Magnificamide, a β-Defensin-Like Peptide from the Mucus of the Sea Anemone Heteractis magnifica, Is a Strong Inhibitor of Mammalian α-Amylases

 ,
, 

Abstract
1. Introduction
2. Results
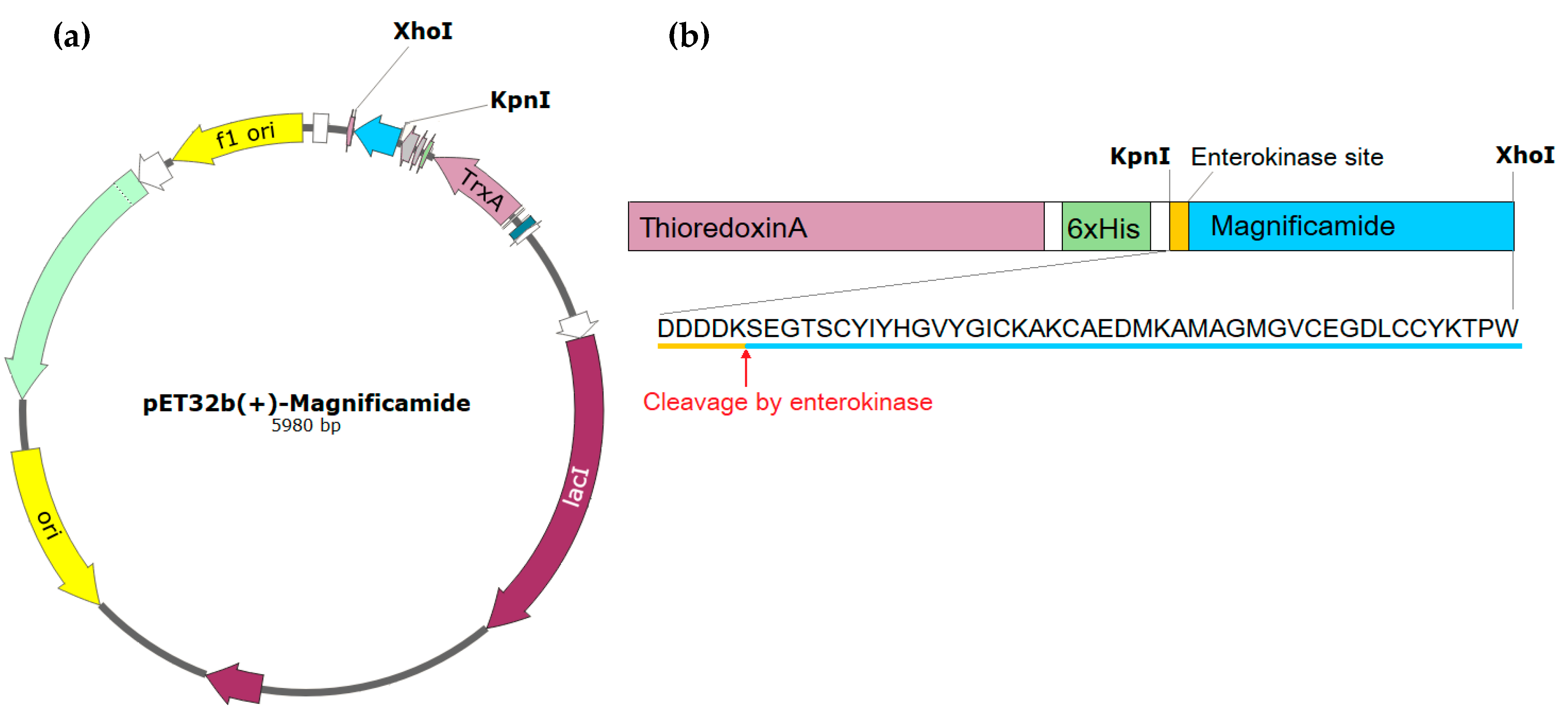
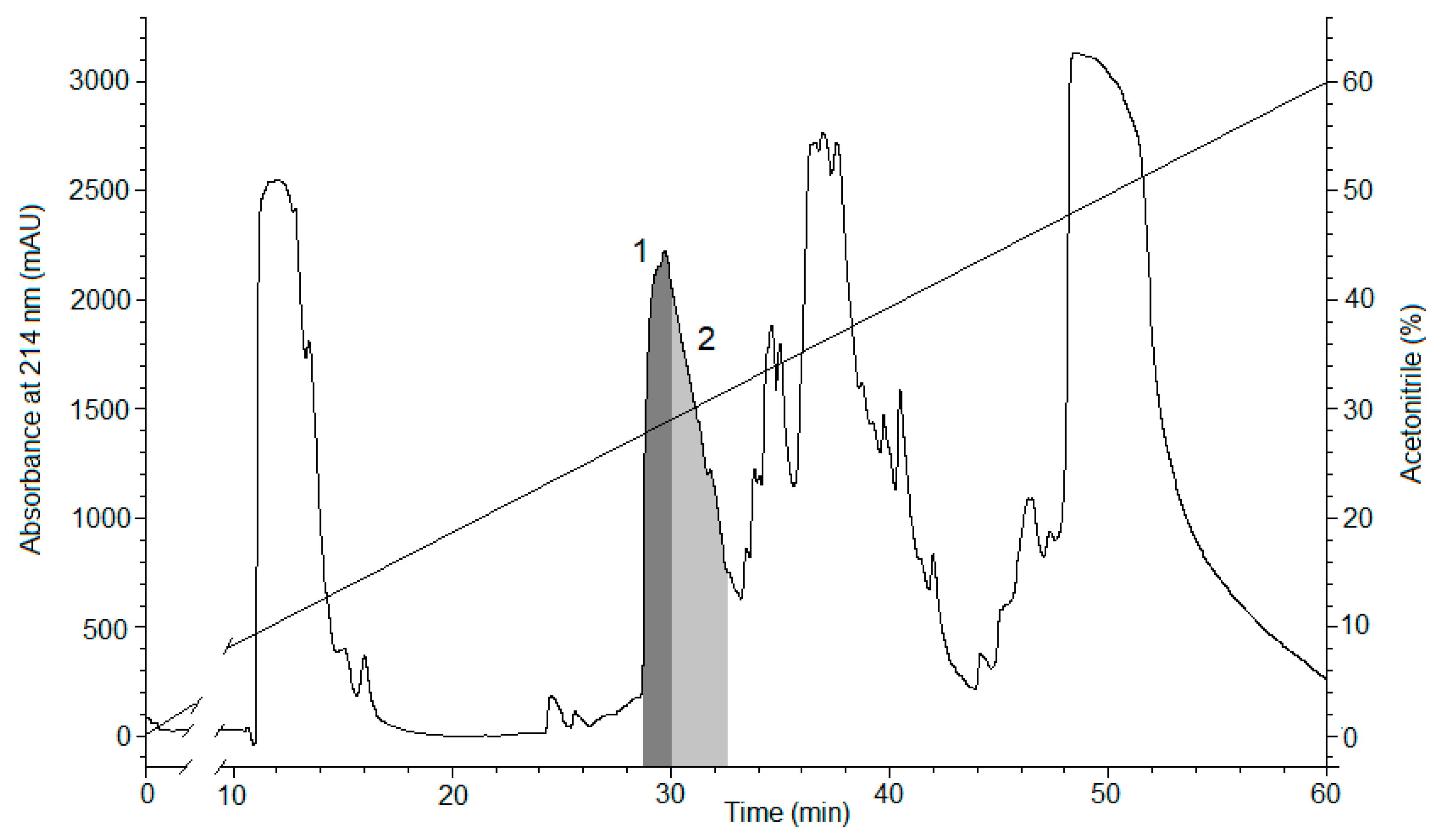
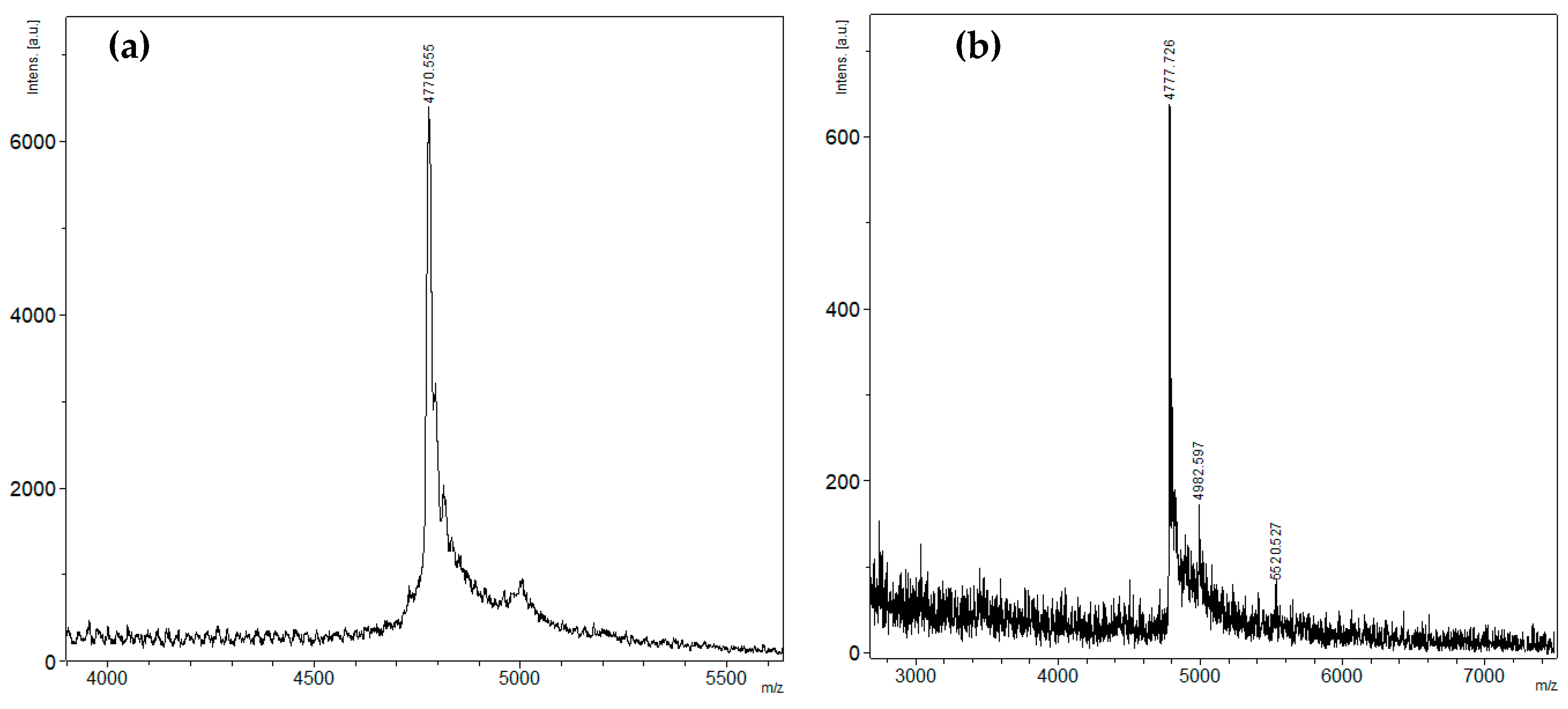
2.1. Peptide Expression and Purification
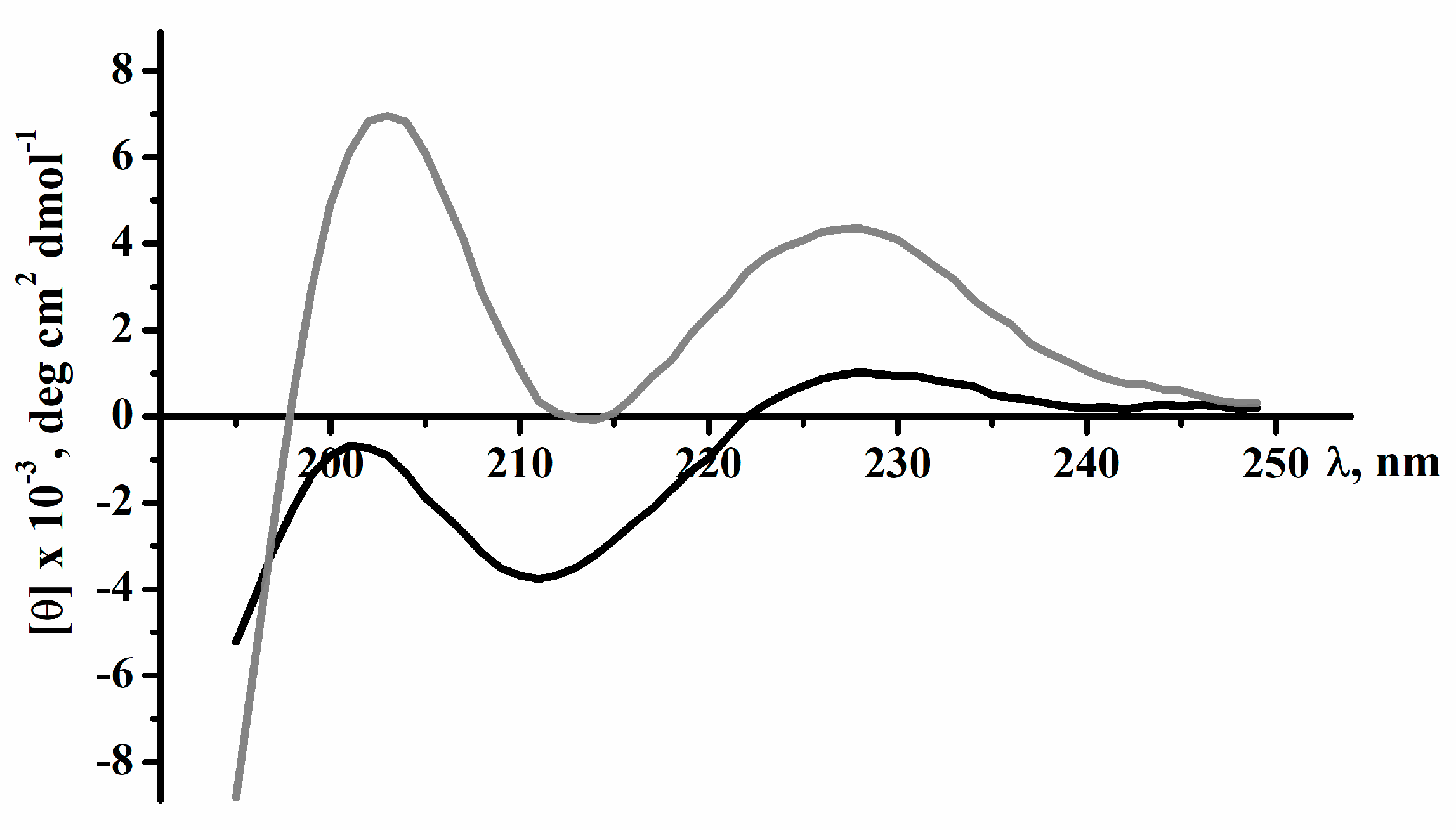
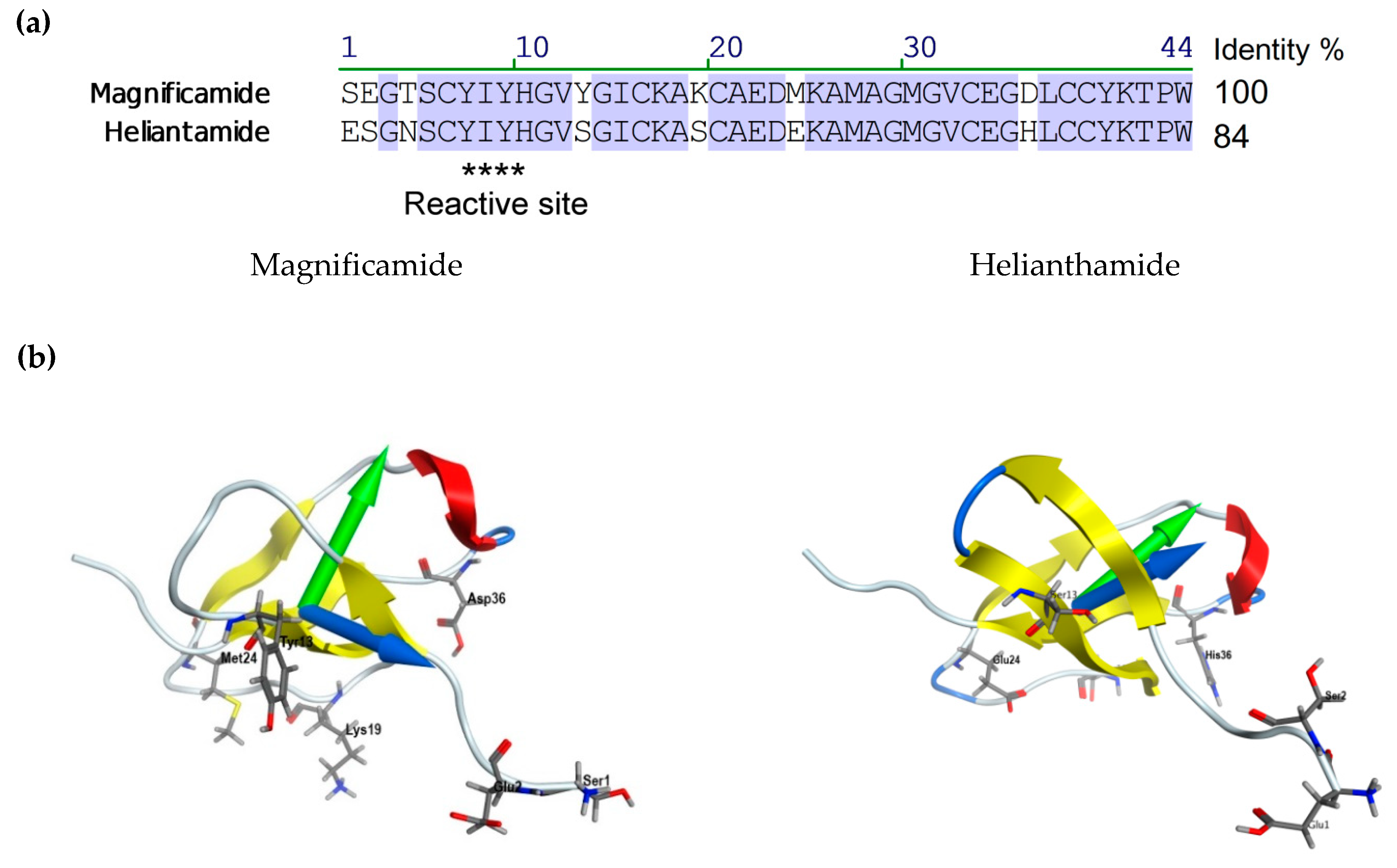
2.2. Secondary Structure of Peptides
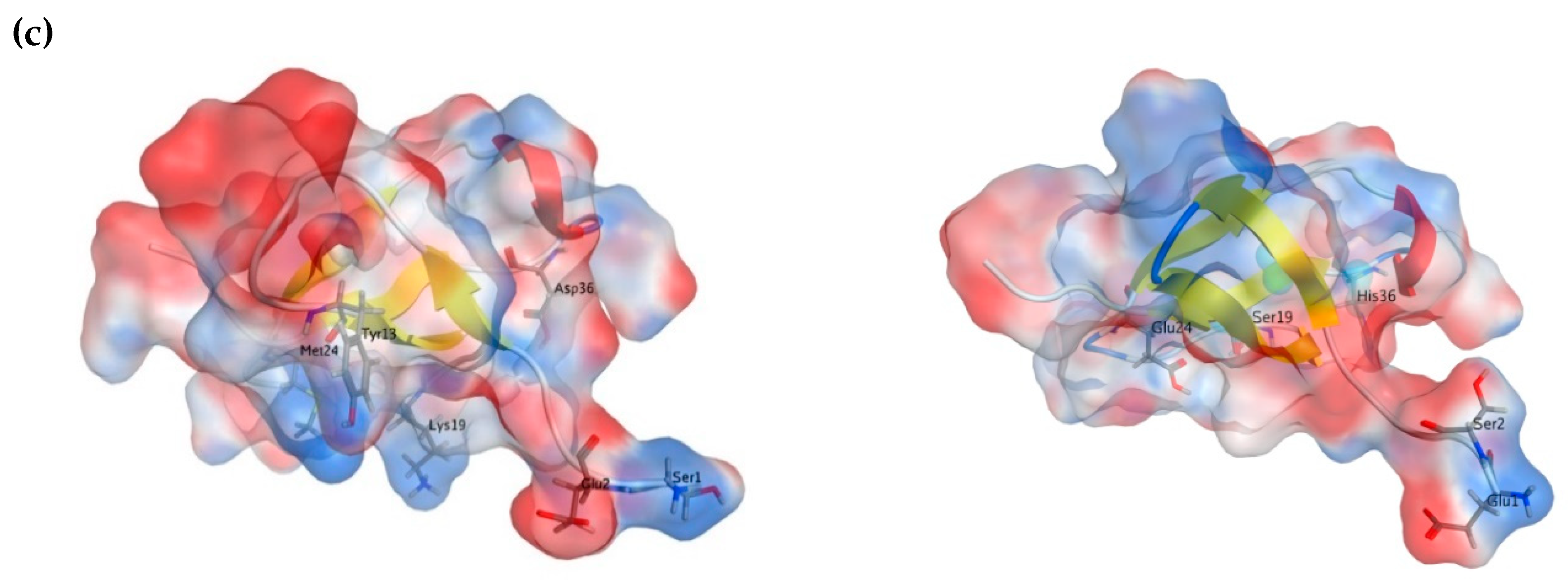
2.3. Molecular Modeling
2.4. Study of Antimicrobial Activity
2.5. Study of Channel Modulating Activity
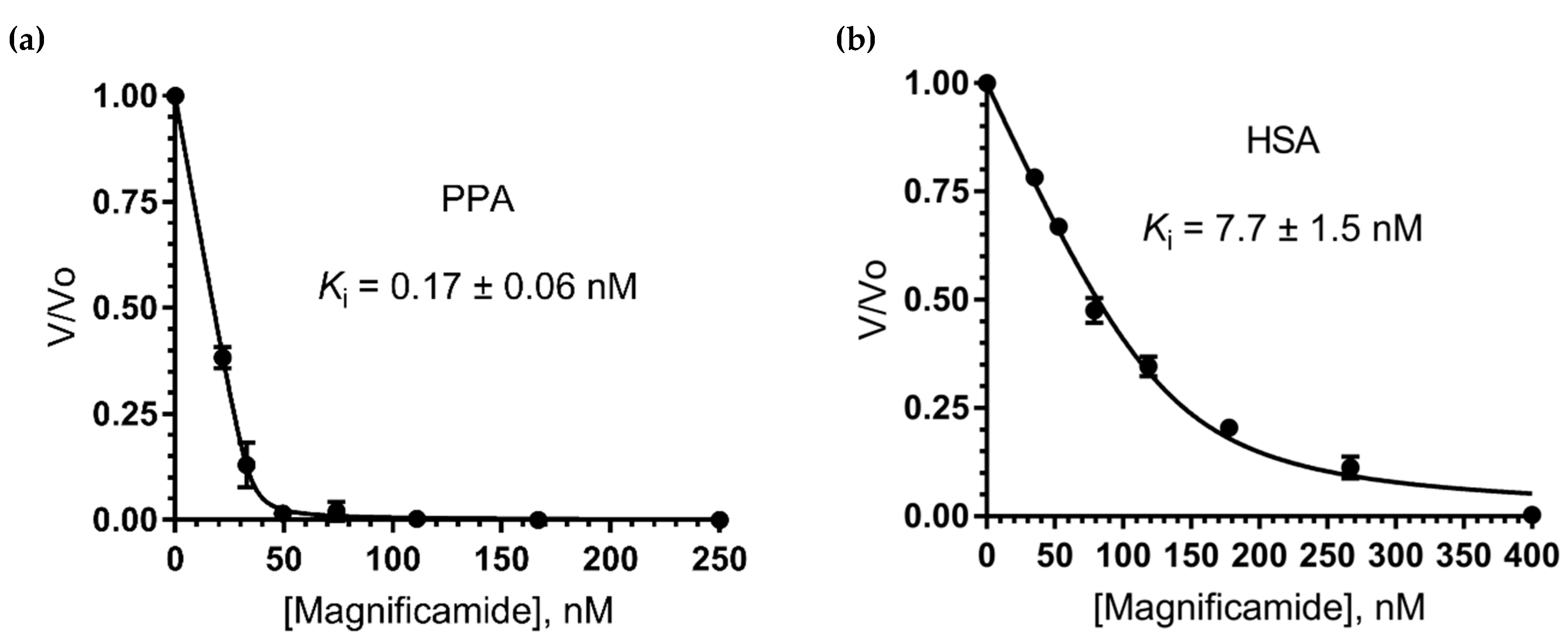
2.6. Study of Mammalian α-Amylase Inhibition
3. Discussion
4. Materials and Methods
4.1. Obtaining the Recombinant Magnificamide
4.2. Mass Spectrometry Analysis
4.3. Assay of Porcine Pancreatic α-Amylase Inhibitory Activity
4.4. Circular Dichroism Spectroscopy
4.5. Homology Modeling
4.6. In Vitro Antimicrobial Activity Assay
4.7. Electrophysiology
4.8. Determination of Ki Against Porcine Pancreatic and Human Saliva α-Amylases
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alam, U.; Asghar, O.; Azmi, S. General aspects of diabetes mellitus. Handb. Clin. Neurol. 2014, 126, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Chawla, S.; Guchhait, P. Type-2 diabetes: Current understanding and future perspectives. IUBMB Life 2015, 67, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Aye, T.; Levitsky, L.L. Type 2 diabetes: An epidemic disease in childhood. Curr. Opin. Pediatr. 2003, 15, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Temneanu, O.R.; Trandafir, L.M.; Purcarea, M.R. Type 2 diabetes mellitus in children and adolescents: A relatively new clinical problem within pediatric practice. J. Med. Life 2016, 9, 235–239. [Google Scholar] [PubMed]
- Adeghate, E.; Schattner, P.; Dunn, E. An update on the etiology and epidemiology of diabetes mellitus. Ann. N. Y. Acad. Sci. 2006, 1084, 1–29. [Google Scholar] [CrossRef]
