Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy



Abstract
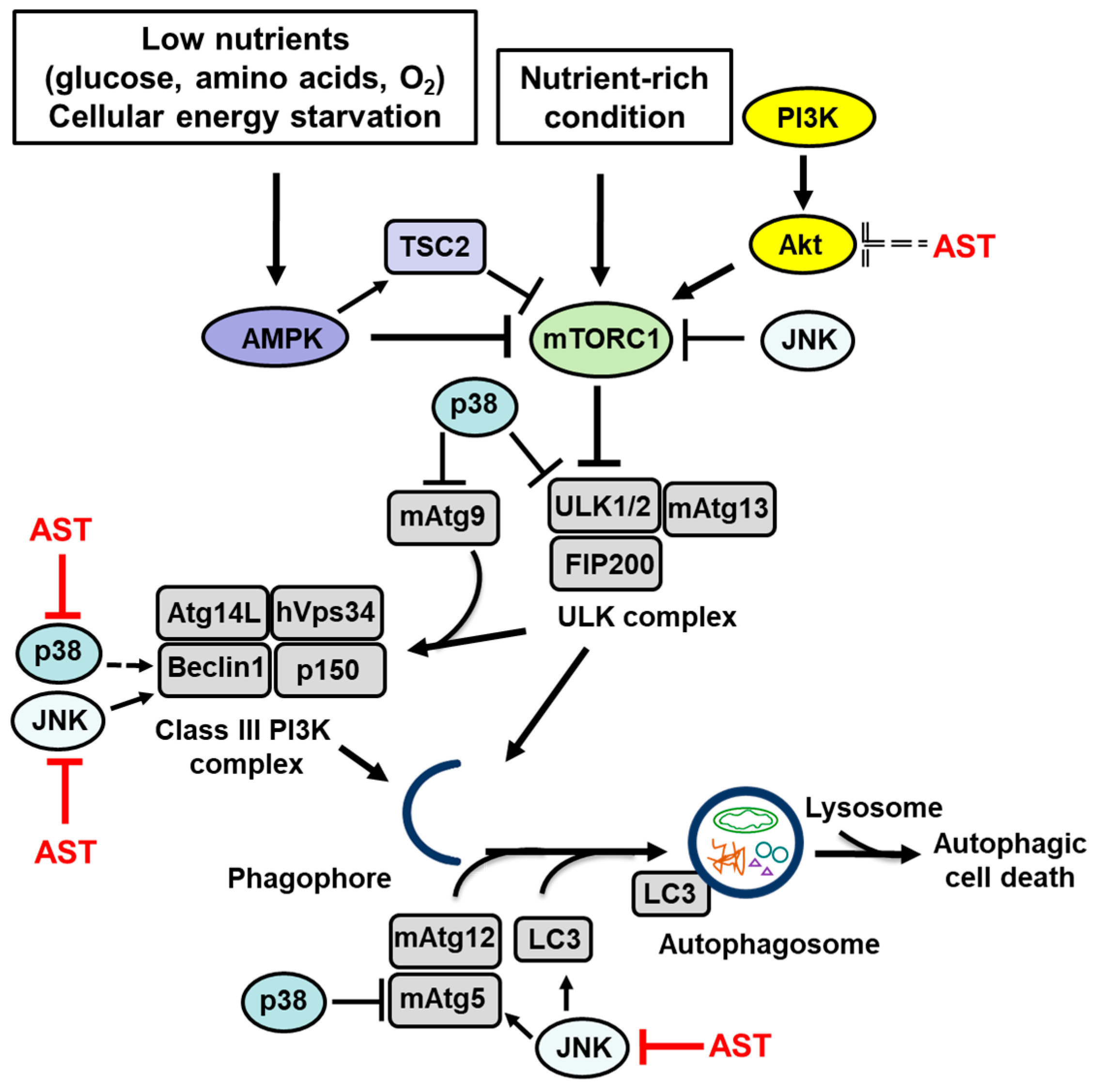
1. The Autophagy Machinery
2. Signaling Pathways That Regulate Autophagy
2.1. AMP-Activated Protein Kinase (AMPK)
2.2. Phosphatidylinositol 3-Kinase (PI3K)/Cellular Homolog of Murine Thymoma Virus akt8 Oncogene (Akt)
2.3. c-Jun N-Terminal Kinase (JNK)
2.4. p38
3. The Effect of Astaxanthin on the Signaling Pathways That Regulate Autophagy
3.1. Astaxnathin and AMPK Signaling
3.2. Astaxanthin and PI3K/Akt Signaling
3.3. Astaxanthin and JNK Signaling
3.4. Astaxanthin and p38 Signaling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Nair, U.; Geng, J.; Klionsky, D.J. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 2008, 19, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.S.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Re, A.L.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovana, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488. [Google Scholar] [CrossRef] [PubMed]
- Sou, Y.S.; Waguri, S.; Iwata, J.I.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Lindmo, K.; Stenmark, H. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 2006, 119, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two Distinct Vps34 Phosphatidylinositol 3–Kinase complexes function in autophagy and carboxypeptidase Y Sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Obara, K.; Noda, T.; Niimi, K.; Ohsumi, Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells 2008, 13, 537–547. [Google Scholar] [CrossRef]
- Panaretou, C.; Domin, J.; Cockcroft, S.; Waterfield, M.D. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase substrate presentation by phosphatidylinositol transfer protein to the p150; PtdIns 3-kinase complex. J. Biol. Chem. 1997, 272, 2477–2485. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672. [Google Scholar] [CrossRef]
- Obara, K.; Sekito, T.; Ohsumi, Y. Assortment of phosphatidylinositol 3-kinase complexes—Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol. Biol. Cell 2006, 17, 1527–1539. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Kanae, S.N.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385. [Google Scholar] [CrossRef]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Kimura, N.; Tokunaga, C.; Dalal, S.; Richardson, C.; Yoshino, K.I.; Hara, K.; Kemp, B.E.; Witters, L.A.; Mimura, O.; Yonezawa, K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells 2003, 8, 65–79. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572. [Google Scholar] [CrossRef]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132. [Google Scholar] [CrossRef]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef]
- Degtyarev, M.; De Mazière, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; Van Dijik, S.; Wu, H.; Gray, D.C.; et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef]
- Wu, H.; Wang, M.C.; Bohmann, D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech. Dev. 2009, 126, 624–637. [Google Scholar] [CrossRef]
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006, 84, 448–454. [Google Scholar] [CrossRef]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef]
- Guan, P.; Sun, Z.M.; Wang, N.; Zhou, J.; Luo, L.F.; Zhao, Y.S.; Ji, E.S. Resveratrol prevents chronic intermittent hypoxia-induced cardiac hypertrophy by targeting the PI3K/AKT/mTOR pathway. Life Sci. 2019, 233, 116748. [Google Scholar] [CrossRef]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol. Med. Rep. 2013, 8, 1011–1016. [Google Scholar] [CrossRef]
- Chen, J.; Cui, Y.; Zhang, N.; Yao, X.; Wang, Z.; Yang, L. Oleanolic acid attenuated diabetic mesangial cell injury by activation of autophagy via miRNA-142-5p/PTEN signaling. Cytotechnology 2019. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef]
- Wu, J.; Hu, G.; Dong, Y.; Ma, R.; Yu, Z.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Matrine induces Akt/mTOR signalling inhibition-mediated autophagy and apoptosis in acute myeloid leukaemia cells. J. Cell. Mol. Med. 2017, 21, 1171–1181. [Google Scholar] [CrossRef]
- Gao, L.; Wang, Z.; Lu, D.; Huang, J.; Liu, J.; Hong, L. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef]
- Ding, R.; Zhang, C.; Zhu, X.; Cheng, H.; Zhu, F.; Xu, Y.; Liu, Y.; Wen, L.; Cao, J. ROS-AKT-mTOR axis mediates autophagy of human umbilical vein endothelial cells induced by cooking oil fumes-derived fine particulate matters in vitro. Free Rad. Biol. Med. 2017, 113, 452–460. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Z.; Li, J.; Chen, L.; Tang, W. High glucose induces autophagy of MC3T3-E1 cells via ROS-AKT-mTOR axis. Mol. Cell. Endocrinol. 2016, 429, 62–72. [Google Scholar] [CrossRef]
