Bioinformatics for Marine Products: An Overview of Resources, Bottlenecks, and Perspectives

 , , , ,
, , , , 

Abstract
1. Introduction
2. Bioinformatics Applications and Resources in Marine Omics
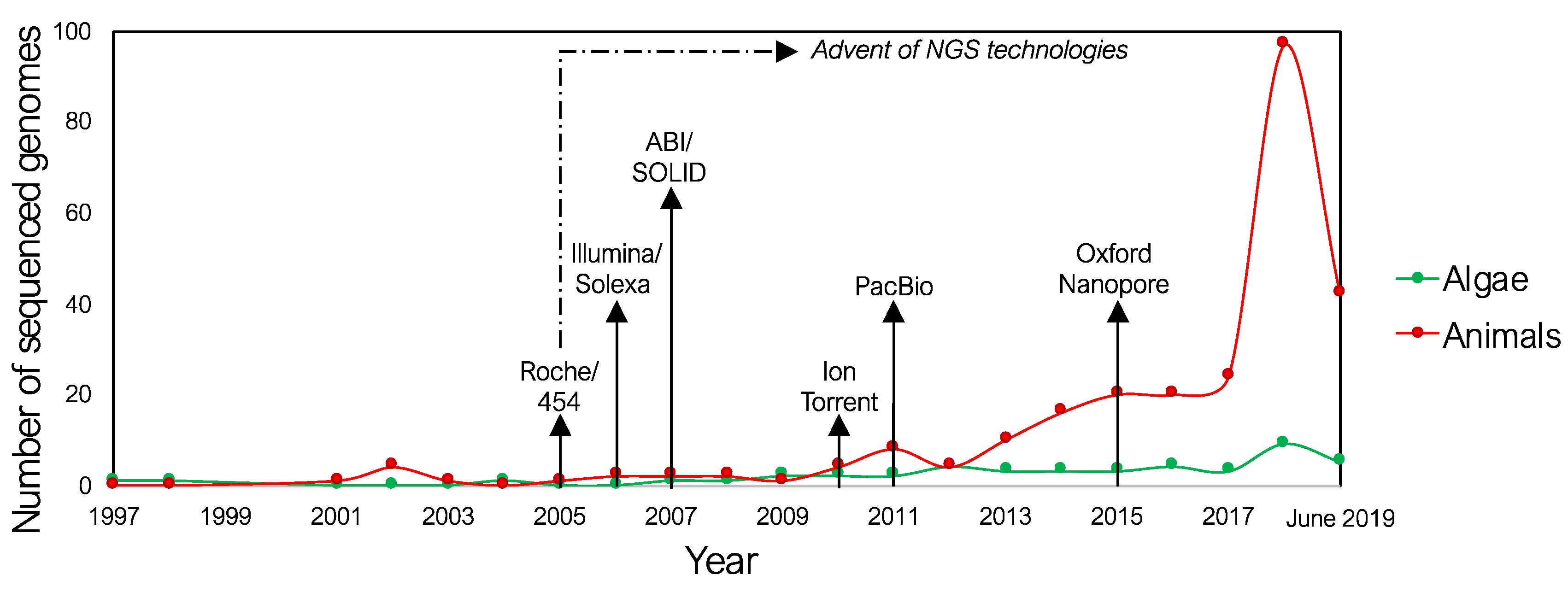
2.1. Genomics and Transcriptomics
2.2. Metagenomics and Metatranscriptomics
2.3. Proteomics and Structural Biology
2.4. Metabolomics
3. Bottlenecks and Perspectives
3.1. Bottlenecks
3.2. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Danovaro, R.; Corinaldesi, C.; Dell’Anno, A.; Fuhrman, J.A.; Middelburg, J.J.; Noble, R.T.; Suttle, C.A. Marine viruses and global climate change. FEMS Microbiol. Rev. 2011, 35, 993–1034. [Google Scholar] [CrossRef] [PubMed]
- Argulis, L.; Schwartz, K.V. Five Kingdoms: An Illustrated Guide to the Phyla of Life on Earth; Freeman WH and Company: New York, NY, USA, 1982. [Google Scholar]
- Macdougall, J.D. A Short History of Planet Earth; John Wiley (Ed.): New York, NY, USA, 1996; p. 274. [Google Scholar]
- Bernhard, J.M.; Kormas, K.; Pachiadaki, M.G.; Rocke, E.; Beaudoin, D.J.; Morrison, C.; Visscher, P.T.; Cobban, A.; Starczak, V.R.; Edgcomb, V.P. Benthic protists and fungi of Mediterranean deep hypsersaline anoxic basin redoxcline sediments. Front. Microbiol. 2014, 5, 605. [Google Scholar] [CrossRef] [PubMed]
- CAREX. Roadmap for Research on Life in Extreme Environment. Available online: http://commercialspace.pbworks.com/f/2011.01+CAREX_Roadmap_Final.pdf (accessed on 20 August 2019).
- Chen, L.; DeVries, A.L.; Cheng, C.-H.C. Convergent evolution of antifreeze glycoproteins in Antarctic notothenioid fish and Arctic cod. Proc. Natl. Acad. Sci. USA 1997, 94, 3817. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C.; Tangherlini, M.; Luna, G.M.; Dell’Anno, A. Extracellular DNA can preserve the genetic signatures of present and past viral infection events in deep hypersaline anoxic basins. Proc. R. Soc. B Biol. Sci. 2014, 281, 20133299. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Gambi, C.; Dell’Anno, A.; Corinaldesi, C.; Pusceddu, A.; Neves, R.C.; Kristensen, R.M. The challenge of proving the existence of metazoan life in permanently anoxic deep-sea sediments. BMC Biol. 2016, 14, 43. [Google Scholar] [CrossRef]
- Danovaro, R.; Molari, M.; Corinaldesi, C.; Dell’Anno, A. Macroecological drivers of archaea and bacteria in benthic deep-sea ecosystems. Sci. Adv. 2016, 2, e1500961. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, N.; Jollivet, D.; Boutet, I.; Brélivet, Y.; Busso, D.; Da Silva, C.; Gaill, F.; Higuet, D.; Hourdez, S.; Knoops, B.; et al. Insights into metazoan evolution from Alvinella pompejana cDNAs. BMC Genom. 2010, 11, 634. [Google Scholar] [CrossRef]
- Hu, Y.; Ghigliotti, L.; Vacchi, M.; Pisano, E.; Detrich, H.W.; Albertson, R.C. Evolution in an extreme environment: Developmental biases and phenotypic integration in the adaptive radiation of antarctic notothenioids. BMC Evol. Biol. 2016, 16, 142. [Google Scholar] [CrossRef]
- Jimeno, J.; Faircloth, G.; Sousa-Faro, J.M.F.; Scheuer, P.; Rinehart, K. New Marine Derived Anticancer Therapeutics—A Journey from the Sea to Clinical Trials. Mar. Drugs 2004, 2, 14–29. [Google Scholar] [CrossRef]
- Zeppilli, D.; Leduc, D. Biodiversity and ecology of meiofauna in extreme and changing environments. Mar. Biodivers. 2018, 48, 1–4. [Google Scholar] [CrossRef]
- Barone, G.; Rastelli, E.; Corinaldesi, C.; Tangherlini, M.; Danovaro, R.; Dell’Anno, A. Benthic deep-sea fungi in submarine canyons of the Mediterranean Sea. Prog. Oceanogr. 2018, 168, 57–64. [Google Scholar] [CrossRef]
- Lauritano, C.; Andersen, J.H.; Hansen, E.; Albrigtsen, M.; Escalera, L.; Esposito, F.; Helland, K.; Hanssen, K.Ø.; Romano, G.; Ianora, A. Bioactivity Screening of Microalgae for Antioxidant, Anti-Inflammatory, Anticancer, Anti-Diabetes, and Antibacterial Activities. Front. Mar. Sci. 2016, 3, 68. [Google Scholar] [CrossRef]
- Cherry, P.; Yadav, S.; Strain, C.R.; Allsopp, P.J.; McSorley, E.M.; Ross, R.P.; Stanton, C. Prebiotics from Seaweeds: An Ocean of Opportunity? Mar. Drugs 2019, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Gentile, A.; Orefice, I.; Ianora, A.; Bruno, A.; Noonan, D.M.; Sansone, C.; Albini, A.; Brunet, C. Microalgal Derivatives as Potential Nutraceutical and Food Supplements for Human Health: A Focus on Cancer Prevention and Interception. Nutrients 2019, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Overland, M.; Mydland, L.T.; Skrede, A. Marine macroalgae as sources of protein and bioactive compounds in feed for monogastric animals. J. Sci. Food Agric. 2019, 99, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Singh, S.; Agrawal, C.; Yadav, S.; Rai, R.; Rai, L.C. Chapter 10—Role of Algae as a Biofertilizer. In Algal Green Chemistry; Rastogi, R.P., Madamwar, D., Pandey, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 189–200. [Google Scholar]
- López-Mosquera, M.E.; Fernández-Lema, E.; Villares, R.; Corral, R.; Alonso, B.; Blanco, C. Composting fish waste and seaweed to produce a fertilizer for use in organic agriculture. Procedia Environ. Sci. 2011, 9, 113–117. [Google Scholar] [CrossRef]
- Mohan, K.; Ravichandran, S.; Muralisankar, T.; Uthayakumar, V.; Chandirasekar, R.; Seedevi, P.; Abirami, R.G.; Rajan, D.K. Application of marine-derived polysaccharides as immunostimulants in aquaculture: A review of current knowledge and further perspectives. Fish Shellfish Immunol. 2019, 86, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Liu, G.-L.; Lu, Y.; Jiang, H.; Chi, Z.-M. Bio-products produced by marine yeasts and their potential applications. Bioresour. Technol. 2016, 202, 244–252. [Google Scholar] [CrossRef]
- Maeda, Y.; Yoshino, T.; Matsunaga, T.; Matsumoto, M.; Tanaka, T. Marine microalgae for production of biofuels and chemicals. Curr. Opin. Biotechnol. 2018, 50, 111–120. [Google Scholar] [CrossRef]
- Swain, M.R.; Natarajan, V.; Krishnan, C. Chapter Nine—Marine Enzymes and Microorganisms for Bioethanol Production. In Advances in Food and Nutrition Research; Kim, S.-K., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 80, pp. 181–197. [Google Scholar]