- Scheen, A.J. Is there a role for alpha-glucosidase inhibitors in the prevention of type 2 diabetes mellitus? Drugs 2003, 63, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, J.-L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; STOP-NIDDM Trail Research Group. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Wu, H.; Liu, J.; Lou, Q.; Liu, J.; Shen, L.; Zhang, M.; Lv, X.; Gu, M.; Guo, X. Comparative assessment of the efficacy and safety of acarbose and metformin combined with premixed insulin in patients with type 2 diabetes mellitus. Medicine 2017, 96, e7533. [Google Scholar] [CrossRef]
- Yoon, S.H.; Robyt, J.F. Study of the inhibition of four alpha amylases by acarbose and its 4IV-α-maltohexaosyl and 4IV-α-maltododecaosyl analogues. Carbohydr. Res. 2003, 338, 1969–1980. [Google Scholar] [CrossRef]
- Poongunran, J.; Kumudu, H.; Perera, I.; Fernando, I.T.; Sivakanesan, R.; Fujimoto, Y. Bioassay-guided fractionation and identification of α-amylase inhibitors from Syzygium cumini leaves. Pharm. Biol. 2017, 55, 206–211. [Google Scholar] [CrossRef]
- Williams, L.K.; Zhang, X.; Caner, S.; Tysoe, C.; Nguyen, N.T.; Wicki, J.; Williams, D.E.; Coleman, J.; McNeill, J.H.; Yuen, V.; et al. The amylase inhibitor montbretin A reveals a new glycosidase inhibition motif. Nat. Chem. Biol. 2015, 11, 691–696. [Google Scholar] [CrossRef]
- Svensson, B.; Fukuda, K.; Nielsen, P.K.; Bønsager, B.C. Proteinaceous alpha-amylase inhibitors. Biochim. Biophys. Acta 2004, 1696, 145–156. [Google Scholar] [CrossRef]
- Vértesy, L.; Oeding, V.; Bender, R.; Zepf, K.; Nesemann, G. Tendamistat (HOE 467), a tight-binding alpha-amylase inhibitor from Streptomyces tendae 4158. Isolation, biochemical properties. Eur. J. Biochem. 1984, 141, 505–512. [Google Scholar] [CrossRef]
- Yokose, K.; Ogawa, K.; Sano, T.; Watanabe, K.; Maruyama, H.B.; Suhara, Y. New alpha-amylase inhibitor, trestatins. I. Isolation, characterization and biological activities of trestatins A, B and C. J. Antibiot. 1983, 36, 1157–1165. [Google Scholar] [CrossRef]
- Wiegand, G.; Epp, O.; Huber, R. The crystal structure of porcine pancreatic alpha-amylase in complex with the microbial inhibitor Tendamistat. J. Mol. Biol. 1995, 247, 99–110. [Google Scholar] [CrossRef]
