Abstract
Nine new angucycline glycosides designated urdamycins N1–N9 (1–9), together with two known congener urdamycins A (10) and B (11), were obtained from a mangrove-derived Streptomyces diastaticus subsp. SCSIO GJ056. The structures of new compounds were elucidated on the basis of extensive spectroscopic data analysis. The absolute configurations of 6–9 were assigned by electronic circular dichroism calculation method. Urdamycins N7 (7) and N8 (8) represent the first naturally occurring (5R, 6R)-angucycline glycosides, which are diastereomers of urdamycins N6 (6) and N9 (9), respectively.
1. Introduction
The angucycline group members are type II polyketide derived metabolites obtained exclusively from actinomycetes [1,2,3]. Angucycline compounds exhibited various bioactivities, including antitumor, cytostatic, enzyme inhibition, antibacterial, antiviral, and inhibition of platelet aggregation function [4,5,6,7].
During the course of searching for novel anti-infective and antitumor agents from the marine environment, we found that the chemical profile of strain SCSIO GJ056 cultivated in AM2 medium revealed an array of secondary metabolites showing typical UV/VIS absorptions, which were similar to those of angucyclines/anthracyclines. Subsequent solvent extraction and isolation procedures led to the purification and structure elucidation of nine new angucycline glycosides, named urdamycins N1–N9 (1–9), together with two known urdamycins A (10) and B (11). Urdamycins N7 (7) and N8 (8) represent the first naturally occurring (5R, 6R)-angucycline glycosides. Herein, we report the fermentation, isolation, and structure elucidation of these compounds.
2. Results and Discussion
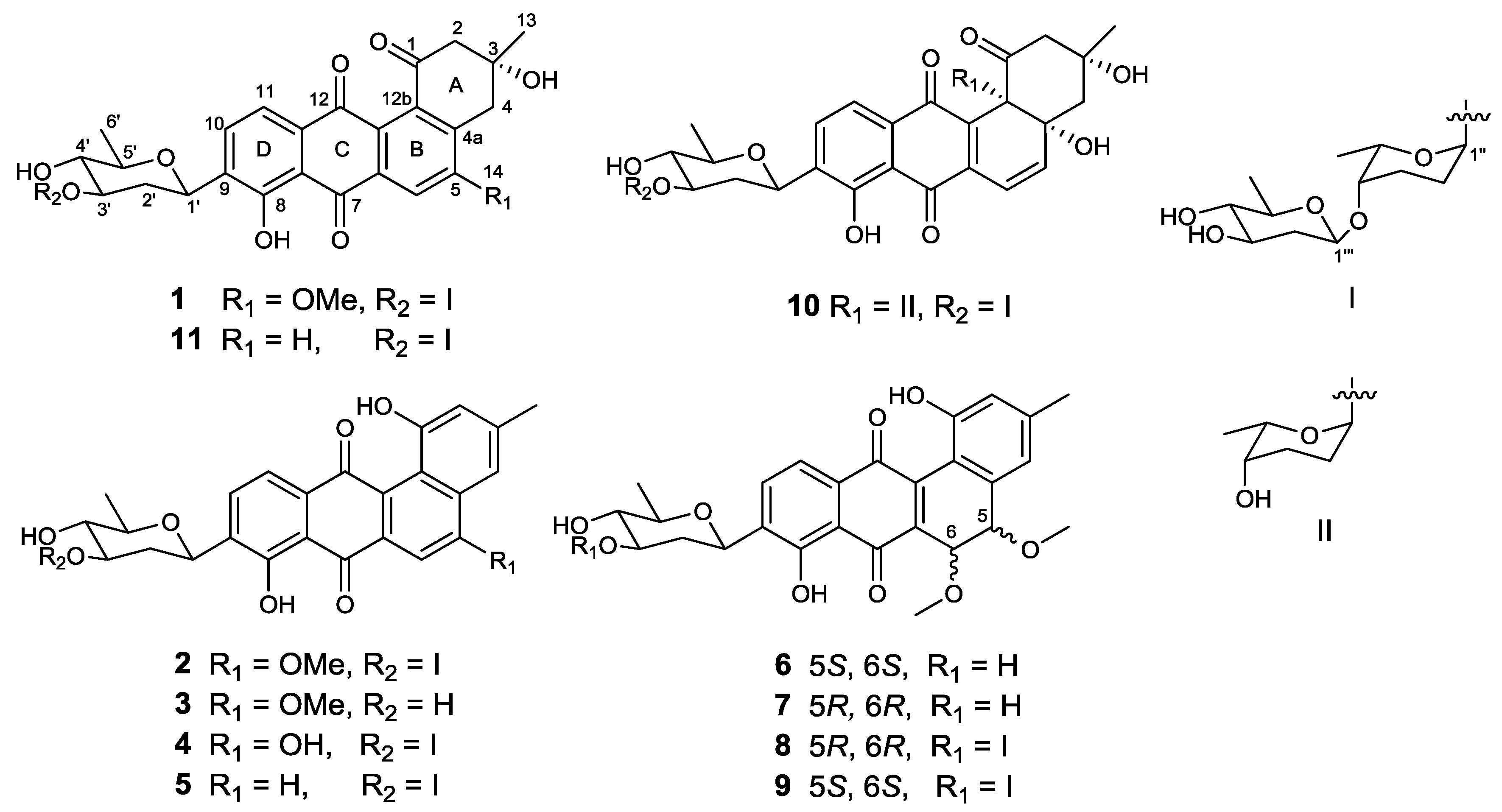
The strain SCSIO GJ056 was fermented (15 L) and the fermentation broth was extracted with butanone. The extract was subjected to repetitive silica gel column chromatography, followed by preparative HPLC purification to yield compounds 1–11 (Figure 1). The known urdamycins A (10) and B (11) were identified by comparisons of MS, 1H, and 13C NMR spectroscopic data with those previously reported [8].