- Corinaldesi, C.; Barone, G.; Marcellini, F.; Dell’Anno, A.; Danovaro, R. Marine Microbial-Derived Molecules and Their Potential Use in Cosmeceutical and Cosmetic Products. Mar. Drugs 2017, 15, 118. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.E.; Kim, K.H.; Kang, N.J. Beneficial Effects of Marine Algae-Derived Carbohydrates for Skin Health. Mar. Drugs 2018, 16, 459. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Anil, S.; Kim, S.K.; Shim, M.S. Marine Fish Proteins and Peptides for Cosmeceuticals: A Review. Mar. Drugs 2017, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Paniagua-Michel, J.; Rosales, A. Marine bioremediation-A sustainable biotechnology of petroleum hydrocarbons biodegradation in coastal and marine environments. J. Bioremediation Biodegredation 2015, 6, 1. [Google Scholar]
- Costa, S.S.; Miranda, A.L.; de Morais, M.G.; Costa, J.A.V.; Druzian, J.I. Microalgae as source of polyhydroxyalkanoates (PHAs)—A review. Int. J. Biol. Macromol. 2019, 131, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Rottacker, E.C.; Kastovsky, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komarek, J.; Gerwick, W.H. Moorea producens gen. nov. sp. nov. and Moorea bouillonii comb. nov. tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Microbiol. 2012, 62 Pt 5, 1171–1178. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Labes, A.; Wiese, J. Bio-mining the microbial treasures of the ocean: New natural products. Biotechnol. Adv. 2011, 29, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef]
- Long, S.; Sousa, E.; Kijjoa, A.; Pinto, M.M. Marine Natural Products as Models to Circumvent Multidrug Resistance. Molecules 2016, 21, 892. [Google Scholar] [CrossRef]
- Xiong, Z.Q.; Wang, J.F.; Hao, Y.Y.; Wang, Y. Recent advances in the discovery and development of marine microbial natural products. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef]
- Xiong, Z.-Q.; Zhang, Z.-P.; Li, J.-H.; Wei, S.-J.; Tu, G.-Q. Characterization of Streptomyces padanus JAU4234, a producer of actinomycin X2, fungichromin, and a new polyene macrolide antibiotic. Appl. Environ. Microbiol. 2012, 78, 589–592. [Google Scholar] [CrossRef]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Badawy, M.E.I.; Rabea, E.I. Current Applications in Food Preservation Based on Marine Biopolymers. In Polymers for Food Applications; Gutiérrez, T.J., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 609–650. [Google Scholar]
- Venugopal, V. Marine Polysaccharides: Food Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Appeltans, W.; Decock, W.; Vanhoorne, B.; Hernandez, F.; Bouchet, P.; Boxshall, G.; Fauchald, K.; Gordon, D.; Poore, G.; Van Soest, R. The World Register of Marine Species: An Authoritative, Open-Access Web-Resource for All Marine Species. Available online: http://marinespecies.org/ (accessed on 20 August 2019).
- Bisby, F.; Roskov, Y.; Culham, A.; Orrell, T.; Nicolson, D.; Paglinawan, L.; Bailly, N.; Appeltans, W.; Kirk, P.; Bourgoin, T.; et al. Species 2000 & ITIS Catalogue of Life, 2012 Annual Checklist. Available online: www.catalogueoflife.org/col/ (accessed on 20 August 2019).
- Patterson, D.; Mozzherin, D.; Shorthouse, D.P.; Thessen, A. Challenges with using names to link digital biodiversity information. Biodivers. Data J. 2016, 4, e8080. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.; Kroh, A.; Ahyong, S.; Bailly, N.; Boyko, C.B.; Brandão, S.N.; Gofas, S.; Hooper, J.N.A.; Hernandez, F.; Holovachov, O.; et al. World Register of Marine Species. Available online: http://www.marinespecies.org (accessed on 20 August 2019).
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.; Worm, B. How many species are there on Earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.R.; Cinquegrani, M. Firms Plunge into the Sea. Marine Biotechnology Industry, a First Investigation. Front. Mar. Sci. 2016, 2, 124. [Google Scholar] [CrossRef]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Leclère, V.; Weber, T.; Jacques, P.; Pupin, M. Bioinformatics Tools for the Discovery of New Nonribosomal Peptides. Methods Mol. Biol. 2016, 1401, 209–232. [Google Scholar] [PubMed]
- Lorente, A.; Makowski, K.; Albericio, F.; Álvarez, M. Bioactive marine polyketides as potential and promising drugs. Ann. Mar. Biol. Res. 2014, 1, 1–10. [Google Scholar]
- Carteni, F.; Bonanomi, G.; Giannino, F.; Incerti, G.; Vincenot, C.E.; Chiusano, M.L.; Mazzoleni, S. Self-DNA inhibitory effects: Underlying mechanisms and ecological implications. Plant Signal. Behav. 2016, 11, e1158381. [Google Scholar] [CrossRef]
- Mazzoleni, S.; Bonanomi, G.; Incerti, G.; Chiusano, M.L.; Termolino, P.; Mingo, A.; Senatore, M.; Giannino, F.; Carteni, F.; Rietkerk, M.; et al. Inhibitory and toxic effects of extracellular self-DNA in litter: A mechanism for negative plant-soil feedbacks? New Phytol. 2015, 205, 1195–1210. [Google Scholar] [CrossRef]
- Mazzoleni, S.; Carteni, F.; Bonanomi, G.; Senatore, M.; Termolino, P.; Giannino, F.; Incerti, G.; Rietkerk, M.; Lanzotti, V.; Chiusano, M.L. Inhibitory effects of extracellular self-DNA: A general biological process? New Phytol. 2015, 206, 127–132. [Google Scholar] [CrossRef]
- Liang, X.; Luo, D.; Luesch, H. Advances in exploring the therapeutic potential of marine natural products. Pharmacol. Res. 2019, 147, 104373. [Google Scholar] [CrossRef] [PubMed]
- Iskar, M.; Zeller, G.; Zhao, X.M.; van Noort, V.; Bork, P. Drug discovery in the age of systems biology: The rise of computational approaches for data integration. Curr. Opin. Biotechnol. 2012, 23, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, P.A. What is the relevance of bioinformatics to pharmacology? Trends Pharmacol. Sci. 2003, 24, 434–439. [Google Scholar] [CrossRef]