- Borsello, T.; Croquelois, K.; Hornung, J.P.; Clarke, P.G. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 2003, 18, 473–485. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef]
- Byun, J.Y.; Yoon, C.H.; An, S.; Park, I.C.; Kang, C.M.; Kim, M.J.; Lee, S.J. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 2009, 30, 1880–1888. [Google Scholar] [CrossRef]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 2006, 177, 5163–5168. [Google Scholar] [CrossRef]
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886. [Google Scholar] [CrossRef]
- Sun, T.; Li, D.D.; Wang, L.L.; Xia, L.P.; Ma, J.G.; Guan, Z.; Feng, G.K.; Zhu, X.F. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J. Transl. Med. 2011, 9, 161. [Google Scholar] [CrossRef]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Chang, Y.G.; Kim, M.K.; Park, W.S.; Lee, J.Y.; Lee, S.Y.; et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology 2012, 56, 644–657. [Google Scholar] [CrossRef]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef]
- Xie, C.M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef]
- Park, K.J.; Lee, S.H.; Kim, T.I.; Lee, H.W.; Lee, C.H.; Kim, E.H.; Jang, J.Y.; Choi, K.S.; Kwon, M.H.; Kim, Y.S. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res. 2007, 67, 7327–7334. [Google Scholar] [CrossRef]
- Park, K.J.; Lee, S.H.; Lee, C.H.; Jang, J.Y.; Chung, J.; Kwon, M.H.; Kim, Y.S. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 2009, 382, 726–729. [Google Scholar] [CrossRef]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef]
- Ye, Y.C.; Yu, L.; Wang, H.J.; Tashiro, S.I.; Onodera, S.; Ikejima, T. TNFα-induced necroptosis and autophagy via supression of the p38–NF-κB survival pathway in L929 cells. J. Pharmacol. Sci. 2011, 117, 160. [Google Scholar] [CrossRef]
- Hu, C.; Zou, M.J.; Zhao, L.; Lu, N.; Sun, Y.J.; Gou, S.H.; Xi, T.; Guo, Q.L. E Platinum, a newly synthesized platinum compound, induces autophagy via inhibiting phosphorylation of mTOR in gastric carcinoma BGC-823 cells. Toxicol. Lett. 2012, 210, 78–86. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Jedinak, A.; Nguyen, H.; Terry, C.; Baldridge, L.A.; Jiang, J.; Sliva, D. Triterpenes from Ganoderma lucidum induce autophagy in colon cancer through the inhibition of p38 mitogen-activated kinase (p38 MAPK). Nutr. Cancer 2010, 62, 630–640. [Google Scholar] [CrossRef]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38α in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693. [Google Scholar] [CrossRef]
- De la Cruz-Morcillo, M.A.; Valero, M.L.L.; Callejas-Valera, J.L.; Arias-Gonzalez, L.; Melgar-Rojas, P.; Galan-Moya, E.M.; Garcia-Gil, E.; Garcia-Cano, J.; Sánchez-Prieto, R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: Implication in resistance. Oncogene 2012, 31, 1073. [Google Scholar] [CrossRef]
- Paillas, S.; Causse, A.; Marzi, L.; De Medina, P.; Poirot, M.; Denis, V.; Vezzio-Vie, N.; Espert, L.; Arzouk, H.; Coquelle, A.; et al. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012, 8, 1098–1112. [Google Scholar] [CrossRef]
- Cui, Q.; Tashiro, S.I.; Onodera, S.; Minami, M.; Ikejima, T. Oridonin induced autophagy in human cervical carcinoma HeLa cells through Ras, JNK, and P38 regulation. J. Pharmacol. Sci. 2007, 105, 317–325. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2009, 298, C542–C549. [Google Scholar] [CrossRef]
- Lv, X.C.; Zhou, H.Y. Resveratrol protects H9c2 embryonic rat heart derived cells from oxidative stress by inducing autophagy: Role of p38 mitogen-activated protein kinase. Can. J. Physiol. Pharmacol. 2012, 90, 655–662. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, J.H.; Lee, G.H.; Kim, H.T.; Lim, J.M.; Chae, S.W.; Chae, J.H.; Kim, H.R. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol. Pharm. Bull. 2010, 33, 545–549. [Google Scholar] [CrossRef]
- Keil, E.; Höcker, R.; Schuster, M.; Essmann, F.; Ueffing, N.; Hoffman, B.; Liebermann, D.A.; Pfeffer, K.; Schulze-Osthoff, K.; Schmitz, I. Phosphorylation of Atg5 by the Gadd45β–MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013, 20, 321. [Google Scholar] [CrossRef]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef]
- Králová, V.; Benešová, S.; Červinka, M.; Rudolf, E. Selenite-induced apoptosis and autophagy in colon cancer cells. Toxicol. Vitr. 2012, 26, 258–268. [Google Scholar] [CrossRef]
- Yang, J.P.; Shin, J.H.; Seo, S.H.; Kim, S.G.; Lee, S.; Shin, E.H. Effects of Antioxidants in Reducing Accumulation of Fat in Hepatocyte. Int. J. Mol. Sci. 2018, 19, 2563. [Google Scholar] [CrossRef]