- Sokočević, A.; Han, S.; Engels, J.W. Biophysical characterization of α-amylase inhibitor Parvulustat (Z-2685) and comparison with Tendamistat (HOE-467). Biochim. Biophys. Acta Proteins Proteomics 2011, 1814, 1383–1393. [Google Scholar] [CrossRef]
- Gonçalves, O.; Dintinger, T.; Blanchard, D.; Tellier, C. Functional mimicry between anti-Tendamistat antibodies and alpha-amylase. J. Immunol. Methods 2002, 269, 29–37. [Google Scholar] [CrossRef]
- Tysoe, C.; Wiliams, L.K.; Keyzers, R.; Nguyen, N.T.; Tarling, C.; Wicki, J.; Goddard-Borger, E.D.; Aguda, A.H.; Perry, S.; Foster, L.J.; et al. Potent human α-amylase inhibition by the β-defensin-like protein helianthamide. ACS Cent. Sci. 2016, 2, 154–161. [Google Scholar] [CrossRef]
- Sintsova, O.; Gladkikh, I.; Chausova, V.; Monastyrnaya, M.; Anastyuk, S.; Chernikov, O.; Yurchenko, E.; Aminin, D.; Isaeva, M.; Leychenko, E.; et al. Peptide fingerprinting of the sea anemone Heteractis magnifica mucus revealed neurotoxins, Kunitz-type proteinase inhibitors and a new β-defensin α-amylase inhibitor. J. Proteom. 2018, 173, 12–21. [Google Scholar] [CrossRef]
- Provencher, S.W.; Glöckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE) 2016.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016. [Google Scholar]
- Cuesta, A.; Meseguer, J.; Esteban, M.Á. Molecular and functional characterization of the gilthead seabream β-defensin demonstrate its chemotactic and antimicrobial activity. Mol. Immunol. 2011, 48, 1432–1438. [Google Scholar] [CrossRef]
- Shafee, T.M.A.; Lay, F.T.; Phan, T.K.; Anderson, M.A.; Hulett, M.D. Convergent evolution of defensin sequence, structure and function. Cell. Mol. Life Sci. 2016, 1–20. [Google Scholar] [CrossRef]
- Dalla Valle, L.; Benato, F.; Maistro, S.; Quinzani, S.; Alibardi, L. Bioinformatic and molecular characterization of beta-defensins-like peptides isolated from the green lizard Anolis Carolinensis. Dev. Comp. Immunol. 2012, 36, 222–229. [Google Scholar] [CrossRef]