- Ortega, S.S.; Cara, L.C.; Salvador, M.K. In silico pharmacology for a multidisciplinary drug discovery process. Drug Metab. Drug Interact. 2012, 27, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Katara, P. Role of bioinformatics and pharmacogenomics in drug discovery and development process. Netw. Modeling Anal. Health Inform. Bioinform. 2013, 2, 225–230. [Google Scholar] [CrossRef]
- Ambrosino, L.; Colantuono, C.; Monticolo, F.; Chiusano, M.L. Bioinformatics Resources for Plant Genomics: Opportunities and Bottlenecks in The -omics Era. Curr. Issues Mol. Biol. 2017, 71–88. [Google Scholar] [CrossRef]
- Esposito, A.; Colantuono, C.; Ruggieri, V.; Chiusano, M.L. Bioinformatics for agriculture in the Next-Generation sequencing era. Chem. Biol. Technol. Agric. 2016, 3, 1–12. [Google Scholar] [CrossRef]
- Trindade, M.; van Zyl, L.J.; Navarro-Fernández, J.; Abd Elrazak, A. Targeted metagenomics as a tool to tap into marine natural product diversity for the discovery and production of drug candidates. Front. Microbiol. 2015, 6, 890. [Google Scholar] [CrossRef] [PubMed]
- Lauritano, C.; Ianora, A. Grand Challenges in Marine Biotechnology: Overview of Recent EU-Funded Projects. In Grand Challenges in Marine Biotechnology; Springer: New York, NY, USA, 2018; pp. 425–449. [Google Scholar]
- Hartmann, E.M.; Durighello, E.; Pible, O.; Nogales, B.; Beltrametti, F.; Bosch, R.; Christie-Oleza, J.A.; Armengaud, J. Proteomics meets blue biotechnology: A wealth of novelties and opportunities. Mar. Genom. 2014, 17, 35–42. [Google Scholar] [CrossRef]
- Lacerda, C.M.; Reardon, K.F. Environmental proteomics: Applications of proteome profiling in environmental microbiology and biotechnology. Brief. Funct. Genom. Proteom. 2009, 8, 75–87. [Google Scholar] [CrossRef]
- Huo, L.; Hug, J.J.; Fu, C.; Bian, X.; Zhang, Y.; Müller, R. Heterologous expression of bacterial natural product biosynthetic pathways. Nat. Prod. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- de Pascale, D.; De Santi, C.; Fu, J.; Landfald, B. The microbial diversity of Polar environments is a fertile ground for bioprospecting. Mar. Genom. 2012, 8, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; O’Leary, N.D.; Kiran, G.S.; Morrissey, J.P.; O’Gara, F.; Selvin, J.; Dobson, A.D. Functional metagenomic strategies for the discovery of novel enzymes and biosurfactants with biotechnological applications from marine ecosystems. J. Appl. Microbiol. 2011, 111, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Parte, S.; Sirisha, V.L.; D’Souza, J.S. Chapter Four—Biotechnological Applications of Marine Enzymes from Algae, Bacteria, Fungi, and Sponges. In Advances in Food and Nutrition Research; Kim, S.-K., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 80, pp. 75–106. [Google Scholar]
- Gross, L. Untapped Bounty: Sampling the Seas to Survey Microbial Biodiversity. PLoS Biol. 2007, 5, e85. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 2004, 304, 66. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Venkatesan, J. Introduction to Marine Biotechnology. In Springer Handbook of Marine Biotechnology; Kim, S.-K., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2015; pp. 1–10. [Google Scholar]
- Medema, M.H.; Fischbach, M.A. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Lin, W.R.; Tan, S.I.; Hsiang, C.C.; Sung, P.K.; Ng, I.S. Challenges and opportunity of recent genome editing and multi-omics in cyanobacteria and microalgae for biorefinery. Bioresour. Technol. 2019, 291, 121932. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122. [Google Scholar] [CrossRef]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, M.; Thumberger, T.; del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [PubMed]
- Saraswathy, N.; Ramalingam, P. 7—Genome sequencing methods. In Concepts and Techniques in Genomics and Proteomics; Woodhead Publishing: Cambridge, UK, 2011; pp. 95–107. [Google Scholar]
- Magi, A.; Benelli, M.; Gozzini, A.; Girolami, F.; Torricelli, F.; Brandi, M.L. Bioinformatics for next generation sequencing data. Genes 2010, 1, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Dehal, P.; Satou, Y.; Campbell, R.K.; Chapman, J.; Degnan, B.; De Tomaso, A.; Davidson, B.; Di Gregorio, A.; Gelpke, M.; Goodstein, D.M.; et al. The Draft Genome of Ciona intestinalis: Insights into Chordate and Vertebrate Origins. Science 2002, 298, 2157. [Google Scholar] [CrossRef]
- Sea Urchin Genome Sequencing, C.; Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C.; Angerer, L.M.; Arnone, M.I.; Burgess, D.R.; et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef]
- Carreras, C.; Ordóñez, V.; Zane, L.; Kruschel, C.; Nasto, I.; Macpherson, E.; Pascual, M. Population genomics of an endemic Mediterranean fish: Differentiation by fine scale dispersal and adaptation. Sci. Rep. 2017, 7, 43417. [Google Scholar] [CrossRef]
- Igarashi, Y.; Zhang, H.; Tan, E.; Sekino, M.; Yoshitake, K.; Kinoshita, S.; Mitsuyama, S.; Yoshinaga, T.; Chow, S.; Kurogi, H.; et al. Whole-Genome Sequencing of 84 Japanese Eels Reveals Evidence against Panmixia and Support for Sympatric Speciation. Genes 2018, 9, 10. [Google Scholar] [CrossRef]
- Malde, K.; Seliussen, B.B.; Quintela, M.; Dahle, G.; Besnier, F.; Skaug, H.J.; Øien, N.; Solvang, H.K.; Haug, T.; Skern-Mauritzen, R.; et al. Whole genome resequencing reveals diagnostic markers for investigating global migration and hybridization between minke whale species. BMC Genom. 2017, 18, 76. [Google Scholar] [CrossRef]
- Xu, S.; Song, N.; Zhao, L.; Cai, S.; Han, Z.; Gao, T. Genomic evidence for local adaptation in the ovoviviparous marine fish Sebastiscus marmoratus with a background of population homogeneity. Sci. Rep. 2017, 7, 1562. [Google Scholar] [CrossRef]
- Xu, S.; Zhao, L.; Xiao, S.; Gao, T. Whole genome resequencing data for three rockfish species of Sebastes. Sci. Data 2019, 6, 97. [Google Scholar] [CrossRef]
- Cochrane, G.; Karsch-Mizrachi, I.; Takagi, T. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2016, 44, D48–D50. [Google Scholar] [CrossRef] [PubMed]
- CRIBI Database. Available online: http://genomes.cribi.unipd.it (accessed on 25 January 2018).