- Aoi, W.; Maoka, T.; Abe, R.; Fujishita, M.; Tominaga, K. Comparison of the effect of non-esterified and esterified astaxanthins on endurance performance in mice. J. Clin. Biochem. Nutr. 2018, 62, 161–166. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, W.; Lee, J.H.; Jeon, S.J.; Choi, Y.H.; Kim, B.W.; Nam, S.W. Astaxanthin inhibits H2O2-mediated apoptotic cell death in mouse neural progenitor cells via modulation of P38 and MEK signaling pathways. J. Microbiol. Biotechnol. 2009, 19, 1355–1363. [Google Scholar] [CrossRef]
- Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxanthin inhibits acetaldehyde-induced cytotoxicity in SH-SY5Y cells by modulating Akt/CREB and p38MAPK/ERK signaling pathways. Mar. Drugs 2016, 14, 56. [Google Scholar] [CrossRef]
- Wen, X.; Huang, A.; Hu, J.; Zhong, Z.; Liu, Y.; Li, Z.; Pan, X.; Liu, Z. Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: Involvement of the Akt/GSK-3β pathway. Neuroscience 2015, 303, 558–568. [Google Scholar] [CrossRef]
- Wang, X.J.; Chen, W.; Fu, X.T.; Ma, J.K.; Wang, M.H.; Hou, Y.J.; Tian, D.C.; Fu, X.Y.; Fan, C.D. Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin: Evidences for mitochondrial dysfunction and signaling crosstalk. Cell Death Discov. 2018, 5, 50. [Google Scholar] [CrossRef]
- Li, Z.; Dong, X.; Liu, H.; Chen, X.; Shi, H.; Fan, Y.; Hou, D.; Zhang, X. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656. [Google Scholar]
- Wang, C.M.; Cai, X.L.; Wen, Q.P. Astaxanthin reduces isoflurane-induced neuroapoptosis via the PI3K/Akt pathway. Mol. Med. Rep. 2016, 13, 4073–4078. [Google Scholar] [CrossRef]
- Qiao, J.; Rong, L.; Wang, Z.; Zhang, M. Involvement of Akt/GSK3β/CREB signaling pathway on chronic omethoate induced depressive-like behavior and improvement effects of combined lithium chloride and astaxanthin treatment. Neurosci. Lett. 2017, 649, 55–61. [Google Scholar] [CrossRef]
- Deng, X.; Wang, M.; Hu, S.; Feng, Y.; Shao, Y.; Xie, Y.; Wu, M.; Chen, Y.; Shi, X. The neuroprotective effect of astaxanthin on pilocarpine-induced status epilepticus in rats. Front. Cell. Neurosci. 2019, 13, 123. [Google Scholar] [CrossRef]
- Fakhri, S.; Dargahi, L.; Abbaszadeh, F.; Jorjani, M. Effects of astaxanthin on sensory-motor function in a compression model of spinal cord injury: Involvement of ERK and AKT signalling pathway. Eur. J. Pain 2019, 23, 750–764. [Google Scholar] [CrossRef]
- Kim, J.H.; Nam, S.W.; Kim, B.W.; Choi, W.; Lee, J.H.; Kim, W.J.; Choi, Y.H. Astaxanthin improves stem cell potency via an increase in the proliferation of neural progenitor cells. Int. J. Mol. Sci. 2010, 11, 5109–5119. [Google Scholar] [CrossRef]
- Kim, J.H.; Nam, S.W.; Kim, B.W.; Kim, W.J.; Choi, Y.H. Astaxanthin improves the proliferative capacity as well as the osteogenic and adipogenic differentiation potential in neural stem cells. Food Chem. Toxicol. 2010, 48, 1741–1745. [Google Scholar] [CrossRef]
- Li, S.; Takahara, T.; Fujino, M.; Fukuhara, Y.; Sugiyama, T.; Li, X.K.; Takahara, S. Astaxanthin prevents ischemia-reperfusion injury of the steatotic liver in mice. PLoS ONE 2017, 12, e0187810. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, J.; Yin, W.; Ding, X. Astaxanthin improves cognitive deficits from oxidative stress, nitric oxide synthase and inflammation through upregulation of PI3K/Akt in diabetes rat. Int. J. Clin. Exp. Pathol. 2015, 8, 6083–6094. [Google Scholar]
- Li, X.; Qi, Z.; Zhao, L.; Yu, Z. Astaxanthin reduces type 2 diabetic-associated cognitive decline in rats via activation of PI3K/Akt and attenuation of oxidative stress. Mol. Med. Rep. 2016, 13, 973–979. [Google Scholar] [CrossRef]
- Guo, S.X.; Zhou, H.L.; Huang, C.L.; You, C.G.; Fang, Q.; Wu, P.; Wang, X.G.; Han, C.M. Astaxanthin attenuates early acute kidney injury following severe burns in rats by ameliorating oxidative stress and mitochondrial-related apoptosis. Mar. Drugs 2015, 13, 2105–2123. [Google Scholar] [CrossRef]
- Fang, Q.; Guo, S.; Zhou, H.; Han, R.; Wu, P.; Han, C. Astaxanthin protects against early burn-wound progression in rats by attenuating oxidative stress-induced inflammation and mitochondria-related apoptosis. Sci. Rep. 2017, 7, 41440. [Google Scholar] [CrossRef]
- Zhang, X.S.; Zhang, X.; Wu, Q.; Li, W.; Zhang, Q.R.; Wang, C.X.; Zhou, X.M.; Li, H.; Shi, J.X.; Zhou, M.L. Astaxanthin alleviates early brain injury following subarachnoid hemorrhage in rats: Possible involvement of Akt/bad signaling. Mar. Drugs 2014, 12, 4291–4310. [Google Scholar] [CrossRef]
- Li, J.; Dai, W.; Xia, Y.; Chen, K.; Li, S.; Liu, T.; Zhang, R.; Wang, J.; Lu, W.; Zhou, Y.; et al. Astaxanthin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells via Inhibition of NF-κB P65 and Wnt/β-catenin in vitro. Mar. Drugs 2015, 13, 6064–6081. [Google Scholar] [CrossRef]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 4433–4444. [Google Scholar] [CrossRef]