- Ovchinnikova, T.V.; Balandin, S.V.; Aleshina, G.M.; Tagaev, A.A.; Leonova, Y.F.; Krasnodembsky, E.D.; Men’shenin, A.V.; Kokryakov, V.N. Aurelin, a novel antimicrobial peptide from jellyfish Aurelia aurita with structural features of defensins and channel-blocking toxins. Biochem. Biophys. Res. Commun. 2006, 348, 514–523. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, S. Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev. Comp. Immunol. 2012, 38, 262–274. [Google Scholar] [CrossRef]
- Tassanakajon, A.; Somboonwiwat, K.; Amparyup, P. Sequence diversity and evolution of antimicrobial peptides in invertebrates. Dev. Comp. Immunol. 2014, 48, 324–341. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Torres, A.M.; Kuchel, P.W. The β-defensin-fold family of polypeptides. Toxicon 2004, 44, 581–588. [Google Scholar] [CrossRef]
- Rodríguez, A.A.; Garateix, A.; Salceda, E.; Peigneur, S.; Zaharenko, A.J.; Pons, T.; Santos, Y.; Arreguín, R.; Ständker, L.; Forssmann, W.G.; et al. PhcrTx2, a new crab-paralyzing peptide toxin from the sea anemone Phymanthus crucifer. Toxins 2018, 10, 72. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B.; Deng, M.; Yuan, Y.; Luo, L.; Peigneur, S.; Xiao, Y.; Liang, S.; Tytgat, J. Drosotoxin, a selective inhibitor of tetrodotoxin-resistant sodium channels. Biochem. Pharmacol. 2010, 80, 1296–1302. [Google Scholar] [CrossRef]
- Zhu, S.; Peigneur, S.; Gao, B.; Umetsu, Y.; Ohki, S.; Tytgat, J. Experimental conversion of a defensin into a neurotoxin: Implications for origin of toxic function. Mol. Biol. Evol. 2014, 31, 546–559. [Google Scholar] [CrossRef]
- Kim, C.-H.; Lee, Y.J.; Go, H.-J.; Oh, H.Y.; Lee, T.K.; Park, J.B.; Park, N.G. Defensin-neurotoxin dyad in a basally branching metazoan sea anemone. FEBS J. 2017, 284, 3320–3338. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Morrison, J.F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta Enzymol. 1969, 185, 269–286. [Google Scholar] [CrossRef]
