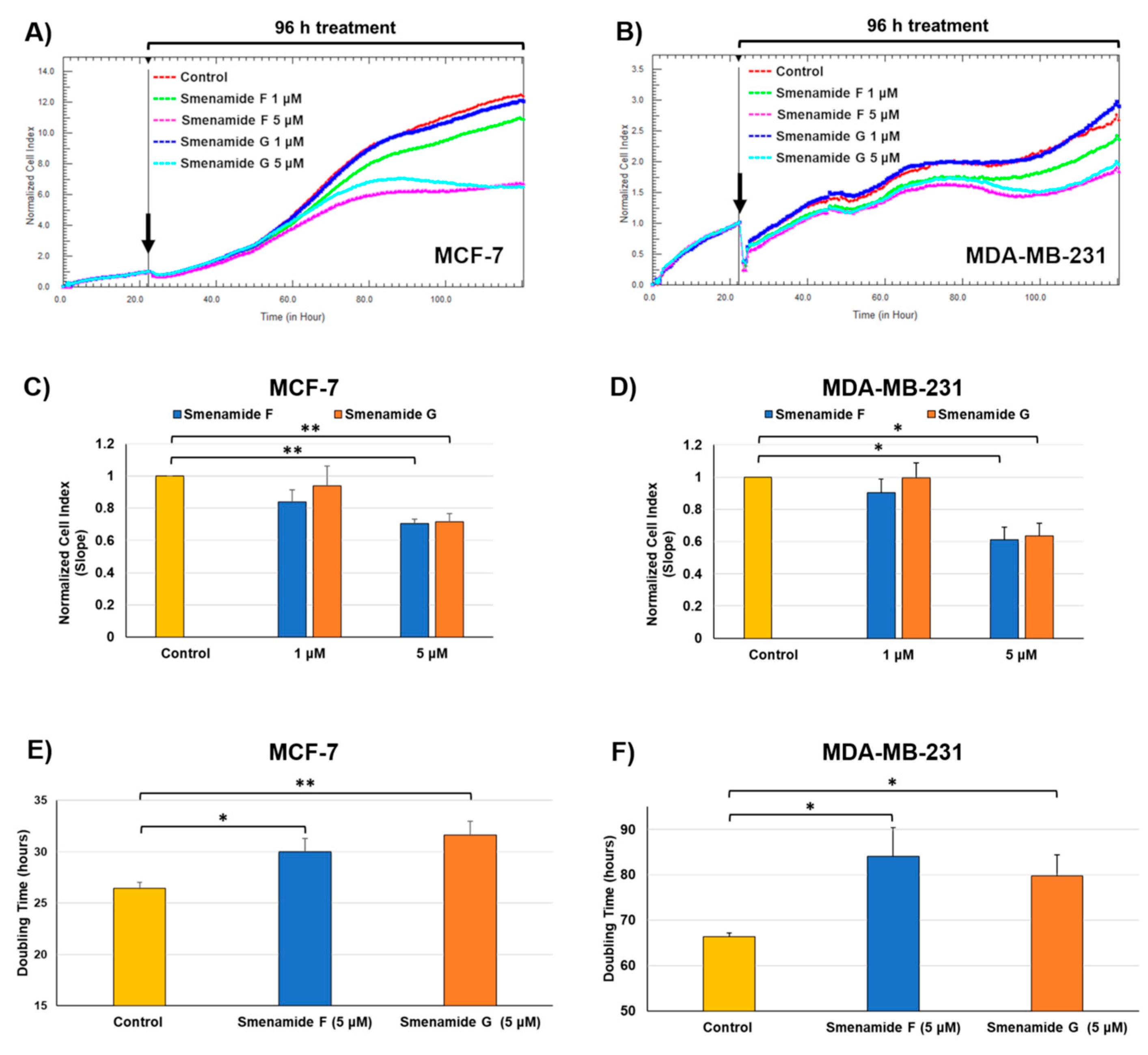
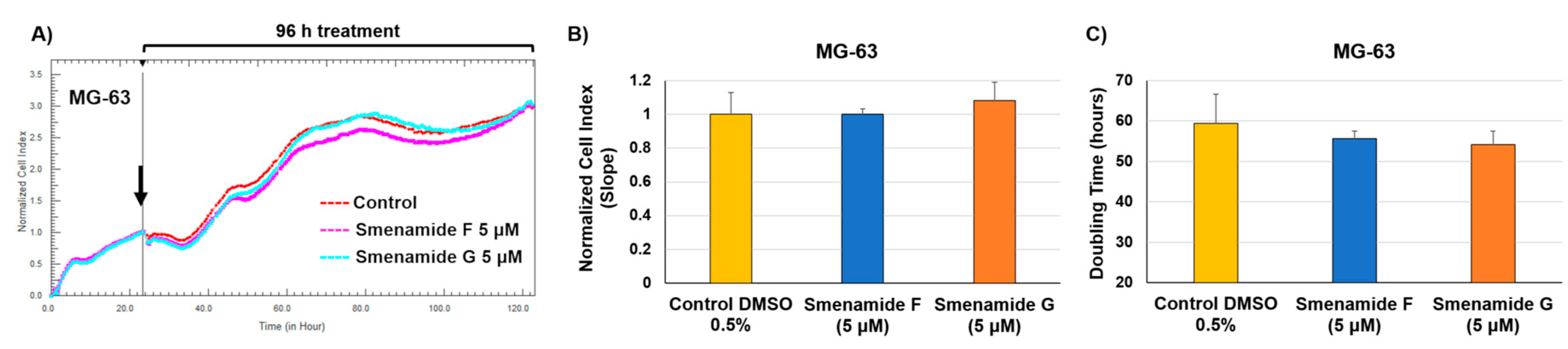
2.3. Antiproliferative Activity of Smenamides F and G
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Collection, Extraction and Isolation
3.2.1. Smenamide F (1)
3.2.2. Smenamide G (2)
3.3. Determination of the Absolute Configuration of the Amino Acids
3.3.1. Ozonolysis and Hydrolysis
3.3.2. Marfey’s Derivatization with D- and L-FDAA
3.3.3. LC-MS Analysis
3.4. Molecular Network
3.5. Cell Culture
3.6. xCELLigence Assays and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gribble, G.W. Biological Activity of Recently Discovered Halogenated Marine Natural Products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef]
- Esposito, G.; Bourguet-Kondracki, M.L.; Mai, L.H.; Longeon, A.; Teta, R.; Meijer, L.; Van Soest, R.; Mangoni, A.; Costantino, V. Chloromethylhalicyclamine B, a marine-derived protein kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016, 79, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides A and B, Chlorinated Peptide/Polyketide Hybrids Containing a Dolapyrrolidinone Unit from the Caribbean Sponge Smenospongia aurea. Evaluation of Their Role as Leads in Antitumor Drug Research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Teta, R.; Miceli, R.; Ceccarelli, L.S.; Della Sala, G.; Camerlingo, R.; Irollo, E.; Mangoni, A.; Pirozzi, G.; Costantino, V. Isolation and Assessment of the in Vitro Anti-Tumor Activity of Smenothiazole A and B, Chlorinated Thiazole-Containing Peptide/Polyketides from the Caribbean Sponge Smenospongia aurea. Mar. Drugs 2015, 13, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Della Sala, G.; Teta, R.; Caso, A.; Bourguet-Kondraki, M.L.; Pawlik, J.R.; Mangoni, A.; Costantino, V. Chlorinated Thiazole-Containing Polyketide-Peptides from the Caribbean Sponge Smenospongia conulosa: Structure Elucidation on Microgram Scale. Eur. J. Org. Chem. 2016, 2871–2875. [Google Scholar] [CrossRef]
- Teta, R.; Della Sala, G.; Esposito, G.; Via, C.W.; Mazzoccoli, C.; Piccoli, C.; Bertin, M.J.; Costantino, V.; Mangoni, A. A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front. 2019, 6, 1762–1774. [Google Scholar] [CrossRef]