- NCBI_Resource_Coordinators, Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- Mashima, J.; Kodama, Y.; Kosuge, T.; Fujisawa, T.; Katayama, T.; Nagasaki, H.; Okuda, Y.; Kaminuma, E.; Ogasawara, O.; Okubo, K.; et al. DNA data bank of Japan (DDBJ) progress report. Nucleic Acids Res. 2016, 44, D51–D57. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Barrett, T.; Clark, K.; Gevorgyan, R.; Gorelenkov, V.; Gribov, E.; Karsch-Mizrachi, I.; Kimelman, M.; Pruitt, K.D.; Resenchuk, S.; Tatusova, T.; et al. BioProject and BioSample databases at NCBI: Facilitating capture and organization of metadata. Nucleic Acids Res. 2012, 40, D57–D63. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update--simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tárraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European Nucleotide Archive. Nucleic Acids Res. 2011, 39, D28–D31. [Google Scholar] [CrossRef]
- Kaminuma, E.; Mashima, J.; Kodama, Y.; Gojobori, T.; Ogasawara, O.; Okubo, K.; Takagi, T.; Nakamura, Y. DDBJ launches a new archive database with analytical tools for next-generation sequence data. Nucleic Acids Res. 2010, 38, D33–D38. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R.; et al. IMG/M v.5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019, 47, D666–D677. [Google Scholar] [CrossRef] [PubMed]
- Mende, D.R.; Letunic, I.; Huerta-Cepas, J.; Li, S.S.; Forslund, K.; Sunagawa, S.; Bork, P. proGenomes: A resource for consistent functional and taxonomic annotations of prokaryotic genomes. Nucleic Acids Res. 2017, 45, D529–D534. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Boguski, M.S.; Lowe, T.M.; Tolstoshev, C.M. dbEST—Database for “expressed sequence tags”. Nat. Genet. 1993, 4, 332–333. [Google Scholar] [CrossRef]
- Clarke, K.; Yang, Y.; Marsh, R.; Xie, L.; Zhang, K.K. Comparative analysis of de novo transcriptome assembly. Sci. China Life Sci. 2013, 56, 156–162. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. Rna 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb Protoc 2015, 2015, 951–969. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.J. DNA Microarray Technology: Devices, Systems, and Applications. Annu. Rev. Biomed. Eng. 2002, 4, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Bostan, H.; Chiusano, M.L. NexGenEx-Tom: A gene expression platform to investigate the functionalities of the tomato genome. BMC Plant Biol. 2015, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, P.; Llorens, C.; Roig, F.J.; Robaina, R.R. Analysis of the Transcriptome of the Red Seaweed Grateloupia imbricata with Emphasis on Reproductive Potential. Mar. Drugs 2018, 16, 490. [Google Scholar] [CrossRef] [PubMed]
- Huerlimann, R.; Wade, N.M.; Gordon, L.; Montenegro, J.D.; Goodall, J.; McWilliam, S.; Tinning, M.; Siemering, K.; Giardina, E.; Donovan, D.; et al. De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome. Sci. Rep. 2018, 8, 13553. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Sun, J.; Xu, T.; Chen, C.; Tian, R.; Qiu, J.-W.; Qian, P.-Y. De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genom. 2018, 19, 394. [Google Scholar] [CrossRef]
- Lauritano, C.; De Luca, D.; Ferrarini, A.; Avanzato, C.; Minio, A.; Esposito, F.; Ianora, A. De novo transcriptome of the cosmopolitan dinoflagellate Amphidinium carterae to identify enzymes with biotechnological potential. Sci. Rep. 2017, 7, 11701. [Google Scholar] [CrossRef]
- Onimaru, K.; Tatsumi, K.; Shibagaki, K.; Kuraku, S. A de novo transcriptome assembly of the zebra bullhead shark, Heterodontus zebra. Sci. Data 2018, 5, 180197. [Google Scholar] [CrossRef]
- Roncalli, V.; Cieslak, M.C.; Sommer, S.A.; Hopcroft, R.R.; Lenz, P.H. De novo transcriptome assembly of the calanoid copepod Neocalanus flemingeri: A new resource for emergence from diapause. Mar. Genom. 2018, 37, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lauritano, C.; De Luca, D.; Amoroso, M.; Benfatto, S.; Maestri, S.; Racioppi, C.; Esposito, F.; Ianora, A. New molecular insights on the response of the green alga Tetraselmis suecica to nitrogen starvation. Sci. Rep. 2019, 9, 3336. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Zhu, Y.; Sun, Y.; Zhao, T.; Huang, Y.; Shi, Q. High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 3901. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Elucidation of the molecular envenomation strategy of the cone snail Conus geographus through transcriptome sequencing of its venom duct. Bmc Genom. 2012, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Peng, C.; Zhu, Y.; Fan, C.; Jiang, H.; Chen, J.; Cao, Y.; Shi, Q. High-Throughput Identification and Analysis of Novel Conotoxins from Three Vermivorous Cone Snails by Transcriptome Sequencing. Mar. Drugs 2019, 17, 193. [Google Scholar] [CrossRef] [PubMed]
- Rivera-de-Torre, E.; Palacios-Ortega, J.; Gavilanes, J.G.; Martínez-del-Pozo, Á.; García-Linares, S. Pore-Forming Proteins from Cnidarians and Arachnids as Potential Biotechnological Tools. Toxins 2019, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Huang, Y.; Baumann, K.; Fry, B.G.; Shi, Q. From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Mar. Drugs 2017, 15, 103. [Google Scholar] [CrossRef]
- Kumar, A.; Sørensen, J.L.; Hansen, F.T.; Arvas, M.; Syed, M.F.; Hassan, L.; Benz, J.P.; Record, E.; Henrissat, B.; Pöggeler, S.; et al. Genome Sequencing and analyses of Two Marine Fungi from the North Sea Unraveled a Plethora of Novel Biosynthetic Gene Clusters. Sci. Rep. 2018, 8, 10187. [Google Scholar] [CrossRef]
- Morlighem, J.-É.R.L.; Huang, C.; Liao, Q.; Braga Gomes, P.; Daniel Pérez, C.; De Brandão Prieto-da-Silva, Á.R.; Ming-Yuen Lee, S.; Rádis-Baptista, G. The Holo-Transcriptome of the Zoantharian Protopalythoa variabilis (Cnidaria: Anthozoa): A Plentiful Source of Enzymes for Potential Application in Green Chemistry, Industrial and Pharmaceutical Biotechnology. Mar. Drugs 2018, 16, 207. [Google Scholar] [CrossRef]
- Smith, D.R.M.; Uria, A.R.; Helfrich, E.J.N.; Milbredt, D.; van Pee, K.H.; Piel, J.; Goss, R.J.M. An Unusual Flavin-Dependent Halogenase from the Metagenome of the Marine Sponge Theonella swinhoei WA. ACS Chem. Biol. 2017, 12, 1281–1287. [Google Scholar] [CrossRef]
- Sarian, F.D.; Janecek, S.; Pijning, T.; Ihsanawati; Nurachman, Z.; Radjasa, O.K.; Dijkhuizen, L.; Natalia, D.; van der Maarel, M.J. A new group of glycoside hydrolase family 13 alpha-amylases with an aberrant catalytic triad. Sci. Rep. 2017, 7, 44230. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Marine microorganisms as a promising and sustainable source of bioactive molecules. Marine Environ. Res. 2017, 128, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Amos, G.C.A.; Awakawa, T.; Tuttle, R.N.; Letzel, A.-C.; Kim, M.C.; Kudo, Y.; Fenical, W.; Moore, B.S.; Jensen, P.R. Comparative transcriptomics as a guide to natural product discovery and biosynthetic gene cluster functionality. Proc. Natl. Acad. Sci. USA 2017, 114, E11121. [Google Scholar] [CrossRef] [PubMed]
- Gorson, J.; Ramrattan, G.; Verdes, A.; Wright, E.M.; Kantor, Y.; Rajaram Srinivasan, R.; Musunuri, R.; Packer, D.; Albano, G.; Qiu, W.G.; et al. Molecular Diversity and Gene Evolution of the Venom Arsenal of Terebridae Predatory Marine Snails. Genome Biol. Evol. 2015, 7, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 2015, 109. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Walker, D.L.; Bhagwate, A.V.; Baheti, S.; Smalley, R.L.; Hilker, C.A.; Sun, Z.; Cunningham, J.M. DNA methylation profiling: Comparison of genome-wide sequencing methods and the Infinium Human Methylation 450 Bead Chip. Epigenomics 2015, 7, 1287–1302. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-Length mRNA-Seq from single cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Brozovic, M.; Dantec, C.; Dardaillon, J.; Dauga, D.; Faure, E.; Gineste, M.; Louis, A.; Naville, M.; Nitta, K.R.; Piette, J.; et al. ANISEED 2017: Extending the integrated ascidian database to the exploration and evolutionary comparison of genome-scale datasets. Nucleic Acids Res. 2018, 46, D718–D725. [Google Scholar] [CrossRef]