- Kowshik, J.; Nivetha, R.; Ranjani, S.; Venkatesan, P.; Selvamuthukumar, S.; Veeravarmal, V.; Nagini, S. Astaxanthin inhibits hallmarks of cancer by targeting the PI3K/NF-κΒ/STAT3 signalling axis in oral squamous cell carcinoma models. IUBMB Life 2019. [Google Scholar] [CrossRef]
- Ko, J.C.; Chen, J.C.; Wang, T.J.; Zheng, H.Y.; Chen, W.C.; Chang, P.Y.; Lin, Y.W. Astaxanthin down-regulates Rad51 expression via inactivation of AKT kinase to enhance mitomycin C-induced cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharmacol. 2016, 105, 91–100. [Google Scholar] [CrossRef]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, W.; Li, Y.; Hu, L.; Dai, Y.; Chen, J.; Xu, S.; Xu, X.; Jiang, H. Astaxanthin ameliorates cerulein-induced acute pancreatitis in mice. Int. Immunopharmacol. 2018, 56, 18–28. [Google Scholar] [CrossRef]
- Shen, M.; Chen, K.; Lu, J.; Cheng, P.; Xu, L.; Dai, W.; Wang, F.; He, L.; Zhang, Y.; Chengfen, W.; et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-1 expression and autophagy. Mediat. Inflamm. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Chitchumroonchokchai, C.; Bomser, J.A.; Glamm, J.E.; Failla, M.L. Xanthophylls and α-tocopherol decrease UVB-induced lipid peroxidation and stress signaling in human lens epithelial cells. J. Nutr. 2004, 134, 3225–3232. [Google Scholar] [CrossRef]
- Sakai, S.; Nishida, A.; Ohno, M.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; Andoh, A. Astaxanthin, a xanthophyll carotenoid, prevents development of dextran sulphate sodium-induced murine colitis. J. Clin. Biochem. Nutr. 2019, 64, 66–72. [Google Scholar] [CrossRef]
- Li, D.; Tong, W.; Liu, D.; Zou, Y.; Zhang, C.; Xu, W. Astaxanthin mitigates cobalt cytotoxicity in the MG-63 cells by modulating the oxidative stress. BMC Pharmacol. Toxicol. 2017, 18, 58. [Google Scholar] [CrossRef]
- Ishiki, M.; Nishida, Y.; Ishibashi, H.; Wada, T.; Fujisaka, S.; Takikawa, A.; Urakaze, M.; Sasaoka, T.; Usui, I.; Tobe, K. Impact of divergent effects of astaxanthin on insulin signaling in L6 cells. Endocrinology 2013, 154, 2600–2612. [Google Scholar] [CrossRef]
- Li, M.Y.; Sun, L.; Niu, X.T.; Chen, X.M.; Tian, J.X.; Kong, Y.D.; Wang, G.Q. Astaxanthin protects lipopolysaccharide-induced inflammatory response in Channa argus through inhibiting NF-κB and MAPKs signaling pathways. Fish Shellfish Immunol. 2019, 86, 280–286. [Google Scholar] [CrossRef]
- Yaghooti, H.; Mohammadtaghvaei, N.; Mahboobnia, K. Effects of palmitate and astaxanthin on cell viability and proinflammatory characteristics of mesenchymal stem cells. Int. Immunopharmacol. 2019, 68, 164–170. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Bi, J.; Gu, J.; Deng, Y.; Liu, C. Astaxanthin pretreatment attenuates acetaminophen-induced liver injury in mice. Int. Immunopharmacol. 2017, 45, 26–33. [Google Scholar] [CrossRef]
- Bhuvaneswari, S.; Yogalakshmi, B.; Sreeja, S.; Anuradha, C.V. Astaxanthin reduces hepatic endoplasmic reticulum stress and nuclear factor-κB-mediated inflammation in high fructose and high fat diet-fed mice. Cell Stress Chaperones 2014, 19, 183–191. [Google Scholar] [CrossRef]
- Kitahara, A.; Takahashi, K.; Morita, N.; Murashima, T.; Onuma, H.; Sumitani, Y.; Tanaka, T.; Kondo, T.; Hosaka, T.; Ishida, H. The novel mechanisms concerning the inhibitions of palmitate-induced proinflammatory factor releases and endogenous cellular stress with astaxanthin on MIN6 β-cells. Mar. Drugs 2017, 15, 185. [Google Scholar] [CrossRef]
- Abdelzaher, L.A.; Imaizumi, T.; Suzuki, T.; Tomita, K.; Takashina, M.; Hattori, Y. Astaxanthin alleviates oxidative stress insults-related derangements in human vascular endothelial cells exposed to glucose fluctuations. Life Sci. 2016, 150, 24–31. [Google Scholar] [CrossRef]
- Palozza, P.; Torelli, C.; Boninsegna, A.; Simone, R.; Catalano, A.; Mele, M.C.; Picci, N. Growth-inhibitory effects of the astaxanthin-rich alga Haematococcus pluvialis in human colon cancer cells. Cancer Lett. 2009, 283, 108–117. [Google Scholar] [CrossRef]
- Li, J.; Xia, Y.; Liu, T.; Wang, J.; Dai, W.; Wang, F.; Zheng, Y.; Chen, K.; Li, S.; Abudumijiti, H.; et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS ONE 2015, 10, e0120440. [Google Scholar] [CrossRef]
- Chen, J.C.; Wu, C.H.; Peng, Y.S.; Zheng, H.Y.; Lin, Y.C.; Ma, P.F.; Yen, T.C.; Chen, T.Y.; Lin, Y.W. Astaxanthin enhances erlotinib-induced cytotoxicity by p38 MAPK mediated xeroderma pigmentosum complementation group C (XPC) down-regulation in human lung cancer cells. Toxicol. Res. 2018, 7, 1247–1256. [Google Scholar] [CrossRef]
- Ikeda, Y.; Tsuji, S.; Satoh, A.; Ishikura, M.; Shirasawa, T.; Shimizu, T. Protective effects of astaxanthin on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1730–1740. [Google Scholar] [CrossRef]