- Monastyrnaya, M.; Leychenko, E.; Isaeva, M.; Likhatskaya, G.; Zelepuga, E.; Kostina, E.; Trifonov, E.; Nurminski, E.; Kozlovskaya, E. Actinoporins from the sea anemones, tropical Radianthus macrodactylus and northern Oulactis orientalis: Comparative analysis of structure–function relationships. Toxicon 2010, 56, 1299–1314. [Google Scholar] [CrossRef]
- Leychenko, E.; Isaeva, M.; Tkacheva, E.; Zelepuga, E. Multigene Family of Pore-forming Toxins from Sea Anemone Heteractis Crispa. Mar. Drugs 2018, 16, 183. [Google Scholar] [CrossRef]
- Valle, A.; Alvarado-Mesén, J.; Lanio, M.E.; Álvarez, C.; Barbosa, J.A.R.G.; Pazos, I.F. The multigene families of actinoporins (part I): Isoforms and genetic structure. Toxicon 2015, 103, 176–187. [Google Scholar] [CrossRef]
- Kalina, R.; Gladkikh, I.; Dmitrenok, P.; Chernikov, O.; Koshelev, S.; Kvetkina, A.; Kozlov, S.; Kozlovskaya, E.; Monastyrnaya, M. New APETx-like peptides from sea anemone Heteractis crispa modulate ASIC1a channels. Peptides 2018, 104, 41–49. [Google Scholar] [CrossRef]
- Monastyrnaya, M.; Peigneur, S.; Zelepuga, E.; Sintsova, O.; Gladkikh, I.; Leychenko, E.; Isaeva, M.; Tytgat, J.; Kozlovskaya, E. Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor. Mar. Drugs 2016, 14, 229. [Google Scholar] [CrossRef]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvåg, K.; et al. Peptide from sea anemone Metridium senile affects transient receptor potential ankyrin-repeat 1 (TRPA1) function and produces analgesic effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef]
- Moran, Y.; Weinberger, H.; Sullivan, J.C.; Reitzel, A.M.; Finnerty, J.R.; Gurevitz, M. Concerted Evolution of Sea Anemone Neurotoxin Genes Is Revealed through Analysis of the Nematostella vectensis Genome. Mol. Biol. Evol. 2008, 25, 737–747. [Google Scholar] [CrossRef]
- García-Fernández, R.; Peigneur, S.; Pons, T.; Alvarez, C.; González, L.; Chávez, M.A.; Tytgat, J. The kunitz-type protein ShPI-1 inhibits serine proteases and voltage-gated potassium channels. Toxins 2016, 8, 1–17. [Google Scholar] [CrossRef]
- Norton, R.S.; Chandy, K.G. Venom-derived peptide inhibitors of voltage-gated potassium channels. Neuropharmacology 2017. [Google Scholar] [CrossRef]
