2. Results and Discussion
The dichloromethane crude extract of
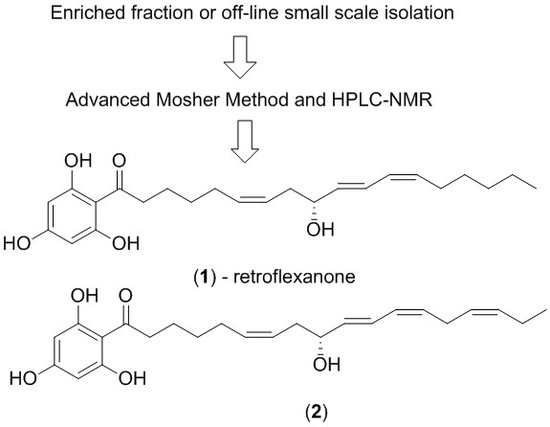
C. retroflexa was subjected to silica gel flash chromatography to yield retroflexanone (
1), and a structurally related phloroglucinol analogue (
2) in an enriched fraction (
Scheme 1). The enriched fraction consisted of an approximate 3:1 ratio of the compounds (
2) and (
1) respectively. This enriched fraction was subjected to a complete 2D NMR analysis (via HPLC-NMR) whereby acquisition of the 2D NMR spectra for both compounds was achieved in the stop-flow mode. This permitted each compound to be structurally assigned. This approach would allow for rapid structure assignment of the Mosher esters based only on the
1H NMR comparison of the Mosher esters to the natural compound.
The enriched fraction was subjected to the advanced Mosher method using α-methoxy-α-trifluoromethylphenylacetyl chloride (MPTA-Cl). As there was a greater amount of (2) present in the fraction, it was suspected that this would be derivatised preferentially over retroflexanone (1).
The enriched fraction was reacted with dry pyridine and (
R)-(−)-MTPA-Cl and (
S)-(+)-MTPA-Cl (α-methoxy-α-trifluoromethylphenylacetyl chloride) respectively to prepare the Mosher esters, paying particular attention to the fact that the (
R)-(−)-MTPA-Cl gives the (
S)-MTPA ester and vice versa [
3]. Esterification yielded the diastereoisomeric (
S)-MTPA and (
R)-MTPA esters. The
1H NMR chemical shift differences between the MTPA esters [Δδ
SR = δ
s − δ
R] were established by comparing the
1H NMR spectra of the esters to that of the original
1H NMR spectrum of retraflexaonone, and are given in
Table 1.
Each reaction was subsequently analysed individually via HPLC-NMR which revealed the presence of a new peak in the HPLC chromatogram, corresponding to the Mosher esters of the structurally related phloroglucinol analogue (
2a and
2b) respectively. The absolute configuration of (
2) has previously been determined using the Horeau, and ozonolysis methods [
12]. Analysis and interpretation of the
1H NMR data for this compound provided a means to assess whether the formation of Mosher esters formed in a mixture could unequivocally provide the correct absolute configuration. By observing which of the proton chemical shifts and therefore which substituent is affected by the formation of each derivative, the absolute configuration of the secondary alcohol is able to be established. The
1H NMR chemical shift differences between the MTPA esters [Δδ
SR = δ
s − δ
R] were established for compound (
2) (see
Table 1). Despite some of the δ
SR values being unresolved or solvent suppressed in the HPLC-NMR analysis, the values that could be obtained were allocated within the same substituent based on their sign (positive or negative) and this permitted the absolute configuration of (
2) to be deduced as being
R, which was in agreement with the previous findings (refer to the retroflexanone example for complete details of the analysis and the supporting information file). With the methodology secured, the determination of retraflexonanone (
1) was targeted.