- Wang, H.Q.; Sun, X.B.; Xu, Y.X.; Zhao, H.; Zhu, Q.Y.; Zhu, C.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, C.Y.; Chiou, J.Y.; Peng, R.Y.; Peng, C.H. Astaxanthine secured apoptotic death of PC12 cells induced by β-amyloid peptide 25–35: Its molecular action targets. J. Med. Food 2010, 13, 548–556. [Google Scholar] [CrossRef]
- Yang, X.; Guo, A.L.; Pang, Y.P.; Cheng, X.J.; Xu, T.; Li, X.R.; Liu, J.; Zhang, Y.; Liu, Y. Astaxanthin attenuates environmental tobacco smoke-induced cognitive deficits: A Critical Role of p38 MAPK. Mar. Drugs 2019, 17, 24. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Jena, G.B. Astaxanthin intervention ameliorates cyclophosphamide-induced oxidative stress, DNA damage and early hepatocarcinogenesis in rat: Role of Nrf2, p53, p38 and phase-II enzymes. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 696, 69–80. [Google Scholar] [CrossRef]
- Fakhri, S.; Dargahi, L.; Abbaszadeh, F.; Jorjani, M. Astaxanthin attenuates neuroinflammation contributed to the neuropathic pain and motor dysfunction following compression spinal cord injury. Brain Res. Bull. 2018, 143, 217–224. [Google Scholar] [CrossRef]
- Chen, W.P.; Xiong, Y.; Shi, Y.X.; Hu, P.F.; Bao, J.P.; Wu, L.D. Astaxanthin reduces matrix metalloproteinase expression in human chondrocytes. Int. Immunopharmacol. 2014, 19, 174–177. [Google Scholar] [CrossRef]
- Li, J.; Wang, F.; Xia, Y.; Dai, W.; Chen, K.; Li, S.; Liu, T.; Zheng, Y.; Wang, J.; Lu, W.; et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar. Drugs 2015, 13, 3368–3387. [Google Scholar] [CrossRef]


{kind=link}
| Experimental Model | Regulation of Signaling Mediators | Regulation of Autophagy | Ref. | |
|---|---|---|---|---|
| AMPK | Oxygen deprivation of immortalized mouse embryo fibroblasts | AMPK activation Tuberous sclerosis complex 2 (TSC2) activation decreased phosphorylation of mTORC1 substrates | Autophagy ↑ (LC3 conversion, LC3 accumulation, autophagosome formation) | [35] |
| Treatment of human breast epithelial cells with TRAIL | Transforming growth factor beta-activated kinase 1 (TAK1)-mediated AMPK activation | Autophagy ↑ (LC3II accumulation) | [36] | |
| Resveratrol treatment in chronic myelogenous leukemia cells | JNK activation c-jun phosphorylation AMPK activation decreased phosphorylation of mTOR and its substrates | Autophagy ↑ (p62 degradation, LC3II accumulation) | [37] | |
| Increased free cytosolic Ca2+ in MCF-7 breast cancer cells | AMPK activation | Autophagy ↑ (autophagosome formation) | [38] | |
| Treatment with AMPK activator AICAR | AMPK activation mTOR inhibition unc-51-like kinase (ULK1) activation | Autophagy ↑ (LC3II accumulation, p62 degradation | [39] | |
| Glucose starvation | AMPK activation ULK1 phosphorylation by AMPK ULK1 activation | Autophagy ↑ | [40,41] | |
| PI3K/Akt | Knockdown of Akt isoforms in cancer cell lines | Akt inhibition | Autophagy ↑ (acidic vesicular organelle accumulation, autophagosome formation) | [43] |
| Starvation | PI3K/Akt inactivation mTOR inactivation | Autophagy ↑ (autophagosome formation, autolysosomal vesicle formation, Atg1 expression, Atg8 accumulation) | [45,46] | |
| Treatment of atherosclerotic vascular smooth muscle cells with insulin-like growth factor 1 (IGF-1) | Akt activation | Autophagy ↓ (no autophagic vacuoles) | [47] | |
| IL-13 stimulation in HT-29 cells | PI3K stimulation Akt activation | Autophagy ↓ | [48] | |
| Ceramide treatment in HT-29 cells | inhibition of Akt activation | Autophagy ↑ (proteolysis, autophagic vacuole accumulation, increased Beclin 1 expression) | [49] | |
| Increased ceramide pool in breast cancer MCF-7 cells by tamoxifen treatment | inhibition of Akt activation | Autophagy ↑ (increased Beclin 1 expression) | [49] | |
| Resveratrol treatment in rat hearts exposed to chronic intermittent hypoxia | decreased PI3K expression decreased Akt activation decreased mTOR activation | Autophagy ↑ (increased LC3 expression, increased LC3II/LC3Ⅰ ratio, decreased p62 expression) | [50] | |
| Brain-derived neurotrophic factor (BDNF) treatment in hypoxic-ischemic brain injury model | decreased Akt activation decreased mTOR activation | Autophagy ↑ (LC3II conversion, LC3II aggregation) | [51] | |
| Oleanolic acid treatment in diabetic nephropathy model | decreased PI3K expression decreased Akt activation decreased mTOR activation | Autophagy ↑ (increased LC3Ⅰ and LC3II expression, decreased p62 expression) | [52] | |
| Treatment of malignant glioma cells with PI3K inhibitor LY294002 or Akt inhibitor UCN-01 | inhibition of Akt activation decreased phosphorylation of mTOR substrates | Autophagy ↑ (autophagic vacuole formation, acidic vesicular organelle accumulation) | [53] | |