- Prentis, P.; Pavasovic, A.; Norton, R. Sea Anemones: Quiet Achievers in the Field of Peptide Toxins. Toxins 2018, 10, 36. [Google Scholar] [CrossRef]
- Mitchell, M.L.; Shafee, T.; Papenfuss, A.T.; Norton, R.S. Evolution of cnidarian trans-defensins: Sequence, structure and exploration of chemical space. Proteins Struct. Funct. Bioinform. 2019, 87, 551–560. [Google Scholar] [CrossRef]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef]
- Diochot, S.; Loret, E.; Bruhn, T.; Béress, L.; Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 2003, 64, 59–69. [Google Scholar] [CrossRef]
- Diochot, S.; Schweitz, H.; Béress, L.; Lazdunski, M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J. Biol. Chem. 1998, 273, 6744–6749. [Google Scholar] [CrossRef]
- Sachkova, M.Y.; Singer, S.A.; Macrander, J.; Reitzel, A.M.; Peigneur, S.; Tytgat, J.; Moran, Y. The birth and death of toxins with distinct functions: A case study in the sea anemone Nematostella. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef]
- Wilcoxss, G.R.; Foghsli, H.; Nortonsgii, R.S. Refined Structure in Solution of the Sea Anemone Neurotoxin ShI. J. Biol. Chem. 1993, 268, 24707–24719. [Google Scholar]
- Gwynn, J.P.; Douglas, C.W.I. Comparison of amylase-binding proteins in oral streptococci. FEMS Microbiol. Lett. 1994, 124, 373–379. [Google Scholar] [CrossRef][Green Version]
- Homoki, J.; Gyémánt, G.; Balogh, P.; Stündl, L.; Bíró-Molnár, P.; Paholcsek, M.; Váradi, J.; Ferenc, F.; Kelentey, B.; Nemes, J.; et al. Sour cherry extract inhibits human salivary α-amylase and growth of Streptococcus mutans (a pilot clinical study). Food Funct. 2018, 9, 4008–4016. [Google Scholar] [CrossRef]
- Forester, S.C.; Gu, Y.; Lambert, J.D. Inhibition of starch digestion by the green tea polyphenol, (-)-epigallocatechin-3-gallate. Mol. Nutr. Food Res. 2012, 56, 1647–1654. [Google Scholar] [CrossRef]
- Barroso, H.; Ramalhete, R.; Domingues, A.; Maci, S. Inhibitory activity of a green and black tea blend on Streptococcus mutans. J. Oral Microbiol. 2018, 10, 1481322. [Google Scholar] [CrossRef]