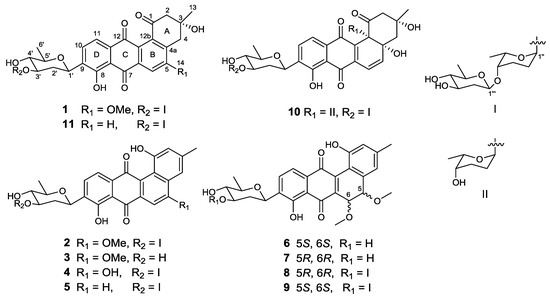
Figure 1.
Structures of compounds 1–11.
Compound 1 was obtained as a yellowish powder. Its molecular formula was determined to be C38H46O14 on the basis of HRESIMS peak at m/z 725.2834 [M − H]−, indicating 16 degrees of unsaturation. The 13C and DEPT NMR spectra of 1 displayed 38 carbon resonances, including five methyls, six methylenes, 14 methines, and 13 nonprotonated carbons. The 1H NMR spectrum showed one chelated hydroxy group signal at δH 12.55 (1H, br s, 8-OH), a pair of ortho-coupled aromatic proton signals at δH 7.81 (d, 7.8 Hz, H-10) and 7.59 (d, 7.8 Hz, H-11), and a singlet aromatic proton signal at δH 7.65 (s, H-6). The HMBC correlations (Figure 2) from H-6 to C-4a, C-5, C-7, and C-12a, from H-11 to C-7a, C-9, and C-12, and from H-10 to C-8, C-9, and C-11a confirmed the existence of the anthraquinone skeleton (rings B, C, and D). Further HMBC correlations of H2-2/C-1, C-12b, C-4; H2-4/C-2, C-4a, C-12b; and H3-13/C-2, C-3, C-4 allowed the assignment of the angular ring (ring A) with a methyl group (CH3-13) substitution at C-3. A methoxy group (OCH3-14) attached at C-5 in ring B was deduced by the HMBC correlation of H3-14/C-5. A hydroxy group linked at C-3 in ring A was inferred based on the 13C NMR chemical shift at δC 71.8. The absolute configuration of C-3 was tentatively deduced to be R, which was identical with that of urdamycinone B and N05WA963D in light of the similar 13C NMR resonances of C-3 and CH3-13, as well as the similar biosynthetic pathway [8,9].