- Esposito, G.; Teta, R.; Della Sala, G.; Pawlik, J.R.; Costantino, V. Isolation of Smenopyrone, a Bis-γ-Pyrone Polypropionate from the Caribbean Sponge Smenospongia aurea. Mar. Drugs 2018, 16, 285. [Google Scholar] [CrossRef]
- Caso, A.; Mangoni, A.; Piccialli, G.; Costantino, V.; Piccialli, V. Studies toward the Synthesis of Smenamide A, an Antiproliferative Metabolite from Smenospongia aurea: Total Synthesis of ent-Smenamide A and 16-epi-Smenamide, A. ACS Omega 2017, 2, 1477–1488. [Google Scholar] [CrossRef]
- Caso, A.; Laurenzana, I.; Lamorte, D.; Trino, S.; Esposito, G.; Piccialli, V.; Costantino, V. Smenamide a analogues. Synthesis and biological activity on multiple myeloma cells. Mar. Drugs 2018, 16, 206. [Google Scholar] [CrossRef]
- Via, C.W.; Glukhov, E.; Costa, S.; Zimba, P.V.; Moeller, P.D.R.; Gerwick, W.H.; Bertin, M.J. The Metabolome of a Cyanobacterial Bloom Visualized by MS/MS-Based Molecular Networking Reveals New Neurotoxic Smenamide Analogs (C, D, and E). Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef]
- Cantrell, T.P.; Freeman, C.J.; Paul, V.J.; Agarwal, V.; Garg, N. Mass Spectrometry-Based Integration and Expansion of the Chemical Diversity Harbored Within a Marine Sponge. J. Am. Soc. Mass Spectrom. 2019, 30, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Della Sala, G.; Saurav, K.; Teta, R.; Bar-Shalom, R.; Mangoni, A.; Steindler, L. Plakofuranolactone as a Quorum Quenching Agent from the Indonesian Sponge Plakortis cf. lita. Mar. Drugs 2017, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Della Sala, G.; Glukhov, E.; Gerwick, L.; Gerwick, W.H.; Mangoni, A.; Costantino, V. Combined LC−MS/MS and Molecular Networking Approach Reveals New Cyanotoxins from the 2014 Cyanobacterial Bloom in Green Lake, Seattle. Environ. Sci. Technol. 2015, 49, 14301–14310. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Guerriero, E.; Teta, R.; Capone, F.; Caso, A.; Sorice, A.; Romano, G.; Ianora, A.; Ruocco, N.; Budillon, A.; et al. Evaluating the effects of an organic extract from the mediterranean sponge Geodia cydonium on human breast cancer cell lines. Int. J. Mol. Sci. 2017, 18, 2112. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Okino, T.; Nogle, L.M.; Marquez, B.L.; Williamson, R.T.; Sitachitta, N.; Berman, F.W.; Murray, T.F.; McGough, K.; Jacobs, R.; et al. Structure, Synthesis, and Biological Properties of Kalkitoxin, a Novel Neurotoxin from the Marine Cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2000, 122, 48. [Google Scholar] [CrossRef]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent 1,5-difluoro-2,4-dinitrobenzene. Carlsberg. Res. Commun. 1984, 49, 591–596. [Google Scholar]
- Dalisay, D.S.; Rogers, E.W.; Edison, A.S.; Molinski, T.F. Trisoxazole macrolides and thiazole-containing cyclic peptides from the nudibranch Hexabranchus sanguineus. J. Nat. Prod. 2009, 72, 732–738. [Google Scholar] [CrossRef]
- Hoffmann, R.W.; Stahl, M.; Schopfer, U.; Frenking, G. Conformation design of hydrocarbon backbones: A modular approach. Chem. Eur. J. 1998, 4, 559–566. [Google Scholar] [CrossRef]
- The Sponge Guide. Available online: http://www.spongeguide.org (accessed on 7 July 2013).






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caso, A.; Esposito, G.; Della Sala, G.; Pawlik, J.R.; Teta, R.; Mangoni, A.; Costantino, V. Fast Detection of Two Smenamide Family Members Using Molecular Networking. Mar. Drugs 2019, 17, 618. https://doi.org/10.3390/md17110618
Caso A, Esposito G, Della Sala G, Pawlik JR, Teta R, Mangoni A, Costantino V. Fast Detection of Two Smenamide Family Members Using Molecular Networking. Marine Drugs. 2019; 17(11):618. https://doi.org/10.3390/md17110618
Chicago/Turabian StyleCaso, Alessia, Germana Esposito, Gerardo Della Sala, Joseph R. Pawlik, Roberta Teta, Alfonso Mangoni, and Valeria Costantino. 2019. "Fast Detection of Two Smenamide Family Members Using Molecular Networking" Marine Drugs 17, no. 11: 618. https://doi.org/10.3390/md17110618
APA StyleCaso, A., Esposito, G., Della Sala, G., Pawlik, J. R., Teta, R., Mangoni, A., & Costantino, V. (2019). Fast Detection of Two Smenamide Family Members Using Molecular Networking. Marine Drugs, 17(11), 618. https://doi.org/10.3390/md17110618