- Kudtarkar, P.; Cameron, R.A. Echinobase: An expanding resource for echinoderm genomic information. Database 2017, 2017, bax074. [Google Scholar] [CrossRef]
- Wang, J.; Kong, L.; Gao, G.; Luo, J. A brief introduction to web-based genome browsers. Brief. Bioinform. 2012, 14, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Dessimoz, C. Phylogenetic and functional assessment of orthologs inference projects and methods. Plos Comput. Biol. 2009, 5, e1000262. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Dessimoz, C. Inferring orthology and paralogy. Methods Mol. Biol. 2012, 855, 259–279. [Google Scholar]
- Ambrosino, L.; Chiusano, M.L. Transcriptologs: A Transcriptome-Based Approach to Predict Orthology Relationships. Bioinform. Biol. Insights 2017, 11, 1–8. [Google Scholar] [CrossRef]
- Dolinski, K.; Botstein, D. Orthology and functional conservation in eukaryotes. Annu. Rev. Genet. 2007, 41, 465–507. [Google Scholar] [CrossRef]
- Gabaldon, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; Koonin, E.V. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002, 18, 619–620. [Google Scholar] [CrossRef]
- Ambrosino, L.; Ruggieri, V.; Bostan, H.; Miralto, M.; Vitulo, N.; Zouine, M.; Barone, A.; Bouzayen, M.; Frusciante, L.; Pezzotti, M.; et al. Multilevel comparative bioinformatics to investigate evolutionary relationships and specificities in gene annotations: An example for tomato and grapevine. BMC Bioinform. 2018, 19, 435. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.C.J.; Purnell, M.A. Genome duplication, extinction and vertebrate evolution. Trends Ecol. Evol. 2005, 20, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Snipen, L.; Liland, K.H. micropan: An R-package for microbial pan-genomics. Bmc Bioinform. 2015, 16, 79. [Google Scholar] [CrossRef]
- Jun, S.R.; Robeson, M.S.; Hauser, L.J.; Schadt, C.W.; Gorin, A.A. PanFP: Pangenome-based functional profiles for microbial communities. BMC Res. Notes 2015, 8, 479. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Zhang, Z.; Zhao, Y.; Sun, C.; Yang, M.; Wang, J.; Liu, Q.; Zhang, B.; Chen, M.; et al. PGAweb: A Web Server for Bacterial Pan-Genome Analysis. Front. Microbiol. 2018, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, C.; Hatte, V.; Andrieu, C.; Barlet, L.; Cologne, A.; Oliveira, R.D.; Ferrato-Berberian, L.; Gardon, H.e.; Lauber, D.; Molinier, M.e.; et al. PanGeneHome: A Web Interface to Analyze Microbial Pangenomes. J. Bioinf. Com. Sys. Bio. 2017, 1, 108. [Google Scholar]
- Rouli, L.; Mbengue, M.; Robert, C.; Ndiaye, M.; La Scola, B.; Raoult, D. Genomic analysis of three African strains of Bacillus anthracis demonstrates that they are part of the clonal expansion of an exclusively pathogenic bacterium. New Microbes New Infect. 2014, 2, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity. Genome Biol. Evol. 2018, 11, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Fondi, M.; Orlandini, V.; Perrin, E.; Maida, I.; de Pascale, D.; Tutino, M.L.; Parrilli, E.; Lo Giudice, A.; Filloux, A.; et al. The pangenome of (Antarctic) Pseudoalteromonas bacteria: Evolutionary and functional insights. BMC Genom. 2017, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Park, C.J.; Andam, C.P. Within-Species Genomic Variation and Variable Patterns of Recombination in the Tetracycline Producer Streptomyces rimosus. Front. Microbiol. 2019, 10, 552. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, J.; Millan-Aguinaga, N.; Zhang, J.J.; O’Neill, E.C.; Ugalde, J.A.; Jensen, P.R.; Mantovani, S.M.; Moore, B.S. Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-directed Genome Mining. ACS Chem. Biol. 2015, 10, 2841–2849. [Google Scholar] [CrossRef]
- Chiusano, M.L. On the Multifaceted Aspects of Bioinformatics in the Next Generation Era: The Run that must keep the Quality. Next Gener. Seq. Applic 2015, 2, e106. [Google Scholar] [CrossRef]
- NCBI. The NCBI Eukaryotic Genome Annotation Pipeline. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/ (accessed on 20 August 2019).
- ENSEMBL. Gene Annotation in Ensembl. Available online: https://www.ensembl.org/info/genome/genebuild/genome_annotation.html (accessed on 20 August 2019).
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef]
- Colantuono, C.; Miralto, M.; Sangiovanni, M.; Ambrosino, L.; Chiusano, M.L. GENOMA: A Multilevel Platform for Marine Biology; PeerJ Preprints: Madera, CA, USA, 2018. [Google Scholar]
- Barone, R.; De Santi, C.; Palma Esposito, F.; Tedesco, P.; Galati, F.; Visone, M.; Di Scala, A.; De Pascale, D. Marine metagenomics, a valuable tool for enzymes and bioactive compounds discovery. Front. Mar. Sci. 2014, 1, 38. [Google Scholar] [CrossRef]
- Madhavan, A.; Sindhu, R.; Parameswaran, B.; Sukumaran, R.K.; Pandey, A. Metagenome Analysis: A Powerful Tool for Enzyme Bioprospecting. Appl. Biochem. Biotechnol. 2017, 183, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. Mmbr 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Béjà, O.; Aravind, L.; Koonin, E.V.; Suzuki, M.T.; Hadd, A.; Nguyen, L.P.; Jovanovich, S.B.; Gates, C.M.; Feldman, R.A.; Spudich, J.L.; et al. Bacterial Rhodopsin: Evidence for a New Type of Phototrophy in the Sea. Science 2000, 289. [Google Scholar] [CrossRef]
- Béjà, O.; Spudich, E.N.; Spudich, J.L.; Leclerc, M.; DeLong, E.F. Proteorhodopsin phototrophy in the ocean. Nature 2001, 411, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zayed, A.A.; Conceicao-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Recent progress and new challenges in metagenomics for biotechnology. Biotechnol. Lett. 2010, 32, 1351–1359. [Google Scholar] [CrossRef]
- Roumpeka, D.D.; Wallace, R.J.; Escalettes, F.; Fotheringham, I.; Watson, M. A Review of Bioinformatics Tools for Bio-Prospecting from Metagenomic Sequence Data. Front Genet 2017, 8, 23. [Google Scholar] [CrossRef]
- Teeling, H.; Glockner, F.O. Current opportunities and challenges in microbial metagenome analysis—A bioinformatic perspective. Brief. Bioinform. 2012, 13, 728–742. [Google Scholar] [CrossRef]
- Bremges, A.; McHardy, A.C. Critical Assessment of Metagenome Interpretation Enters the Second Round. mSystems 2018, 3, 4. [Google Scholar] [CrossRef]
- Dombrowski, N.; Teske, A.P.; Baker, B.J. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat. Commun. 2018, 9, 4999. [Google Scholar] [CrossRef]
- Seitz, K.W.; Dombrowski, N.; Eme, L.; Spang, A.; Lombard, J.; Sieber, J.R.; Teske, A.P.; Ettema, T.J.G.; Baker, B.J. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun. 2019, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [PubMed]
- Machado, H.; Sonnenschein, E.C.; Melchiorsen, J.; Gram, L. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genom. 2015, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.H.; Gregoracci, G.B.; Walter, J.M.; Thompson, C.C.; Thompson, F.L. Metagenomics Sheds Light on the Ecology of Marine Microbes and Their Viruses. Trends Microbiol. 2018, 26, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Cameron Thrash, J.; Temperton, B.; Swan, B.K.; Landry, Z.C.; Woyke, T.; DeLong, E.F.; Stepanauskas, R.; Giovannoni, S.J. Single-cell enabled comparative genomics of a deep ocean SAR11 bathytype. ISME J. 2014, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Tsementzi, D.; Wu, J.; Deutsch, S.; Nath, S.; Rodriguez, R.L.; Burns, A.S.; Ranjan, P.; Sarode, N.; Malmstrom, R.R.; Padilla, C.C.; et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature 2016, 536, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Varrella, S.; Tangherlini, M.; Rastelli, E.; Dell’Anno, A.; Danovaro, R.; Corinaldesi, C. Marine Fungi: Biotechnological Perspectives from Deep-Hypersaline Anoxic Basins. Diversity 2019, 11, 113. [Google Scholar] [CrossRef]
- Orsi, W.D.; Barker Jorgensen, B.; Biddle, J.F. Transcriptional analysis of sulfate reducing and chemolithoautotrophic sulfur oxidizing bacteria in the deep subseafloor. Environ. Microbiol. Rep. 2016, 8, 452–460. [Google Scholar] [CrossRef]