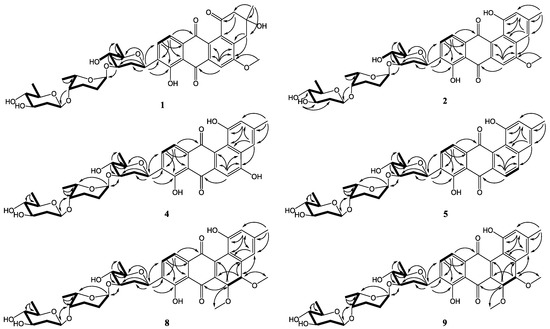
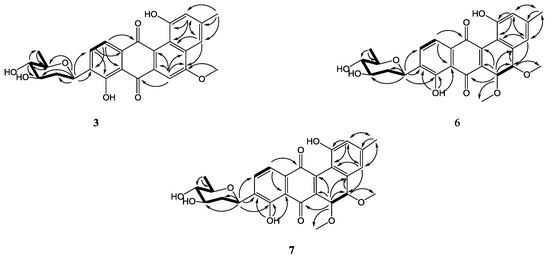
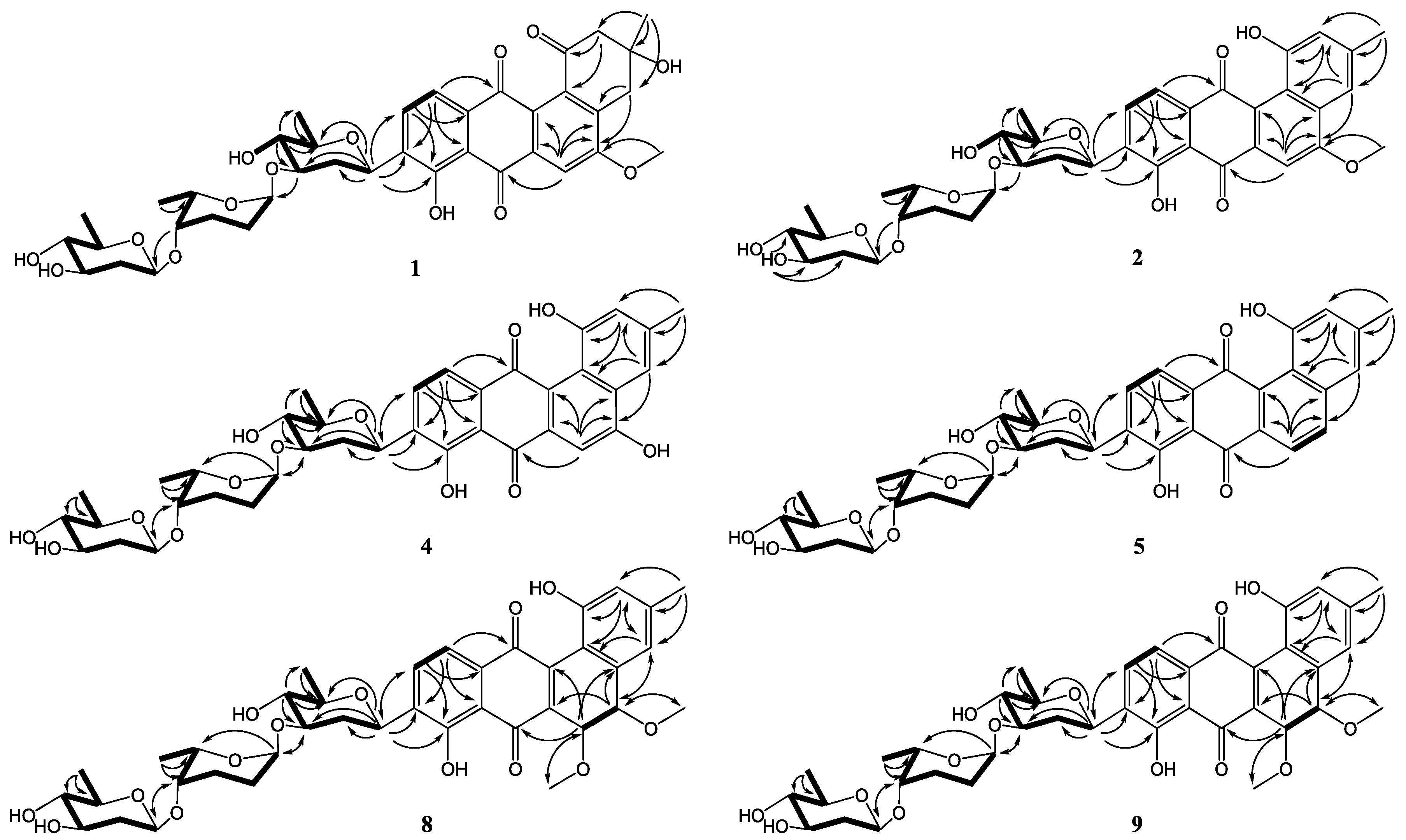
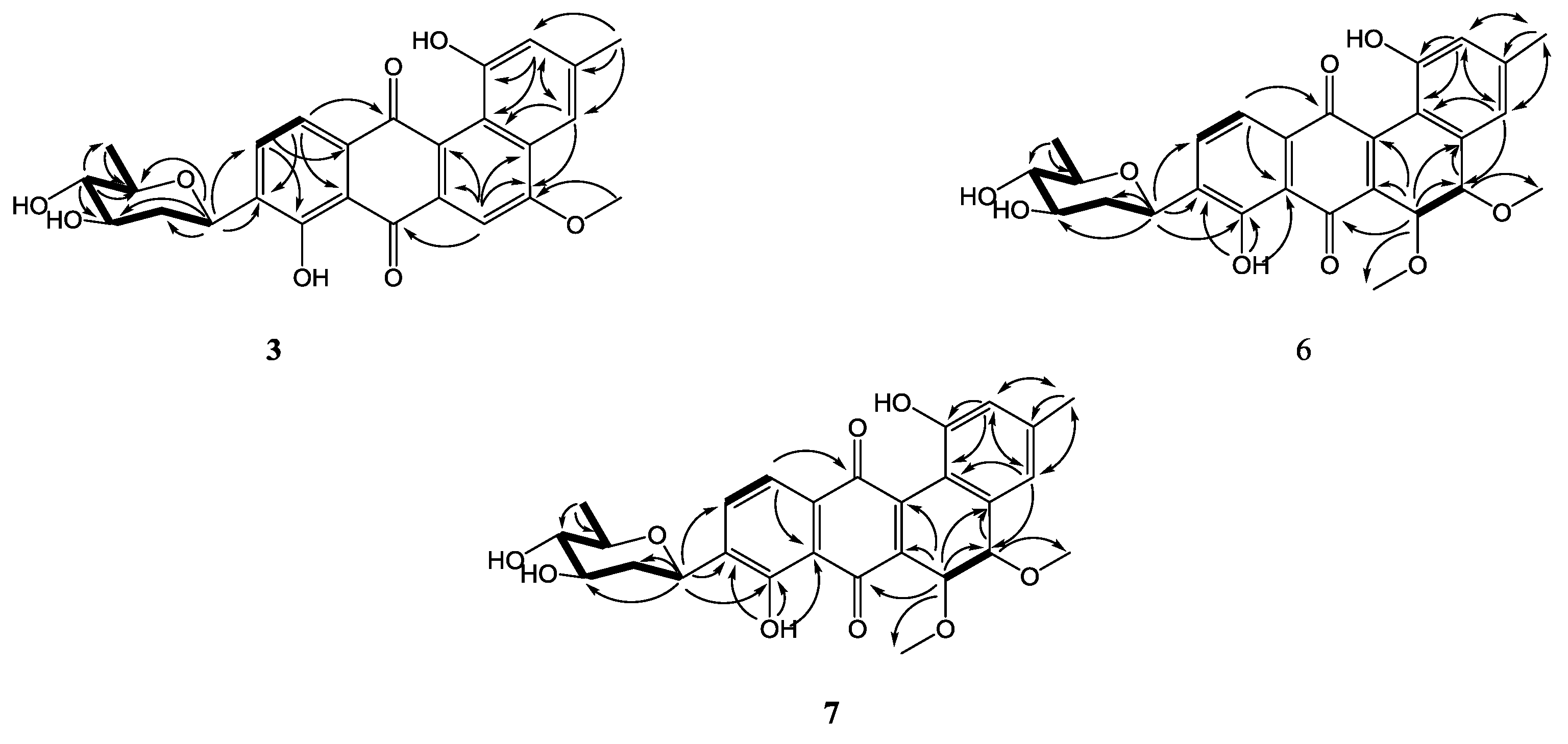
Figure 2.
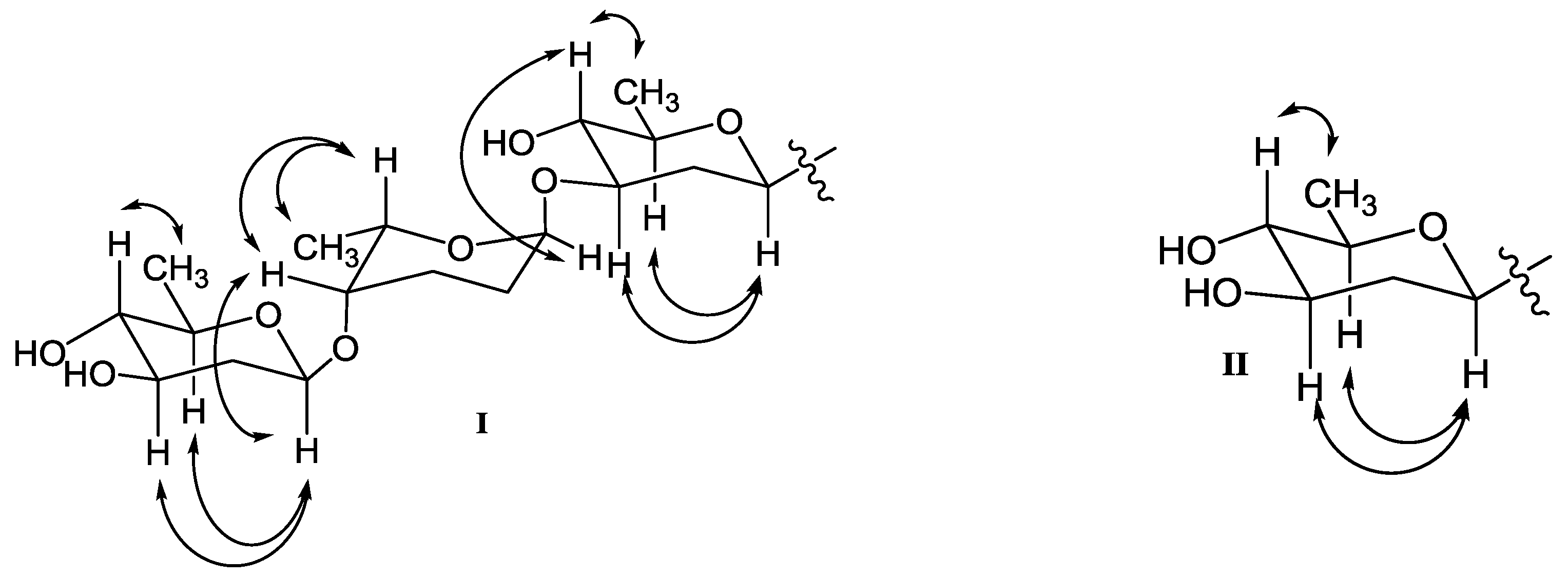
COSY (bold) and selected HMBC (arrow) correlations for 1–9.
In addition, three anomeric methine signals at δH 4.82 and δC 71.1 (CH-1′), δH 4.96 and δC 97.4 (CH-1″), and δH 4.45 and δC 101.5 (CH-1′′′), together with three doublet methyl signals at δH 1.37 (CH3-6′), δH 1.16 (CH3-6′′), and δH 1.23 (CH3-6′′′) revealed the presence of a trisaccharide moiety consisting of three deoxy sugars in 1. The 1H-1H COSY spectrum allowed the full assignment of the sugar moieties from CH-1′ to CH3-6′, from CH-1′′ to CH3-6′′, and from CH-1′′′ to CH3-6′′′. The HMBC correlations of H-3′/C-1′′ and H-4′′/C-1′′′ confirmed the existence of a β-olivose-(1→4)-α-rhodinose-(1→3)-β-olivosyl unit. This trisaccharide was connected with the aglycone at C-9 based on the