| Matrine treatment in acute myeloid leukemia cells | decreased Akt activation decreased mTOR activation decreased phosphorylation of mTOR substrates | Autophagy ↑ (p62 degradation, LC3II accumulation) | [54] | |
| Paeonol treatment in ovarian cancer cells | decreased Akt activation decreased mTOR activation decreased phosphorylation of mTOR substrates | Autophagy ↑ (increased LC3II expression, p62 degradation, autophagosome formation, LC3-labled autophagic vacuolation) | [55] | |
| Akt activation in HeLa cells in normal or starvation condition | Akt activation | Autophagy ↓ (Beclin 1 phosphorylation, inhibition of ClassⅢPI3K signaling) | [56] | |
| Exposure of human umbilical vein endothelial cells (HUVEC) to cooking oil fumes-derived particle matters | decreased PI3K activation decreased Akt activation decreased mTOR activation | Autophagy ↑ (autophagosome formation, increased LC3 puncta, increased Beclin 1 expression, increased LC3II/LC3Ⅰ ratio) | [57] | |
| High glucose exposure | decreased Akt activation decreased mTOR activation | Autophagy ↑ (increased LC3II expression, increased Beclin 1 expression, p62 degradation) | [58] | |
| JNK | Treatment with neurotoxic N-methyl-d-aspartate (NMDA) | JNK activation increased c-jun phosphorylation increased c-fos expression | Autophagy ↑ (autophagic vacuole formation) | [59] |
| Treatment with caspase inhibitor z-VAD | JNK activation Atg7 expression Beclin 1 expression | Autophagy ↑ (autophagic vacuole formation) | [60] | |
| Oncogenic H-ras infection | JNK-activating kinase, MAP kinase kinase 7 (MKK7) phosphorylation JNK activation increased Atg5 expression | Autophagy ↑ (acidic vacuole formation, LC3II accumulation, increased LC3 puncta, LC3 and LAMP-1 colocalization) | [61] | |
| Activation of CD4+ T cells | JNK signaling activation | Autophagy ↑ (increased LC3 puncta) | [62] | |
| Treatment of ceramide in cancer cells or neural progenitor cells | JNK activation increased c-jun phosphorylation | Autophagy ↑ (increased LC3 puncta, LC3II accumulation, autophagic vacuole formation, acidic vesicular organelle accumulation, increased expression of LC3, increased expression of Beclin 1) | [63,64] | |
| Ectopic expression of histone deacetylase 6 (HDAC6) in liver cancer cell lines | JNK activation increased c-jun phosphorylation | Autophagy ↑ (autophagic vacuole formation, LC3II accumulation, increased Beclin 1 expression) | [65] | |
| Exposure of human tumor cells to 1,3-dibutyl-2-thiooxo-imidazolidine-4,5-dione(C1) | JNK activation increased total and phosphorylated c-jun expression | Autophagy ↑ (increased LC3II expression, increased LC3II puncta, autophagosome formation, autophagic vacuole formation, Atg5-Atg7 conjugation) | [66] | |
| Bufalin treatment in human colon cancer cells | JNK2 activation | Autophagy ↑ (increased LC3 puncta, LC3II conversion, increased Atg5 expression, increased Beclin 1 expression) | [67] | |
| Nutrient starvation | JNK activation Multisite phosphorylation of Bcl-2 Beclin 1 dissociation | Autophagy ↑ | [70] | |
| Ceramide treatment in cancer cell lines | JNK1 activation Multisite phosphorylation of Bcl-2 Beclin 1 dissociation | Autophagy ↑ | [71] | |
| Human single-chain fragment variable, HW-1, treatment on cancer cells | JNK activation Beclin 1 expression | Autophagy ↑ (autophagic vesicle accumulation, increased LC3 puncta) | [72,73] | |
| p38 | LPS stimulation in microglia | p38α activation ULK1 phosphorylation ULK1-Atg13 complex disruption | Autophagy ↓ (decreased LC3II expression, increased p62 expression, decreased autophagosome number) | [74] |
| TNF-α treatment in murine fibroblast L929 cells | decreased p38 activation decreased NF-κB | Autophagy ↓ (autophagic vacuole formation, LC3 puncta, LC3II expression, Beclin 1 expression) | [75] | |
| E Platinum treatment in gastric carcinoma cells | decreased Akt activation decreased p38 activation decreased mTOR activation | Autophagy ↑ (increased LC3 puncta, increased LC3II/LC3Ⅰratio, autolysosome formation, increased expression of lysosomal markers LAMP-1 and cathepsin D) | [76] | |
| Tumor treatment with Ganoderma Lucidum triterpenes | p38α signaling activation | Autophagy ↑ (autophagic vacuole formation, increased LC3 expression, increased Beclin 1 expression) | [77] | |
| Treatment of colon cancer HCT116 cells with 5-fluorouracil | p38 activation | Autophagy ↓ | [79] | |
| Treatment of HCT116 cells with active metabolite of irinotecan | MAPK14/P38α kinase activation | Autophagy ↑ (increased LC3II expression, autophagic vacuole formation, punctated LC3) | [80] | |