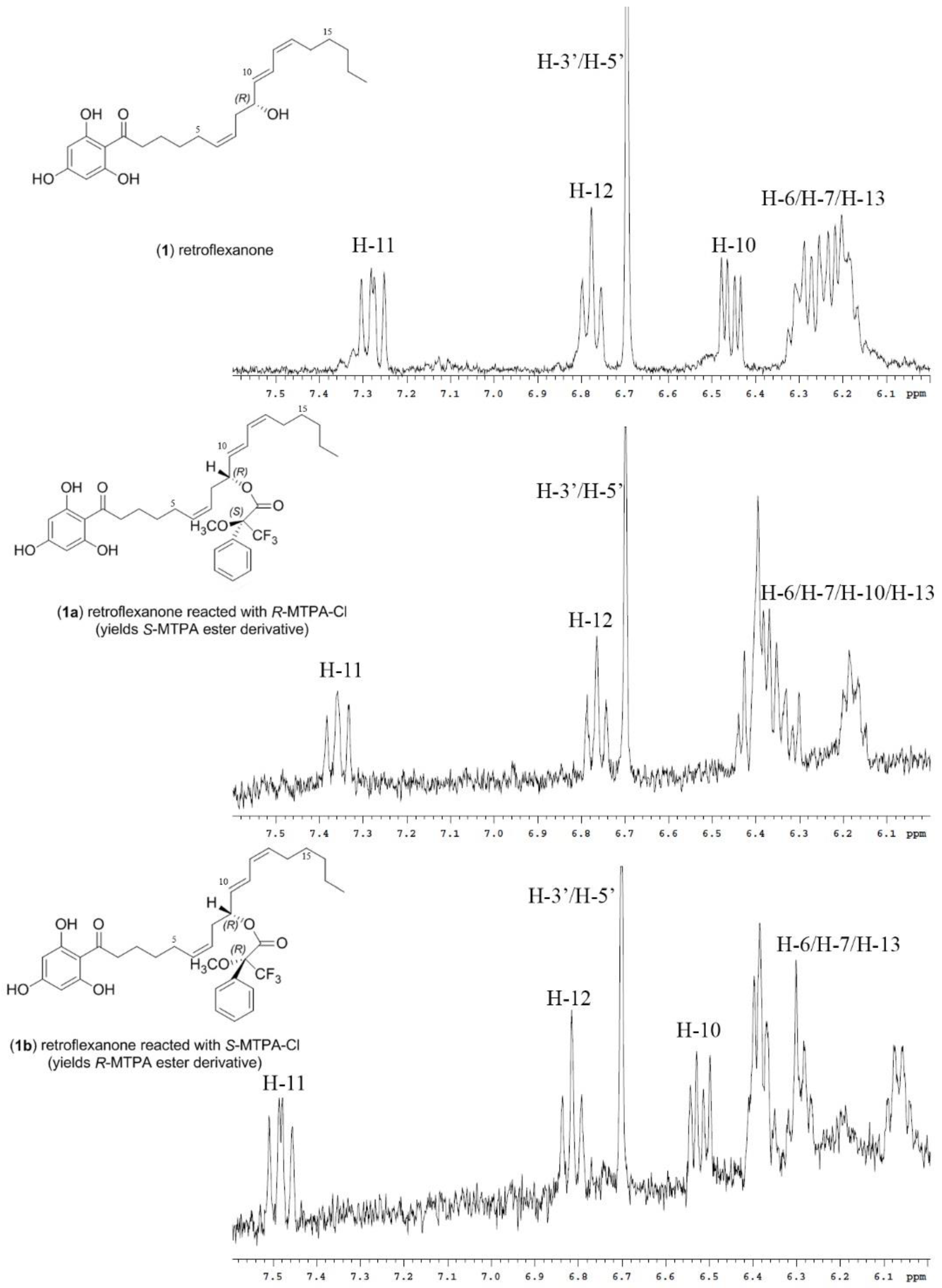
It was noted during the HPLC-NMR analysis of the advanced Mosher reactions, that both retroflexanone (1) and the structurally related phloroglucinol (2) were still present in the enriched fraction, and that neither had completely reacted with the Mosher reagents. Moreover the Mosher esters of retroflexanone (1) were not observed. The reaction was carried out on a number of occasions using slightly different reaction conditions, but each time the Mosher esters of retroflexanone (1) were not observed.