HMBC correlations of H-1′/C-9 and H-9/C-1′. The relative configuration of the trisaccharide moiety was deduced by 1H-1H coupling constants (Table 1) and NOE experiment (Figure 3). Detailed comparisons showed that the 1H and 13C NMR spectroscopic data of the sugar units were almost identical with those in urdamycin A [10,11]. Thus, the structure of 1 was determined and named urdamycin N1.

Table 1.
The 1H and 13C NMR data of compounds 1–3 (δ in ppm, J in Hz).
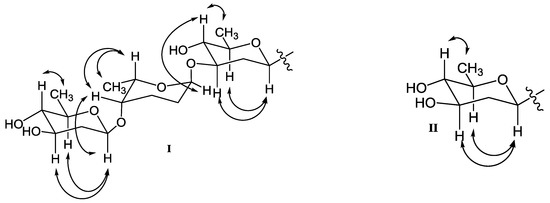
Figure 3.
Key NOESY correlations of β-olivose-(1→4)-α-rhodinose-(1→3)-β-olivose in 1, 2, 4, 5, 8, and 9 (I), and β-olivose in 3, 6 and 7 (II).
Compound 2, isolated as a dark red powder, has the molecular formula of C38H44O13 on the basis of HRESIMS peak at m/z 707.2708 [M − H]−, showing 17 degrees of unsaturation and an 18 amu less than that of compound 1. An obvious red shift on the UV-VIS spectrum of 2 relative to that of 1 indicated an additional conjugated system in 2. The 13C and DEPT NMR data of 2 displayed 38 carbon signals attributable to five methyls, four methylenes, 16 methines, and 13 nonprotonated carbons. The 1H and HSQC NMR spectra suggested three singlet olefnic proton signals at δH 7.58 (H-4), δH 7.54 (H-6), and δH 6.99 (H-2), and a pair of ortho-coupled aromatic proton signals at δH 7.82 (d, 7.6 Hz, H-10) and 7.68 (d, 7.6 Hz, H-11). Comparing the 1H and 13C NMR spectroscopic data to those of 1 revealed that 2 possessed a similar core structure with that of 1. The difference between 2 and 1 was the aromatization of ring A in 2, which supported by the HMBC correlations from CH3-13 to C-2, C-3, and C-4, from H-2 to C-1, C-4, and C-12b, and from H-4 to C-2, C-4a, and C-12b. Compound 2 possessed the same trisaccharide moiety with 1 according to similar 1H and 13C NMR signals in aliphatic area. The structure of 2 was elucidated as shown in Figure 1 by detailed analysis of 2D NMR spectra data.