| Oridonin treatment in human cervical carcinoma HeLa cells | Increased p38 expression Increased JNK expression Increased p38 phosphorylation Increased JNK phosphorylation | Autophagy ↑ (autophagic vacuole formation, increased LC3II expression, increased Beclin 1 expression) | [81] | |
| LPS stimulation in skeletal muscle | p38 activation | Autophagy ↑ (expression of Beclin 1, Atg7, Atg12) | [82] | |
| H2O2 stimulation in myotubes | p38 activation | Autophagy ↑ (increased expression of Atg7) | [82] | |
| Resveratrol treatment in H2O2-stimulated embryonic rat heart-derived cells | p38 activation | Autophagy ↑ (autophagosome formation, increased LC3II expression, increased Beclin 1 expression) | [83] | |
| ER stress induced by pharmarcologic agents | p38 activation | Autophagy ↑ (autophagic vacuole formation, autophagosome formation, increased Beclin 1 expression) | [84] | |
| Transfection of NIH/3T3 fibroblasts with Gadd45β | p38 activation | Inhibition of the autophagic flux (increased LC3 puncta, decreased autolysosomal degradation, Atg5 phosphorylation) | [85] | |
| Activation of p38 in starved cells | p38α kinase activation loss of membrane bound p38IP inhibition of p38IP – mAtg9 interaction | Autophagy ↓ (autophagosome formation, increased LC3II expression, p62 degradation) | [86] | |
| Selenite treatment on colon cancer cells | p38 activation JNK activation | Autophagy ↑ (autophagic vacuole formation, p62 degradation, increased Beclin 1, Lamp-1 and cathepsin D expression, LC3II conversion) | [87] |
| Experimental Model | Astaxanthin Dose | Regulation of Signaling Mediators | Notable Results | Ref. |
|---|---|---|---|---|
| Oleic acid-induced hepatic steatosis | 10 μM | increased p-AMPK/AMPK ratio | decreased cell death reduced cell damage | [88] |
| Performance endurance test | 0.02% esterified astaxanthin from Haematococcus pluvialis | increased total AMPK expression | increased endurance performance | [89] |
| H2O2-stimulation in mouse neural progenitor cells | 10 ng/mL | increased p-Akt expression decreased p-p38 expression | decreased cell death reduced cytotoxicity increased cell proliferation | [90] |
| Acetaldehyde-induced neurotoxicity | 50 ng/mL | increased p-Akt expression decreased p-p38 expression | decreased cell death reduced cytotoxicity | [91] |
| Glutamate-induced cytotoxicity in neurotoxicity | 5 μM | increased p-Akt expression activation Nrf2 | decreased cell death reduced cytotoxicity | [92] |
| Homocysteine-induced neurotoxicity | 5 μM | increased p-Akt expression | decreased cell death reduced cytotoxicity | [93] |
| H2O2 stimulated retinal pigment epithelial cells | 20 μM | increased p-Akt expression activation of Nrf2 | decreased cell death | [94] |
| Isoflurane-induced neurotoxicity | 8 μM 100 mg/kg | increased p-Akt/Akt ratio | decreased cell death reduced cytotoxicity | [95] |
| Chronic organophosphorus pesticide exposure | 50 mg/kg/d | increased p-PI3K expression increased p-Akt expression | reduced cytotoxicity | [96] |
| Pilocarpine-induced status epilepticus | 30 mg/kg | increased p-Akt/Akt ratio | decreased cell death reduced cytotoxicity | [97] |
| Spinal cord injury | 10 μL of 0.2 mM | increased expression of p-Akt | decreased cell death | [98] |
| Neural progenitor/stem cells | 10 ng/mL | activation of PI3K increased expression of p-Akt | increased cell proliferation | [99,100] |
| Hypoxia and reoxygenation-stimulated Kupffer cells | 10 μM | increased p-Akt expression increased mTOR expression | decreased cell death | [101] |
| Hypoxia and reoxygenation-induced ischemia-reperfusion injury | 10 μM | decreased p-JNK expression decreased p-p38 expression | Decreased cell death | [101] |
| Cognitive deficit in diabetic rats | 10, 20, 40 mg/kg | increased PI3K expression increased Akt expression | decreased oxidative cell death | [102] |
| Cognitive deficit in diabetic rats | 50, 100 mg/kg | increased total Akt and p-Akt expression | decreased oxidative cell death | [103] |
| Early acute kidney injury | 20 mg/kg | increased p-Akt expression | decreased cell death | [104] |
| Early burn wound | 5, 10, 20 mg/kg | increased p-Akt expression | decreased cell death | [105] |
| Brain injury post-subarachnoid hemorrhage | 20 μL of 0.1mM | increased p-Akt expression | decreased cell death | [106] |
| Human hepatocellular carcinoma cells | 100, 200, 300 μM | decreased p-Akt/Akt ratio inhibition of NF-κB inhibition of Wnt/β-catenin | inhibition of cell proliferation loss of cell viability | [107] |
| Hamster model of DMBA-induced oral cancer | 15 mg/kg BW | decreased total Akt and p-Akt expression inhibition of NF-κB inhibition of Wnt/β-catenin | inhibition of cell proliferation loss of cell viability | [108] |