An alternative approach was taken where retroflexanone (
1) was rapidly purified from the enriched fraction by semi-preparative reversed phase HPLC. It was eventually established that retroflexanone (
1) would remain intact long enough if kept in dichloromethane or methanol at −80 °C, but that it would degrade rapidly in the presence of chloroform or if kept at room temperature. Retroflexanone (
1) was reacted with dry pyridine and the Mosher reagents (
R)-(−)-MTPA-Cl and (
S)-(+)-MTPA-Cl respectively. Subsequent HPLC-NMR analysis was performed on each of the reactions and this revealed the presence of the new Mosher ester derivatives of retroflexanone (
1a and
1b), together with residual retroflexanone (
1) that had not reacted with the Mosher reagents. Stop-flow HPLC-NMR was carried out on the two Mosher esters (
1a and
1b) to obtain their corresponding
1H NMR spectra. The
1H NMR spectrum (
Figure 1) of retroflexanone (
1) was compared to the spectra obtained for the Mosher esters (
1a and
1b). Characteristic upfield and downfield shifts of the
1H NMR chemical shifts were noted depending on their occurrence on either side of the stereogenic secondary alcohol.
Differences in the proton chemical shifts of
1a and
1b are given in
Table 1. The majority of the Δδ
SR values could be calculated despite some unassigned or solvent suppressed signals. The two diastereomeric MTPA esters for retroflexanone are given in
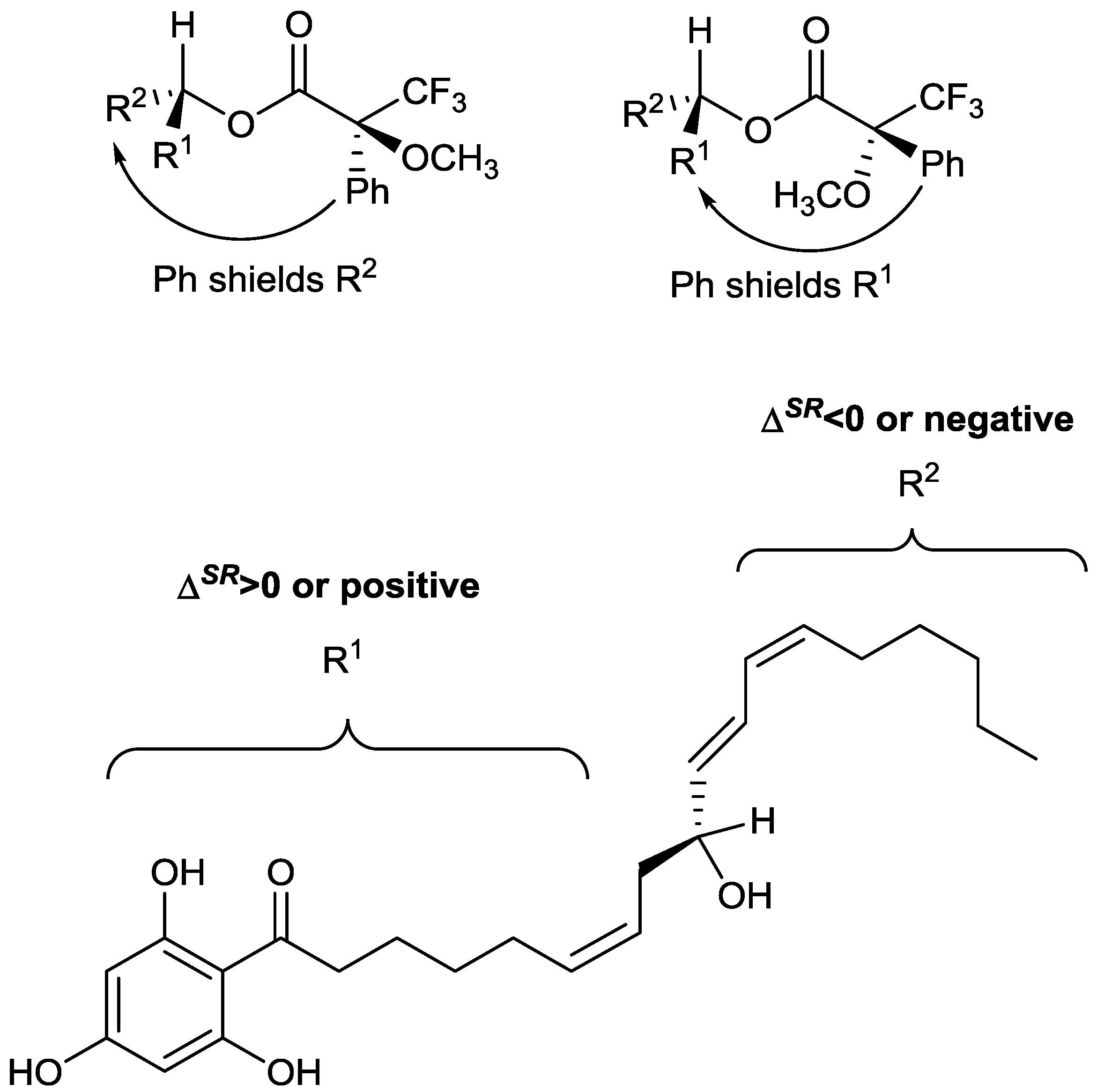
Figure 2, with R
1 and R
2 representing that alkyl chains on either side of the secondary alcohol moiety. It is shown in
Figure 2 that the phenyl group from the MTPA will either shield the R
1 or R
2 group, depending on which diastereomer is present.