Compound 3, a dark green powder, was isolated as minor component from the extract. Its molecular formula of C26H24O8 was determined by the HRESIMS peak at m/z 463.1409 [M − H]−, indicating 15 degrees of unsaturation. Comprehensive analysis of its 1H and 13C NMR spectroscopic data revealed that 3 had the same aglycone with that of 2. However, a set of 1H and 13C resonances ascribed to β-olivose-(1→4)-α-rhodinosyl moiety disappeared, indicating the absence of two sugar units in 3. This is consistent with the HRESIMS data, which showing a C12H20O5 fragment loss relative to 2. Therefore, the structure of 3 was established and named urdamycin N3.
Compound 4 was obtained as a dark green powder. Its molecular formula was determined to be C37H42O13 by the HRESIMS peak at m/z 693.2554 [M − H]−. The 1H and 13C NMR data of 4 were closely similar to those of 2, except that the methoxy signals at δH 4.14, δC 56.6 in 2 were absent. The 13C NMR signal of C-5 shifted from δC 160.3 in 2 to δC 163.6 in 4, indicating the OMe-5 in 2 was replaced by OH-5 in 4. Compound 4 was named urdamycin N4.
Compound 5 was obtained as a red powder. The molecular formula of C37H42O12, as determined by HRESIMS, which was one oxygen atom less than that of 4. The 1H and 13C NMR spectroscopic data were similar with those of 4, except that two pairs of ortho-coupled aromatic signals were observed. Additionally, the 13C NMR signal at δC 163.6 for the oxygen-bearing aromatic C-5 in 4 was replaced by an aromatic methine signal at δC 135.4. Thus, the structure of 5 was determined as 5-demethoxy-urdamycin N2, designated as urdamycin N5.
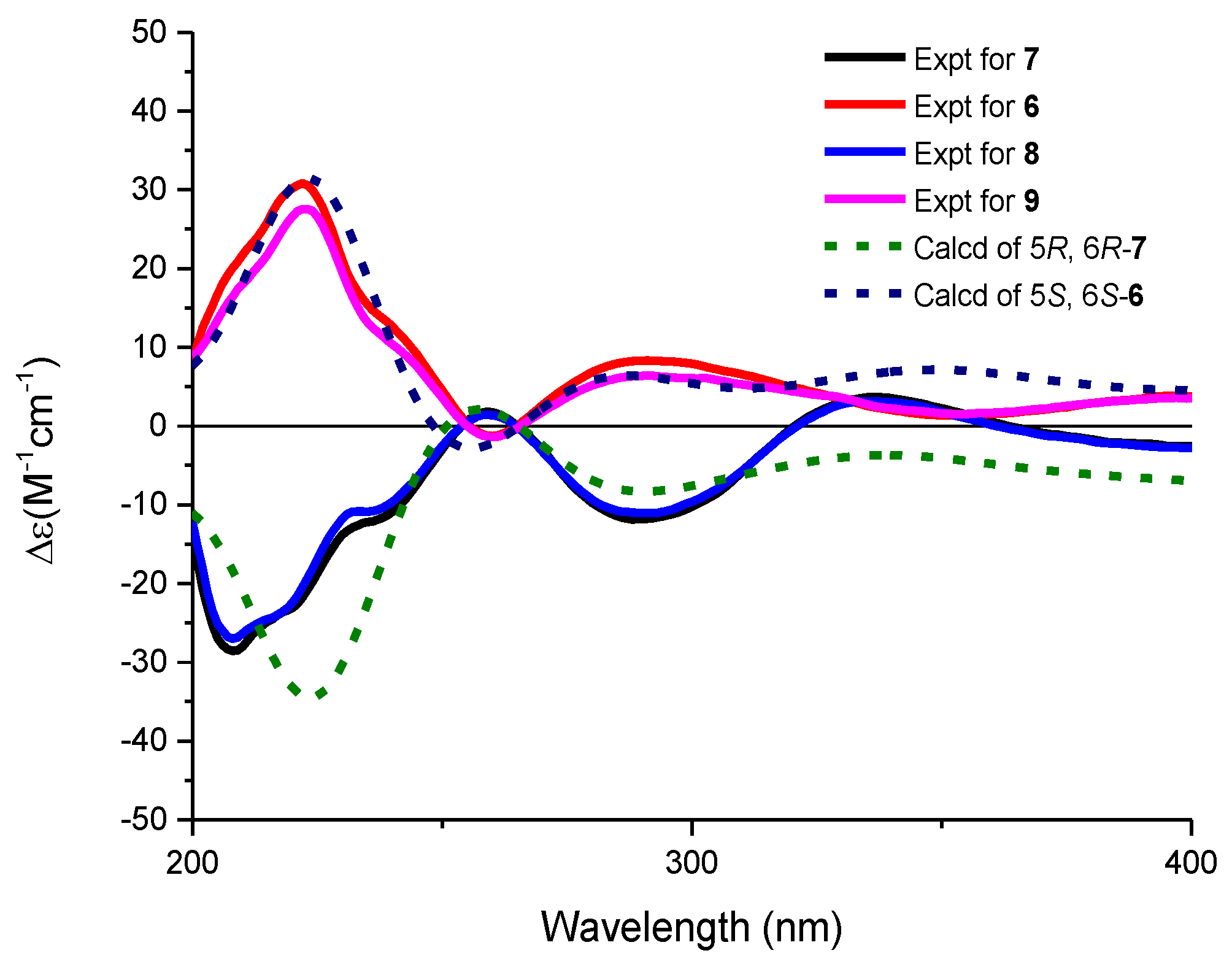
The molecular formulae of compounds 6 and 7 were determined both to be C27H28O9 by HRESIMS, indicating 14 degrees of unsaturation. The 1H and 13C NMR spectroscopic data of 6 were similar with those of 3, except that two aromatic carbon signals at δC 160.5 (C-5) and 100.0 (C-6) in 3 were replaced by two oxygen-bearing methine carbon signals at δC 78.1 (C-5) and 70.2 (C-6). Furthermore, two methoxys were attached