- Lau, M.C.Y.; Harris, R.L.; Oh, Y.; Yi, M.J.; Behmard, A.; Onstott, T.C. Taxonomic and Functional Compositions Impacted by the Quality of Metatranscriptomic Assemblies. Front. Microbiol. 2018, 9, 1235. [Google Scholar] [CrossRef]
- Mitchell, A.; Bucchini, F.; Cochrane, G.; Denise, H.; ten Hoopen, P.; Fraser, M.; Pesseat, S.; Potter, S.; Scheremetjew, M.; Sterk, P.; et al. EBI metagenomics in 2016--An expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Res. 2016, 44, D595–D603. [Google Scholar] [CrossRef]
- Consortium, G.O. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Wilke, A.; Bischof, J.; Gerlach, W.; Glass, E.; Harrison, T.; Keegan, K.P.; Paczian, T.; Trimble, W.L.; Bagchi, S.; Grama, A.; et al. The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2016, 44, D590–D594. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC bioinformatics 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Klemetsen, T.; Raknes, I.A.; Fu, J.; Agafonov, A.; Balasundaram, S.V.; Tartari, G.; Robertsen, E.; Willassen, N.P. The MAR databases: development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2017, 46, D692–D699. [Google Scholar] [CrossRef]
- Robertsen, E.; Denise, H.; Mitchell, A.; Finn, R.; Bongo, L.; Willassen, N. ELIXIR pilot action: Marine metagenomics ? towards a domain specific set of sustainable services [version 1; peer review: 1 approved, 2 approved with reservations]. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Bowler, C.; de Vargas, C.; Gorsky, G.; Karsenti, E.; Wincker, P. Tara Oceans. Tara Oceans studies plankton at planetary scale. Introduction. Science 2015, 348, 873. [Google Scholar] [CrossRef]
- Anderson, R.F.; Mawji, E.; Cutter, G.A.; MEASURES, C.I.; Jeandel, C. GEOTRACES: changing the way we explore ocean chemistry. Oceanography 2014, 27, 50–61. [Google Scholar]
- Biller, S.J.; Berube, P.M.; Dooley, K.; Williams, M.; Satinsky, B.M.; Hackl, T.; Hogle, S.L.; Coe, A.; Bergauer, K.; Bouman, H.A.; et al. Marine microbial metagenomes sampled across space and time. Scientific Data 2018, 5, 180176. [Google Scholar] [CrossRef]
- Karl, D.M.; Church, M.J. Microbial oceanography and the Hawaii Ocean Time-series programme. Nat. Rev. Microbiol. 2014, 12, 699–713. [Google Scholar] [CrossRef]
- Steinberg, D.K.; Carlson, C.A.; Bates, N.R.; Johnson, R.J.; Michaels, A.F.; Knap, A.H. Overview of the US JGOFS Bermuda Atlantic Time-series Study (BATS): A decade-scale look at ocean biology and biogeochemistry. Deep Sea Res. Part II Top. Studies Oceanogr. 2001, 48, 1405–1447. [Google Scholar] [CrossRef]
- Villar, E.; Vannier, T.; Vernette, C.; Lescot, M.; Cuenca, M.; Alexandre, A.; Bachelerie, P.; Rosnet, T.; Pelletier, E.; Sunagawa, S.; et al. The Ocean Gene Atlas: exploring the biogeography of plankton genes online. Nucleic Acids Res. 2018, 46, W289–W295. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. The ISME Journal 2019. [Google Scholar] [CrossRef] [PubMed]
- Tangherlini, M.; Miralto, M.; Colantuono, C.; Sangiovanni, M.; Dell’ Anno, A.; Corinaldesi, C.; Danovaro, R.; Chiusano, M.L. GLOSSary: The GLobal Ocean 16S subunit web accessible resource. BMC Bioinform. 2018, 19, 443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed]
- Tchigvintsev, A.; Tran, H.; Popovic, A.; Kovacic, F.; Brown, G.; Flick, R.; Hajighasemi, M.; Egorova, O.; Somody, J.C.; Tchigvintsev, D.; et al. The environment shapes microbial enzymes: five cold-active and salt-resistant carboxylesterases from marine metagenomes. Appl. Microbiol. Biotechnol. 2015, 99, 2165–2178. [Google Scholar] [CrossRef]
- Han, X.; Hou, L.; Hou, J.; Zhang, Y.; Li, H.; Li, W. Heterologous Expression of a VioA Variant Activates Cryptic Compounds in a Marine-Derived Brevibacterium Strain. Marine Drugs 2018, 16, 191. [Google Scholar] [CrossRef]
- Feder, M.E.; Walser, J.C. The biological limitations of transcriptomics in elucidating stress and stress responses. J. Evol. Biol. 2005, 18, 901–910. [Google Scholar] [CrossRef]
- Tomanek, L. Proteomics to study adaptations in marine organisms to environmental stress. J. Proteom. 2014, 105, 92–106. [Google Scholar] [CrossRef]
- Slattery, M.; Ankisetty, S.; Corrales, J.; Marsh-Hunkin, K.E.; Gochfeld, D.J.; Willett, K.L.; Rimoldi, J.M. Proteomics: A Critical Assessment of an Emerging Technology. J. Nat. Prod. 2012, 75, 1833–1877. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Aslanian, A.; Yates, J.R., 3rd. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, D.; Villard, C.; Lafitte, D. Advances in top-down proteomics for disease biomarker discovery. J. Proteom. 2011, 74, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.E.; McLuckey, S.A. ’Top down’ protein characterization via tandem mass spectrometry. J. Mass Spectrom. 2002, 37, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, A.; Marino, F.; Post, H.; van den Toorn, H.W.P.; Mohammed, S.; Heck, A.J.R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, D.J.; Klima, J.; Gardner, W.; Kelleher, K.J. Adolescent behavioral risk screening and use of health services. Adm. Policy Ment. Health Ment. Health Serv. Res. 2009, 36, 374. [Google Scholar] [CrossRef]
- Forbes, A.J.; Mazur, M.T.; Patel, H.M.; Walsh, C.T.; Kelleher, N.L. Toward efficient analysis of >70 kDa proteins with 100% sequence coverage. Proteomics 2001, 1, 927–933. [Google Scholar] [CrossRef]
- Wu, S.L.; Kim, J.; Hancock, W.S.; Karger, B. Extended Range Proteomic Analysis (ERPA): A new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications-comprehensive analysis of beta-casein and epidermal growth factor receptor (EGFR). J. Proteome Res. 2005, 4, 1155–1170. [Google Scholar] [CrossRef]
- Sidoli, S.; Lin, S.; Karch, K.R.; Garcia, B.A. Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Anal. Chem. 2015, 87, 3129–3133. [Google Scholar] [CrossRef]
- Sidoli, S.; Schwammle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hidalgo, J.C.; Lopez-Bustins, J.A.; Štepánek, P.; Martin-Vide, J.; de Luis, M. Monthly precipitation trends on the Mediterranean fringe of the Iberian Peninsula during the second-half of the twentieth century (1951–2000). Int. J. Climatol. 2009, 29, 1415–1429. [Google Scholar] [CrossRef]
- Taouatas, N.; Drugan, M.M.; Heck, A.J.; Mohammed, S. Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nat. Methods 2008, 5, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Pérez, D.; Campos, A.; Alexei Rodríguez, A.; Turkina, M.V.; Ribeiro, T.; Osorio, H.; Vasconcelos, V.; Antunes, A. Proteomic Analyses of the Unexplored Sea Anemone Bunodactis verrucosa. Mar. Drugs 2018, 16, 42. [Google Scholar] [CrossRef]
- Cassiano, C.; Esposito, R.; Tosco, A.; Zampella, A.; D’Auria, M.V.; Riccio, R.; Casapullo, A.; Monti, M.C. Heteronemin, a marine sponge terpenoid, targets TDP-43, a key factor in several neurodegenerative disorders. Chem. Commun. 2014, 50, 406–408. [Google Scholar] [CrossRef]
- Biass, D.; Dutertre, S.; Gerbault, A.; Menou, J.L.; Offord, R.; Favreau, P.; Stocklin, R. Comparative proteomic study of the venom of the piscivorous cone snail Conus consors. J. Proteom. 2009, 72, 210–218. [Google Scholar] [CrossRef]
- Wase, N.V.; Wright, P.C. Systems biology of cyanobacterial secondary metabolite production and its role in drug discovery. Expert Opin. Drug Discov. 2008, 3, 903–929. [Google Scholar] [CrossRef]
- Knigge, T. Proteomics in Marine Organisms. Proteomics 2015, 15, 3921–3924. [Google Scholar] [CrossRef]
- Uniprot_consortium, UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinformatics 2014, 48, 3.13.1–3.13.16. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Congreve, M.; Murray, C.W.; Blundell, T.L. Structural biology and drug discovery. Drug Discov. Today 2005, 10, 895–907. [Google Scholar] [CrossRef]
- De Santi, C.; Ambrosino, L.; Tedesco, P.; Zhai, L.; Zhou, C.; Xue, Y.; Ma, Y.; de Pascale, D. Identification and characterization of a novel salt-tolerant esterase from a Tibetan glacier metagenomic library. Biotechnol. Prog. 2015, 31, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.J. Toward a molecular understanding of cold activity of enzymes from psychrophiles. Extrem. Life Under Extrem. Cond. 2000, 4, 83–90. [Google Scholar] [CrossRef]