Using the conformations shown in
Figure 2 for the MTPA esters of generic alcohols, the R
1 and R
2 substituents could be assigned. Protons that have positive Δδ
SR values reside within R
1, whereas those with negative values of Δδ
SR belong to the R
2 substituent [
13].
Figure 2 shows that all of the positive Δδ
SR are located on the left hand side, while the negative Δδ
SR are located on the right hand side of the molecule. Specifically protons with positive Δδ
SR values (those in R
1) are all on one side (front) of the plane of the MTPA moiety, whereas those with negative values are all on the opposite (back) side of that plane. This, in conjunction with the Cahn Ingold Prelog convention, permitted the assignment of the original secondary alcohol absolute configuration to be assigned as
R.
Since the structure of retroflexanone (
1) had previously been successfully and unequivocally characterised by HPLC-NMR [
1], an attempt was made to dissolve pure retroflexanone (
1) in CDCl
3 to obtain conventional NMR data. Despite retroflexanone (
1) being unstable in CDCl
3, this solvent was chosen to make the comparison to other related compounds in the literature easier. 1D and 2D NMR data was successfully obtained, however the methylene protons at position C-2 displayed unexpected splitting patterns. It was expected that these methylene protons would display a typical triplet. However the methylene at position C-2 appeared as a set of non-equivalent protons displaying a multiplet-type splitting. Initially it was thought that retroflexanone (
1) may have degraded, but subsequent interpretation of the NMR data still supported the initial structure. It was proposed that when retroflexanone (
1) and other structurally related compounds are dissolved in CDCl
3, that non-equivalent protons displaying multiplet-type splitting is observed rather than a typical triplet due to a pro-chiral effect created by the carbonyl group at C-1. To test this theory, the structurally related analogue (
2) was placed into CDCl
3 and CD
3OD. The NMR data revealed that in CDCl
3 the methylene protons at position C-2 displayed multiplet-type splitting, but in CD
3OD the methylene protons appear as a typical triplet (see
supporting information section). This confirmed that the appearance of these methylene protons is dependent on the NMR solvent used for acquisition. Detailed NMR data can be found in supporting materials.
3. Materials and Methods
3.1. General Experimental Procedures
All organic solvents used were analytical reagent (AR or GR), UV spectroscopic, or HPLC grades with Milli-Q water also being used. 1H (500 MHz) and 13C (125 MHz) NMR spectra were acquired in CDCl3 and CD3OD on a 500 MHz Agilent DD2 NMR spectrometer, or a 300 MHz Bruker Avance III NMR spectrometer with referencing to solvent signals (δH 7.26; δC 77.0 and, δH 3.31; δC 49.0). Two-dimensional NMR experiments recorded included gradient correlation spectroscopy (gCOSY), heteronuclear single-quantum correlation spectroscopy with adiabatic pulses (HSQCAD), and gradient heteronuclear multiple-bond spectroscopy with adiabatic pulses (gHMBCAD).
3.2. Alga Material
C. retroflexa was collected via SCUBA on 21 April 2010 from Governor Reef (near Indented Head), Port Phillip Bay, Victoria, Australia. The alga was identified by Dr Gerry Kraft (The University of Melbourne) and a voucher specimen (designated the code number 2010-09) is deposited at the School of Science (Applied Chemistry and Environmental Science), RMIT University.