at C-5 and C-6 based on the HMBC correlations of H3-14/C-5 and H3-15/C-6, respectively. Small coupling constants (2.8 Hz) between H-5 and H-6 revealed a trans configuration of H-5 and H-6, indicating an (5R, 6R) or (5S, 6S) configuration of 6. To determine the absolute configurations of 6, comparisons of the experimental and ECD spectra using a time-dependent density functional theory (TDDFT) were employed. Comparison of the experimental and calculated CD spectra (Figure 4) established the absolute configuration as (5S, 6S) for 6, which were the same as those of PMO70747, PD116740, and TAN-1085 [12,13,14,15,16,17]. Compound 7 possessed the same planar structure with that of 6, as deduced by the COSY and HMBC spectra (Figure 2). However, the experimental and calculated CD spectra of 7 showed cotton effects totally opposite to those of 6, respectively, inferring the (5R, 6R) configuration for 7 (Figure 4). Compounds 6 and 7 were named urdamycins N6 and N7, respectively.
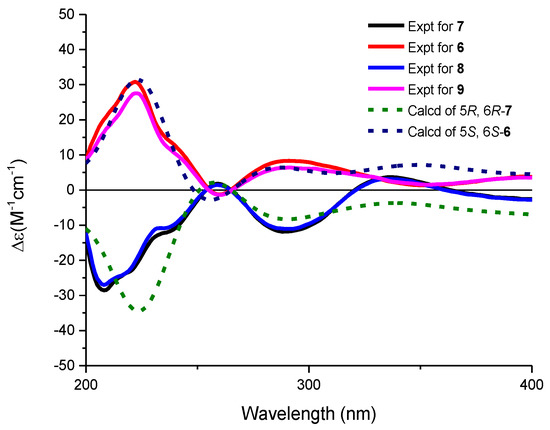
Figure 4.
Experimental and calculated ECD spectra of 6–9 in methanol.
Compounds 8 and 9 were isolated as red powder, both had the same molecular formula of C39H48O14 as determined by HRESIMS. The 1H and 13C NMR spectroscopic data of 8 and 9 resembled those of 2, except that two aromatic carbon signals at δC 160.3 (C-5) and 99.8 (C-6) in 2 were replaced by two oxygen-bearing methine carbon signals at δC 80.0 (C-5) and 67.4 (C-6) in 8, and at δC 78.1 (C-5) and 70.2 (C-6) in 9. Two methoxys were attached at C-5 and C-6 in 8 and 9 based on the HMBC correlations of H3-14/C-5 and H3-15/C-6, respectively. Small coupling constants of H5/H6 revealed the both trans configurations in compounds 8 and 9. The absolute configurations were determined to be (5R, 6R) for 8 and (5S, 6S) for 9 by comparing their CD curve to those of 7 and 6, respectively (Figure 4). Finally, compounds 8 and 9 were elucidated and named urdamycin N8 and urdamycin N9, respectively. Compounds 7 and 8 represent the first naturally occurring (5R, 6R)-angucycline metabolite.