| Oral squamous cell carcinoma | 400 μM 15 mg/kg | inhibition of PI3K decreased p-Akt expression inhibition of NF-κB/STAT3 | loss of cell viability enhanced cytotoxicity | [109] |
| Human non-small cell lung cancer cells | 20 μM | inactivation of Akt kinase | loss of cell viability enhanced cytotoxicity | [110] |
| High fat diet-induced nonalcoholic fatty liver disease model | 30 mg/kg | decreased p-Akt/Akt ratio decreased p-GSK3 expression | induction of hepatic autophagy | [111] |
| Cerulein-induced acute pancreatitis | 20, 40 mg/kg | increased p-STAT3 expression increased expression of Bcl-2 decreased expression Beclin 1 | inhibition of cerulein-induced autophagy | [112] |
| Bile duct ligation-induced liver fibrosis | 40, 80 mg/kg | decreased NF-κB expression and activation | Inhibition of hepatic autophagy | [113] |
| UVB irradiated human lens epithelial cells | 2 μmol/L | decreased JNK1 and JNK2 activation decreased p-p38 expression | alleviated oxidative damage | [114] |
| Dextran sulphate sodium-induced colitis TNF-α stimulation in colonic epithelial cell | 0.02, 0.04% | decreased p-JNK expression decreased p-p38 expression | alleviated inflammation | [115] |
| Cobalt-induced cytotoxicity | 1, 10, 20 nM | decreased p-JNK expression decreased p38 expression decreased Akt expression | reduced cytotoxicity | [116] |
| Insulin signaling in skeletal muscle | 5, 10, 20 μM | increased p-Akt expression decreased p-JNK expression | increased glucose uptake | [117] |
| LPS-stimulated inflammation | 50, 100, 200 mg/kg | decreased p-JNK expression decreased p-p38 expression | alleviated inflammation | [118] |
| Palmitate stimulation in mesenchymal stem cells | 10 μM | decreased p-JNK expression decreased p-p38 expression | decreased cell death reduced cytotoxicity | [119] |
| Acetaminophen-induced liver injury | 30, 60 mg/kg/d | decreased total JNK and p-JNK expression decreased p-p38 expression | decreased cell death reduced cytotoxicity | [120] |
| High fructose and high fat diet-fed mice | 2 mg/kg | increased total JNK and p-JNK expression | alleviated inflammation | [121] |
| Palmitate-induced cytotoxicity | 10 μmol/L | decreased p-JNK expression decreased p-Akt expression | alleviated inflammation | [122] |
| Fluctuating high glucose exposure in human vascular endothelial cells | 0.05, 0.1, 0.5 μM | decreased p-JNK expression decreased p-p38 expression | decreased cell death reduced cytotoxicity | [123] |
| Human colon cancer cells | 15, 25 μg/mL Haematococcus pluvialis extract | increased p-JNK expression increased p-p38 expression decreased p-Akt expression | inhibition of cell proliferation loss of cell viability | [124] |
| ConA-induced autoimmune hepatitis | 20, 40 mg/kg | decreased p-JNK expression | Inhibition of hepatic autophagy | [125] |
| Human lung carcinoma cells | 20 μM | increased p-p38 expression | loss of cell viability enhanced cytotoxicity | [126] |
| 6-hydroxydopamine-induced neurotoxicity | 20 μM | decreased p-p38 expression | decreased cell death reduced cytotoxicity | [127] |
| Beta-amyloid-induced neurotoxicity | 5, 10 μM | decreased p-p38 expression | decreased cell death reduced cytotoxicity | [128] |
| Glutamate-induced neurotoxicity | 50 μg/L | decreased p-p38/p38 ratio | decreased cell death reduced cytotoxicity | [129] |
| β-amyloid peptide-induced neurotoxicity | 0.1 μM | decreased p-p38 expression | decreased cell death reduced cytotoxicity | [130] |
| Environmental tobacco smoke-induced cognitive deficits | 40, 80 mg/kg | decreased p-p38 expression | reduced cytotoxicity | [131] |
| Cyclophosphamide-induced hepatocarcinogenesis | 25 mg/kg | decreased p-p38 expression | inhibition of early hepatocarciongenesis | [132] |
| Spinal cord injury | 10 μL of 0.2 mM | decreased p-p38 expression | alleviated neuropathy | [133] |
| IL-1β-induced osteoarthritis in chondrocytes | 10, 50 μM | decreased p-p38 expression | lower MMP level | [134] |
| Hepatic ischemia reperfusion injury | 60 mg/kg | decreased p-JNK expression decreased p-p38 expression decreased p-ERK expression | inhibition of hepatic autophagy | [135] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Kim, H. Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy. Mar. Drugs 2019, 17, 546. https://doi.org/10.3390/md17100546
Kim SH, Kim H. Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy. Marine Drugs. 2019; 17(10):546. https://doi.org/10.3390/md17100546
Chicago/Turabian StyleKim, Suhn Hyung, and Hyeyoung Kim. 2019. "Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy" Marine Drugs 17, no. 10: 546. https://doi.org/10.3390/md17100546
APA StyleKim, S. H., & Kim, H. (2019). Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy. Marine Drugs, 17(10), 546. https://doi.org/10.3390/md17100546