3.3. Fractionation of Dichloromethane Crude Extract
The alga (133 g, wet weight) was extracted with 3:1 MeOH/CH2Cl2 (1 L). The crude extract was decanted and concentrated under reduced pressure and sequentially solvent partitioned (triturated) into CH2Cl2 and MeOH soluble extracts, respectively. A portion of the CH2Cl2 crude extract (445 mg) of C. retroflexa was subjected to silica gel flash chromatography. Silica gel flash chromatography was carried out using Davisil LC35Å silica gel (40–60 mesh) using a 20% stepwise solvent elution from 100% petroleum spirits (60–80 °C) to 100% CH2Cl2 to 100% EtOAc and finally to 100% MeOH. The 60% CH2Cl2/EtOAc fraction yielded a mixture which contained retroflexanone (1) and the structurally related phloroglucinol (2) (136.2 mg).
3.4. Advanced Mosher Derivatisation of Phloroglucinol (2) in an Enriched Fraction
The 60% CH2Cl2/EtOAc fraction (1 mg) was dissolved in CH2Cl2 (1 mL) to which (R)-(−)-MTPA-Cl (4.0 mg, 16 µmol) or alternatively (S)-(+)-MTPA-Cl (4.5 mg, 18 µmol) was added. Dry pyridine (200 µL, 760 µmol) was then added and the reaction stirred for 4 h. The reaction mixtures were then dried under a stream of nitrogen and reconstituted into HPLC-NMR grade CH3CN.
3.5. HPLC Purification of Retroflexanone
Semi-preparative HPLC was carried out on a Dionex P680 (solvent delivery module) equipped with a Dionex UVD340U PDA detector and a Foxy Jr. automated fraction collector. An isocratic HPLC method (85% CH3CN/H2O) was employed using a Phenomenex Luna (2) 100 Å C18 250 × 10 mm (5 µm) HPLC column. The automatic fraction collector was programmed to collect the compounds based on their retention times at a flow rate of 4.0 mL/min. A portion of the 60% CH2Cl2/EtOAc fraction was subjected to reversed phase HPLC to yield retroflexanone (1) (2.8 mg) and the phloroglucinol (2) (6.0 mg).
3.6. Advanced Mosher Reaction of Retroflexanone
Retroflexanone (1.4 mg, 3.5 µmol) was dissolved in CH2Cl2 (1 mL) to which (R)-(−)-MTPA-Cl (50 mg, 198 µmol, 57 equiv.) or alternatively (S)-(+)-MTPA-Cl (50 mg, 198 µmol, 57 equiv.) was added. Dry pyridine (300 µL, 1140 µmol, 325 equiv.) was then added and the reaction stirred for 4 h. The reaction mixtures were then dried under a stream of nitrogen and reconstituted into HPLC-NMR grade CH3CN.
3.7. HPLC-NMR Analysis
For HPLC-NMR details, please refer to Brkljaca and Urban [
14]. For stop-flow HPLC-NMR modes, 50-μL injections of the reconstituted samples in CH
3CN were injected onto an Agilent Eclipse Plus C
18 (150 × 4.6) 5-µ column using a solvent composition of 75% CH
3CN/D
2O at a flow rate of 1 mL/min. In the stop-flow HPLC-NMR mode WET1D NMR experiments were acquired.
3.8. On-Line NMR Data Obtained via HPLC-NMR
Retroflexanone or 9R-hydroxy-1-(2,4,6-trihydroxyphenyl)-6Z,10E,12Z-Octadecatrien-1-one reacted with (R)-(−)-MTPA-Cl yielding the S-MTPA ester (1a); 1H NMR (500 MHz, 75% CH3CN/D2O) δ 8.28 (5H, m, MTPA-aromatic), 7.36 (1H, dd, J = 11.5, 13.0 Hz, H-11), 6.76 (1H, dd, J = 11.0, 11.5 Hz, H-12), 6.70 (2H, s, H-3′/H-5′), 6.14−6.45 (4H, m, H-6/H-7/H-10/H-13), 4.83 (1H, m, H-9), 4.35 (3H, s, MTPA-OCH3), 3.85 (2H, t, J = 7.5 Hz, H-2), 3.33 (2H, m, H-8), 2.44 (2H, m, H-3), 2.18 (2H, m, H-4), 2.10 (2H, m, H-17), 1.70 (3H, t, J = 7.0 Hz, H-18), ND (11H, H-5/H-14/H-15/H-16,/2′-OH/4′-OH/6′-OH), ND indicates signal not detected.