- Gladkikh, I.; Monastyrnaya, M.; Leychenko, E.; Zelepuga, E.; Chausova, V.; Isaeva, M.; Anastyuk, S.; Andreev, Y.; Peigneur, S.; Tytgat, J.; et al. Atypical reactive center Kunitz-type inhibitor from the sea anemone Heteractis crispa. Mar. Drugs 2012, 10, 1545–1565. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]
- Kuzmic, P.; Elrod, K.C.; Cregar, L.M.; Sideris, S.; Rai, R.; Janc, J.W. High-Throughput Screening of Enzyme Inhibitors: Simultaneous Determination of Tight-Binding Inhibition Constants and Enzyme Concentration. Anal. Biochem. 2000, 50, 45–50. [Google Scholar] [CrossRef][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | α-Helix | β-Structure | β-Turn | Unordered Structure | ||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | I | II | III | |||
| Magnificamide | 0.0 | 1.7 | 1.7 | 21.4 | 13.7 | 35.1 | 24.3 | 38.9 |
| r-Magnificamide | 0.1 | 1.1 | 1.2 | 18.2 | 14.7 | 32.9 | 22.2 | 43.7 |
| Helianthamide | 19 | 32 | 18 | 31 | ||||
| r-Helianthamide | 11 | 33 | 23 | 33 | ||||
| Physico-Chemical Characteristics | Magnificamide | Helianthamide (PDB ID 4XON) |
|---|---|---|
| Radius of hydration (Å) | 10.25 | 10.21 |
| Hydrophilic surface area (Å2) | 2009.0 | 1976.3 |
| Hydrophobic surface area (Å2) | 1553.0 | 1275.4 |
| WDW volume (Å3) | 3727.1 | 4157.9 |
| Isoelectric point | 5.81 | 5.33 |
| Charge | −1.21 | −1.82 |
| Dipole moment (D) | 161.86 | 96.07 |
| Hydrophobic moment | 145.7 | 165.9 |
| Organisms | r-Magnificamide (1, 5, 10, 20 µM) | |
|---|---|---|
| Gram-positive | Staphylococcus aureus ATCC 21027 | Not active |
| Bacillus subtilis ATCC6633 | Not active | |
| Gram-negative | Escherichia coli VKPM B-7935 | Not active |
| Pseudomonas aeruginosa ATCC 27853 | Not active | |
| Fungi | Candida albicans KMM 455 | Not active |
| Channels | r-Magnificamide (10 µM) | |
|---|---|---|
| Voltage-gated potassium channels | Kv1.1, Kv1.2, Kv1.3, Kv1.4, Kv1.5, Kv1.6, Kv2.1, Kv3.1, Kv4.2, Kv10.1, hERG, Shaker * | Not active |
| Voltage-gated sodium channels | Nav1.2, Nav1.4, Nav1.5, Nav1.6, Nav1.8, BgNav1 * | Not active |
| Inhibitor name | Source | Mr, Da | Ki, M | Enzyme | Reference |
|---|---|---|---|---|---|