- De Santi, C.; Tedesco, P.; Ambrosino, L.; Altermark, B.; Willassen, N.P.; de Pascale, D. A New Alkaliphilic Cold-Active Esterase from the Psychrophilic Marine Bacterium Rhodococcus sp.: Functional and Structural Studies and Biotechnological Potential. Appl. Biochem. Biotechnol. 2014. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bhattacharya, D. Does inclusion of residue-residue contact information boost protein threading? Proteins 2019, 87, 596–606. [Google Scholar] [CrossRef]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Delarue, M.; Koehl, P. Combined approaches from physics, statistics, and computer science for ab initio protein structure prediction: Ex unitate vires (unity is strength)? F1000Res 2018, 7, 1125. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Nishiyama, M.; Kitao, A. Molecular dynamics simulation of proteins under high pressure: Structure, function and thermodynamics. Biochim. Et Biophys. Acta. Gen. Subj. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Lohning, A.E.; Levonis, S.M.; Williams-Noonan, B.; Schweiker, S.S. A Practical Guide to Molecular Docking and Homology Modelling for Medicinal Chemists. Curr. Top. Med. Chem. 2017, 17, 2023–2040. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.F.; Wasim, A.A.; Iqbal, S.; Khan, M.A.; Lateef, M.; Iqbal, L. Synthesis, lipoxygenase inhibition activity and molecular docking of oxamide derivative. Pak. J. Pharm. Sci. 2019, 32 (Suppl. 3), 1253–1259. [Google Scholar]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. Blind docking of 260 protein-ligand complexes with EADock 2.0. J. Comput. Chem. 2009, 30, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Axelsson, H.; Almqvist, H.; Dahlgren, B.; Jonsson, M.; Lundback, T. Quantitative Interpretation of Intracellular Drug Binding and Kinetics Using the Cellular Thermal Shift Assay. Biochemistry 2018, 57, 6715–6725. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: a review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Manjasetty, B.A.; Büssow, K.; Panjikar, S.; Turnbull, A.P. Current methods in structural proteomics and its applications in biological sciences. 3 Biotech 2012, 2, 89–113. [Google Scholar] [CrossRef]
- Chandramouli, K.; Qian, P.-Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum Genom. Proteom. 2009, 2009, 239204. [Google Scholar] [CrossRef]
- Yalamanchili, H.K.; Wan, Y.W.; Liu, Z. Data Analysis Pipeline for RNA-seq Experiments: From Differential Expression to Cryptic Splicing. Curr. Protoc. Bioinformatics 2017, 59, 11.15.1–11.15.21. [Google Scholar] [CrossRef]
- Cruickshank, D.W. Remarks about protein structure precision. Acta Crystallogr. Sect. DBiol. Crystallogr. 1999, 55 Pt 3, 583–601. [Google Scholar] [CrossRef]
- Bundy, J.G.; Willey, T.L.; Castell, R.S.; Ellar, D.J.; Brindle, K.M. Discrimination of pathogenic clinical isolates and laboratory strains of Bacillus cereus by NMR-based metabolomic profiling. FEMS Microbiol. Lett. 2005, 242, 127–136. [Google Scholar] [CrossRef]
- Baltar, F.; Bayer, B.; Bednarsek, N.; Deppeler, S.; Escribano, R.; Gonzalez, C.E.; Hansman, R.L.; Mishra, R.K.; Moran, M.A.; Repeta, D.J.; et al. Towards Integrating Evolution, Metabolism, and Climate Change Studies of Marine Ecosystems. Trends Ecol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brierley, A.S.; Kingsford, M.J. Impacts of Climate Change on Marine Organisms and Ecosystems. Curr. Biol. 2009, 19, R602–R614. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, T.; Heer, D.; Begemann, B.; Zamboni, N. High-Throughput, Accurate Mass Metabolome Profiling of Cellular Extracts by Flow Injection–Time-of-Flight Mass Spectrometry. Anal. Chem. 2011, 83, 7074–7080. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Sekar, K.; Zamboni, N.; Sauer, U. Frontiers of high-throughput metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, Y.; Refaat, J.; Abdelmohsen, U.R.; Othman, E.M.; Stopper, H.; Fouad, M.A. Metabolomic profiling and biological investigation of the marine sponge-derived bacterium Rhodococcus sp. UA13. Phytochem. Anal. 2018, 29, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Amiri Moghaddam, J.; Crüsemann, M.; Alanjary, M.; Harms, H.; Dávila-Céspedes, A.; Blom, J.; Poehlein, A.; Ziemert, N.; König, G.M.; Schäberle, T.F. Analysis of the Genome and Metabolome of Marine Myxobacteria Reveals High Potential for Biosynthesis of Novel Specialized Metabolites. Sci. Rep. 2018, 8, 16600. [Google Scholar] [CrossRef]
- Oppong-Danquah, E.; Parrot, D.; Blümel, M.; Labes, A.; Tasdemir, D. Molecular Networking-Based Metabolome and Bioactivity Analyses of Marine-Adapted Fungi Co-cultivated With Phytopathogens. Front. Microbiol. 2018, 9, 2072. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef]
- Karp, P.D.; Latendresse, M.; Paley, S.M.; Krummenacker, M.; Ong, Q.D.; Billington, R.; Kothari, A.; Weaver, D.; Lee, T.; Subhraveti, P.; et al. Pathway Tools version 19.0 update: Software for pathway/genome informatics and systems biology. Brief. Bioinform. 2016, 17, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Latendresse, M.; Caspi, R. The pathway tools pathway prediction algorithm. Stand. Genom. Sci. 2011, 5, 424–429. [Google Scholar] [CrossRef] [PubMed]
- The_Royal_Society_of_Chemistry, Editorial: ChemSpider--a tool for Natural Products research. Nat. Prod. Rep. 2015, 32, 1163–1164. [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2014, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Barupal, D.K.; Fan, S.; Fiehn, O. Integrating bioinformatics approaches for a comprehensive interpretation of metabolomics datasets. Curr. Opin. Biotechnol. 2018, 54, 1–9. [Google Scholar] [CrossRef]
- Meier, R.; Ruttkies, C.; Treutler, H.; Neumann, S. Bioinformatics can boost metabolomics research. J. Biotechnol. 2017, 261, 137–141. [Google Scholar] [CrossRef]
- Riekeberg, E.; Powers, R. New frontiers in metabolomics: From measurement to insight. F1000Research 2017, 6, 1148. [Google Scholar] [CrossRef]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metab. Off. J. Metab. Soc. 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, R.; Jouffray, J.-B.; Wabnitz, C.C.C.; Sundström, E.; Österblom, H. Corporate control and global governance of marine genetic resources. Sci. Adv. 2018, 4, eaar5237. [Google Scholar] [CrossRef] [PubMed]
- Bourlat, S.J.; Borja, A.; Gilbert, J.; Taylor, M.I.; Davies, N.; Weisberg, S.B.; Griffith, J.F.; Lettieri, T.; Field, D.; Benzie, J.; et al. Genomics in marine monitoring: New opportunities for assessing marine health status. Mar. Pollut. Bull. 2013, 74, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.E.; Amaral-Zettler, L.A.; Fautin, D.G.; Paulay, G.; Rynearson, T.A.; Sosik, H.M.; Stachowicz, J.J. Envisioning a Marine Biodiversity Observation Network. BioScience 2013, 63, 350–361. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Mohanty, A.K.; Kumar, M.; Sreenivasamurthy, S.K.; Dey, G.; Nirujogi, R.S.; Pinto, S.M.; Madugundu, A.K.; Patil, A.H.; Advani, J.; et al. Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes. Genome Res. 2017, 27, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, R.N.; Demko, A.M.; Patin, N.V.; Kapono, C.A.; Donia, M.S.; Dorrestein, P.; Jensen, P.R. Detection of Natural Products and Their Producers in Ocean Sediments. Appl. Environ. Microbiol. 2019, 85, e02830-e18. [Google Scholar] [CrossRef]
- Gurevitch, J.; Koricheva, J.; Nakagawa, S.; Stewart, G. Meta-analysis and the science of research synthesis. Nature 2018, 555, 175. [Google Scholar] [CrossRef]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.B.; et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 2017, 14, 1023. [Google Scholar] [CrossRef]


{kind=link}
| Name | Section | Website |
|---|---|---|
| Scientific literature | ||