Retroflexanone or 9R-hydroxy-1-(2,4,6-trihydroxyphenyl)-6Z,10E,12Z-Octadecatrien-1-one reacted with (S)-(+)-MTPA-Cl yielding the R-MTPA ester (1b); 1H NMR (500 MHz, 75% CH3CN/D2O) δ 8.28 (5H, m, MTPA-aromatic), 7.48 (1H, dd, J = 11.0, 15.5 Hz, H-11), 6.82 (1H, dd, J = 10.5, 11.5 Hz, H-12), 6.70 (2H, s, H-3′/H-5′), 6.52 (1H, dd, J = 7.5, 15.5, H-10), 6.02–6.42 (3H, m, H-6/H-7/H-13), 4.83 (1H, m, H-9), 4.32 (3H, s, MTPA-OCH3), 3.84 (2H, t, J = 7.5 Hz, H-2), 3.26 (2H, m, H-8), 2.43 (2H, m, H-3), 2.16 (2H, m, H-4), 2.10 (2H, m, H-17), 1.70 (3H, m, H-18), ND (11H, H-5/H-14/H-15/H-16/2′-OH/4′-OH/6′-OH), ND indicates signal not detected.
9R-hydroxy-1-(2,4,6-trihydroxy-phenyl)-6Z,10E,12Z,15Z-Octadecatetraen-1-one reacted with (R)-(−)-MTPA-Cl yielding the S-MTPA ester (2a); 1H NMR (500 MHz, 75% CH3CN/D2O) δ 8.27 (5H, m, MTPA-aromatic), 7.38 (1H, dd, J = 11.5, 14.5 Hz, H-11), 6.76 (1H, dd, J = 10.5, 11.5 Hz, H-12), 6.69 (2H, s, H-3′/H-5′), 6.07−6.46 (6H, m, H-6/H-7/H-10/H-13/H-15/H-16), 4.34 (3H, s, MTPA-OCH3), 3.84 (2H, t, J = 7.5 Hz, H-2), 3.65 (2H, m, H-14), 3.35 (1H, m, H-8a), 3.29 (1H, m, H-8b), 2.42 (2H, p, J = 7.5 Hz, H-3), 2.19 (2H, p, J = 7.5 Hz, H-4), 1.75 (3H, t, J = 7.5 Hz, H-18), ND (8H, H-5/H-9/H-17/2′-OH/4′-OH/6′-OH), ND indicates signal not detected.
9R-hydroxy-1-(2,4,6-trihydroxy-phenyl)-6Z,10E,12Z,15Z-Octadecatetraen-1-one reacted with (S)-(+)-MTPA-Cl to yield the R-MTPA ester (2b); 1H NMR (500 MHz, 75% CH3CN/D2O) δ 8.30 (5H, m, MTPA-aromatic), 7.52 (1H, dd, J = 11.0, 15.5 Hz, H-11), 6.82 (1H, dd, J = 10.5, 11.0 Hz, H-12), 6.70 (2H, s, H-3′/H-5′), 6.55 (1H, dd, J = 7.5, 15.5 Hz, H-10), 6.04−6.42 (5H, m, H-6/H-7/H-13/H-15/H-16), 4.32 (3H, s, MTPA-OCH3), 3.84 (2H, t, J = 7.0 Hz, H-2), 3.73 (2H, m, H-14), 3.31 (1H, m, H-8a), 3.24 (1H, m, H-8b), 2.41 (2H, p, J = 7.0 Hz, H-3), 2.15 (2H, p, J = 7.0 Hz, H-4), 1.76 (3H, t, J = 7.0 Hz, H-18), ND (8H, H-5/H-9/H-17/2′-OH/4′-OH/6′-OH), ND indicates signal not detected.