| Peptides | |||||
| Magnificamide | H. magnifica | 4770 | 1.7 × 10−10 7.7 × 10−9 | PPA HSA | |
| Helianthamide | S. helianthus | 4716 | 1 × 10−10 1 × 10−11 | PPA HPA | [18] |
| Tendamistat (HOE-467A) | Streptomyces tendae 4158 | 7958 | 9 × 10−12 | PPA | [13] |
| Parvulustat (Z-2685) | Streptomyces parvullus FH-1641 | 8129 | 2.8 × 10−11 + | PPA HSA | [16] |
| Low molecular compounds | |||||
| Acarbose | Actinomycetes | 646 | 0.6 × 10−6 1.3 × 10−6 | PPA HSA | [9] |
| Montbretin A | Crocosmia sp. | 789 | 8.1 × 10−9 | HPA | [11] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sintsova, O.; Gladkikh, I.; Kalinovskii, A.; Zelepuga, E.; Monastyrnaya, M.; Kim, N.; Shevchenko, L.; Peigneur, S.; Tytgat, J.; Kozlovskaya, E.; et al. Magnificamide, a β-Defensin-Like Peptide from the Mucus of the Sea Anemone Heteractis magnifica, Is a Strong Inhibitor of Mammalian α-Amylases. Mar. Drugs 2019, 17, 542. https://doi.org/10.3390/md17100542
Sintsova O, Gladkikh I, Kalinovskii A, Zelepuga E, Monastyrnaya M, Kim N, Shevchenko L, Peigneur S, Tytgat J, Kozlovskaya E, et al. Magnificamide, a β-Defensin-Like Peptide from the Mucus of the Sea Anemone Heteractis magnifica, Is a Strong Inhibitor of Mammalian α-Amylases. Marine Drugs. 2019; 17(10):542. https://doi.org/10.3390/md17100542
Chicago/Turabian StyleSintsova, Oksana, Irina Gladkikh, Aleksandr Kalinovskii, Elena Zelepuga, Margarita Monastyrnaya, Natalia Kim, Lyudmila Shevchenko, Steve Peigneur, Jan Tytgat, Emma Kozlovskaya, and et al. 2019. "Magnificamide, a β-Defensin-Like Peptide from the Mucus of the Sea Anemone Heteractis magnifica, Is a Strong Inhibitor of Mammalian α-Amylases" Marine Drugs 17, no. 10: 542. https://doi.org/10.3390/md17100542
APA StyleSintsova, O., Gladkikh, I., Kalinovskii, A., Zelepuga, E., Monastyrnaya, M., Kim, N., Shevchenko, L., Peigneur, S., Tytgat, J., Kozlovskaya, E., & Leychenko, E. (2019). Magnificamide, a β-Defensin-Like Peptide from the Mucus of the Sea Anemone Heteractis magnifica, Is a Strong Inhibitor of Mammalian α-Amylases. Marine Drugs, 17(10), 542. https://doi.org/10.3390/md17100542