| MarinLit | Marine natural products literature | http://pubs.rsc.org/marinlit/ |
| Genomics and Transcriptomics | ||
| AmiGO | GO functional annotation repository and analyses services | http://amigo.geneontology.org/amigo |
| Aniseed | Genome browser and multi-omics repository for Ascidiacea | https://www.aniseed.cnrs.fr/aniseed/ |
| ArrayExpress | Next-generation-sequencing (NGS) data repository | https://www.ebi.ac.uk/arrayexpress/ |
| BLAST | Local alignment versus sequence database service | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| CCTop | CRISPR/Cas9 target prediction tool | https://crispr.cos.uni-heidelberg.de/ |
| CHOPCHOP | CRISPR/Cas9 and TALEN target Prediction Tool | http://chopchop.cbu.uib.no/ |
| dbEST | Expressed sequence tag (EST) sequence repository | https://www.ncbi.nlm.nih.gov/nucleotide/ |
| DDBJ | General multi-omics repository and analyses services | https://www.ddbj.nig.ac.jp/index-e.html |
| DRA | General NGS data repository | https://www.ddbj.nig.ac.jp/dra/index-e.html |
| Echinobase | Genome browser and multi-omics repository for Echinoderms | http://www.echinobase.org/Echinobase/ |
| Ensembl | General multi-omics repository and analyses services | https://www.ensembl.org/ |
| Gene Ontology | GO functional annotation repository and analyses services | http://geneontology.org/ |
| IMG/ER | Prokaryotic sequence and function repository | https://img.jgi.doe.gov/cgi-bin/mer/main.cgi |
| JGI | Multi-omics repository and analyses services | https://jgi.doe.gov/ |
| KEGG Genome | Genome sequence repository | https://www.genome.jp/kegg/genome.html |
| LAST | Long sequence alignment service | http://last.cbrc.jp/ |
| Mauve | Genome alignment via homolog blocks detection | http://darlinglab.org/mauve/ |
| MicroPan | Bacterial pangenome analysis library for R environment | https://cran.r-project.org/web/packages/micropan/index.html |
| NCBI | General multi-omics repository and analyses services | https://www.ncbi.nlm.nih.gov/ |
| OIST MGU | Genome browser and analyses services for 19 marine species | https://marinegenomics.oist.jp/ |
| PanFP | Bacterial pangenome-based functional profiles | https://github.com/srjun/PanFP |
| PGAWeb | Bacterial pangenome analyses service | http://pgaweb.vlcc.cn |
| ProGenomes | Prokaryotic sequence and functional repository | http://progenomes.embl.de/ |
| SRA | General NGS data repository | https://www.ncbi.nlm.nih.gov/sra |
| Metagenomics and metatranscriptomics | ||
| dbCAN | Automated carbohydrate-active enzyme annotation | http://bcb.unl.edu/dbCAN2/ |
| EBI Metagenomics | Microbiome sequence repository and analyses services | https://www.ebi.ac.uk/metagenomics/ |
| Geotraces | Marine key trace elements and isotopes data repository | http://www.geotraces.org/ |
| GLOSSary | Marine microbial sequence repository and analyses services | https://bioinfo.szn.it/glossary/ |
| KEGG MGENES | Annotated environmental gene catalog and analyses service | https://www.genome.jp/mgenes |
| Marine Metagenomics Portal | Marine microbiome repository and analyses services | https://mmp.sfb.uit.no/ |
| MG-RAST | Phylogenetic and functional analysis for metagenomics | https://www.mg-rast.org/ |
| Ocean Gene Atlas | Analytical service for marine planktonic organisms | http://tara-oceans.mio.osupytheas.fr/ocean-gene-atlas/ |
| Tara Oceans Database | Expedition specific raw reads sequence repository | https://www.ebi.ac.uk/services/tara-oceans-data |
| Proteomics and structural biology | ||
| AMBER | Molecular dynamics simulation program | http://ambermd.org/ |
| AutoDock | Molecular docking program | http://autodock.scripps.edu/ |
| AutoDock Vina | Multithreading program for molecular docking | http://vina.scripps.edu |
| CHARMM | Molecular dynamics simulations program | https://www.charmm.org/charmm/ |
| Desmond | Molecular dynamics simulations server | https://www.schrodinger.com/desmond |
| DOCK | Molecular docking server | http://dock.compbio.ucsf.edu/ |
| FlexX | Molecular docking server | https://www.biosolveit.de/FlexX/ |
| Glide | Molecular docking server | https://www.schrodinger.com/glide |
| GOLD | Molecular docking program | https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ |
| GROMACS | Molecular dynamics simulations program | http://www.gromacs.org |
| HHpred | Homology modelling server | https://toolkit.tuebingen.mpg.de/#/tools/hhpred |
| I-TASSER | Ab-initio structure prediction server | https://zhanglab.ccmb.med.umich.edu/I-TASSER/ |
| ICM | Molecular docking program | http://www.molsoft.com/docking.html |
| InterPro | Protein function repository and analytical services | https://www.ebi.ac.uk/interpro/ |
| LeDock | Molecular docking program | http://www.lephar.com/download.htm |
| Modeller | Homology modelling program | https://salilab.org/modeller/ |
| MOE-Dock | Molecular docking server | https://www.chemcomp.com/index.htm |
| NAMD | Molecular dynamics simulations program | http://www.ks.uiuc.edu/Research/namd/ |
| OpenMM | Molecular dynamics simulations program | http://openmm.org/ |
| PDB | Protein structure repository | https://www.rcsb.org/ |
| PFAM | Protein family repository | https://pfam.xfam.org/ |
| Phyre2 | Threading and ab-initio structure prediction server | http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index |
| RaptorX | Homology modelling and threading structure prediction server | http://raptorx.uchicago.edu |
| rDock | Molecular docking program | http://rdock.sourceforge.net/ |
| Robetta | Homology modelling and ab-initio structure prediction server | http://www.robetta.org/ |
| Surflex | Molecular docking program | http://www.jainlab.org/downloads.html |
| Swiss-model | Homology modelling server | https://swissmodel.expasy.org |
| SwissDock | Molecular docking server | http://www.swissdock.ch |
| UniProt | Protein sequence and function repository | https://www.uniprot.org/ |
| Metabolomics | ||
| Anti-smash | Annotation and analysis of secondary metabolite biosynthesis | https://antismash.secondarymetabolites.org/#!/start |
| ChemSpider | Compound repository | http://www.chemspider.com/ |
| GNPS | Tandem mass (MS/MS) spectrometry data repository | https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp |
| KEGG | Metabolism data repository and analyses services | https://www.genome.jp/kegg/ |
| MEROPS | Compound repository and analyses services | https://www.ebi.ac.uk/merops/ |
| MetaCyc | Metabolism data repository and analyses services | https://metacyc.org/ |
| NaPDoS | Compound repository and analyses services | https://www.biokepler.org/use_cases/napdos |
| Reactome | Metabolism data repository and analyses services | https://reactome.org/ |
| The Super Natural II database | Compound repository | http://bioinf-applied.charite.de/supernatural_new/index.php |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosino, L.; Tangherlini, M.; Colantuono, C.; Esposito, A.; Sangiovanni, M.; Miralto, M.; Sansone, C.; Chiusano, M.L. Bioinformatics for Marine Products: An Overview of Resources, Bottlenecks, and Perspectives. Mar. Drugs 2019, 17, 576. https://doi.org/10.3390/md17100576
Ambrosino L, Tangherlini M, Colantuono C, Esposito A, Sangiovanni M, Miralto M, Sansone C, Chiusano ML. Bioinformatics for Marine Products: An Overview of Resources, Bottlenecks, and Perspectives. Marine Drugs. 2019; 17(10):576. https://doi.org/10.3390/md17100576
Chicago/Turabian StyleAmbrosino, Luca, Michael Tangherlini, Chiara Colantuono, Alfonso Esposito, Mara Sangiovanni, Marco Miralto, Clementina Sansone, and Maria Luisa Chiusano. 2019. "Bioinformatics for Marine Products: An Overview of Resources, Bottlenecks, and Perspectives" Marine Drugs 17, no. 10: 576. https://doi.org/10.3390/md17100576
APA StyleAmbrosino, L., Tangherlini, M., Colantuono, C., Esposito, A., Sangiovanni, M., Miralto, M., Sansone, C., & Chiusano, M. L. (2019). Bioinformatics for Marine Products: An Overview of Resources, Bottlenecks, and Perspectives. Marine Drugs, 17(10), 576. https://doi.org/10.3390/md17100576