3.9. Off-Line NMR Data
Retroflexanone (1); unstable oil that darkened over time; 1H NMR (500 MHz, CDCl3) δ 6.52 (1H, dd, J = 11.5, 15.0 Hz, H-11), 5.97 (1H, dd, J = 11.0, 11.5 Hz, H-12), 5.88 (2H, s, H-3′/H-5′), 5.70 (1H, dd, J = 6.0, 15.0 Hz, H-10), 5.54 (1H, m, H-6), 5.45 (1H, dt, J = 7.0, 11.0 Hz, H-13), 5.39 (1H, m, H-7), 4.26 (1H, dt, J = 6.0, 6.5 Hz, H-9), 3.14 (1H, m, H-2a), 3.00 (1H, m, H-2b), 2.41 (1H, m, H-8a), 2.31 (1H, m, H-8b), 2.17 (2H, m, H-14), 2.12 (2H, m, H-5), 1.72 (2H, m, H-3), 1.48 (2H, m, H-4), 1.37 (2H, m, H-15), 1.29 (4H, m, H-16/H-17), 0.88 (3H, t, J = 7.0 Hz, H-18), ND (9-OH/2′-OH/4′-OH/6′-OH), ND indicates signal not detected; 13C NMR (125 MHz, CDCl33) δ 134.6 (CH, C-10), 133.5 (CH, C-6), 133.4 (CH, C-13), 127.5 (CH, C-12), 126.1 (CH, C-11), 124.7 (CH, C-7), 95.4 (CH, C-3′/C-5′), 72.5 (CH, C-9), 43.6 (CH2, C-2), 35.2 (CH2, C-8), 31.4 (CH2, C-16), 29.3 (CH2, C-4/C-15), 27.4 (CH2, C-14), 26.8 (CH2, C-5), 24.4 (CH2, C-3), 22.6 (CH2, C-17), 14.1 (CH3, C-18), ND (C-1/C-1′/C-2′/C-4′/C-6′), ND indicates signal not detected.
9R-hydroxy-1-(2,4,6-trihydroxy-phenyl)-6Z,10E,12Z,15Z-Octadecatetraen-1-one (2); unstable oil that darkened over time; 1H NMR (500 MHz, CDCl3) δ 6.55 (1H, dd, J = 11.5, 15.0 Hz, H-11), 5.98 (1H, dd, J = 11.0, 11.0 Hz, H-12), 5.88 (2H, bs, H-3′/H-5′), 5.72 (1H, dd, J = 6.0, 14.5 Hz, H-10), 5.27−5.59 (5H, m, H-6/H-7/H-13/H-15/H-16), 4.27 (1H, dt, J = 6.0, 6.5 Hz, H-9), 3.11 (1H, m, H-2a), 3.00 (1H, m, H-2b), 2.93 (2H, dd, J = 7.0, 7.5 Hz, H-14), 2.39 (1H, m, H-8a), 2.32 (1H, m, H-8b), 2.12 (4H, m, H-5/H-17), 1.71 (2H, m, H-3), 1.48 (2H, p, J = 7.0 Hz, H-4), 0.97 (3H, t, J = 7.5 Hz, H-18), ND (9-OH/2′-OH’4′-OH/6′-OH), ND indicates signal not detected; 1H NMR (300 MHz, CD3OD) δ 6.55 (1H, dd, J = 11.1, 15.0 Hz, H-11), 5.97 (1H, dd, J = 10.8, 11.1 Hz, H-12), 5.83 (2H, s, H-3′/H-5′), 5.68 (1H, dd, J = 6.6, 15.0 Hz, H-10), 5.27−5.55 (5H, m, H-6/H-7/H-13/H-15/H-16), 4.15 (1H, dt, J = 6.3, 6.6 Hz, H-9), 3.06 (2H, t, J = 7.5 Hz, H-2), 2.94 (2H, dd, J = 7.0, 7.5 Hz, H-14), 2.32 (2H, m, H-8), 2.10 (4H, m, H-5/H-17), 1.69 (2H, p, J = 7.5 Hz, H-3), 1.45 (2H, p, J = 7.5 Hz, H-4), 0.98 (3H, t, J = 7.5 Hz, H-18), ND (9-OH/2′-OH’4′-OH/6′-OH), ND indicates signal not detected.







{kind=link}
{kind=link}
{kind=link}
{kind=link}