3. Experimental Section
3.1. General Experimental Procedures
Column chromatography (CC) was performed using silica gel (100–200 mesh; Jiangpeng Silica gel development, Inc., Shandong, China). Thin layer chromatography (TLC) was conducted with precoated glass plates (0.1–0.2 mm; silica gel GF254, 10–40 nm, Jiangpeng, China). HPLC analyses were performed with a 1260 infinity system (Agilent, Santa Clara, CA, USA) using a Phenomenex Prodigy ODS (2) column (150 × 4.6 mm, 5 μm; USA). Semi-preparative HPLC were performed with a Primaide 1110 solvent delivery module equipped with a 1430 photodiode array detector (Hitachi, Tokyo, Japan), using a YMC-Pack ODS-A column (250 mm × 10 mm, 5 μm; YMC Co., Ltd., Kyoto, Japan). UV spectra were recorded on a U-2910 spectrometer (Shimadzu, Kyoto, Japan); IR spectra were obtained on an IRAffinity-1 spectrophotometer (Shimadzu, Kyoto, Japan). CD spectra were measured on a Chirascan circular dichroism spectrometer (Applied Photophysics, Leatherhead, UK). High-resolution mass spectral data were obtained on a MaXis Q-TOF mass spectrometer (Bruker, Billerica, MA, USA). Optical rotations were recorded with an MCP-500 polarimeter (Anton Paar, Graz, Austria). NMR spectra were recorded on a Bruker Avance 500 or a Bruker Avance 700 spectrometer (Bruker, Billerica, MA, USA). Carbon signals and the residual proton signals of DMSO-d6 (δC 39.52 and δH 2.50), CD3OD (δC 49.0 and δH 4.87), and CDCl3 (δC 77.16 and δH 7.26) were used for calibration. Coupling constants (J) are given in Hz.
3.2. Bacterial Materials
Strain SCSIO GJ056 was isolated from a mangrove-derived sediment sample collected in Yalong bay, China. It was identified as Streptomyces diastaticus subsp. on the basis of morphological characteristics and 16S rRNA sequence analysis by comparisons with other sequences in the GenBank database. The DNA sequence has been deposited in GenBank (accession no. MH368281). The strain was preserved at the RNAM Center for Marine Microbiology, South China Sea Institute of Oceanology, Chinese Academy of Sciences and also at the China General Microbiological Culture Collection Center (CGMCC, Beijing, China), CGMCC No. 13648.
3.3. Fermentation, Extraction, and Isolation of the Compounds
A portion of spore and mycelium mixture of SCSIO GJ056 grown on ISP4 medium agar plates were inoculated into 50 mL modified AM2 medium (0.5% soybean flour, 0.5% soluble starch, 0.2% yeast extract, 0.2% peptone, 2.0% glucose, 0.05% KH2PO4, 0.05% MgSO4·7H2O, 0.4% NaCl, 0.2% CaCO3, 3.0% sea salt (Guangdong Province Salt Industry Group, Guangzhou, China), pH 7.2 before sterilization) in 250 mL Erlenmeyer flasks, and were incubated at 28 °C on a rotary shaker at 200 rpm for 1.5 days as the seed culture. Then the 50 mL of culture solution was transferred into a 1 L Erlenmeyer flask and then incubated at 28 °C, 200 rpm for seven days. On the seventh day, the entire culture broth (15 L) was harvested and centrifuged to yield the mycelial cake and liquid broth. The liquid broth was extracted with butanone for three times, and the mycelial cake was extracted using 1 L of acetone for three times. The combined organic layers were dried under vacuum to yield a residue. The residue was subjected to silica gel CC using gradient elution with CHCl3 and MeOH mixtures (100:0, 99:1, 97:3, 95:5, 90:10, 80:20, and 50:50, v/v) to give seven fractions (Fr.A1–Fr.A7). Fr.A1 and Fr.A2 were combined after HPLC analysis and purified by silica gel CC eluting with petroleum ether and EtOAc mixtures (100:0, 90:10, 80:20, 70:30, 60:40, 40:60, 20:80, 0:100, v/v) to give eight fractions (Fr.B1–Fr.B8). Fr.B4 was subjected to Sephadex LH-20 CC eluted with CHCl3/MeOH (1:1) to obtain 1 (6 mg). Fr.B5 and Fr.B6 were combined and further purified by semi-preparative HPLC with an ODS column to afford 10 (220 mg) at tR 26 min and 11 (70 mg) at tR 30 min. Fractions B1 and B2 were combined and subjected to preparative TLC using CHCl3/MeOH (92:8) to obtain six sub-fractions (Fr.C1–Fr.C6). Fr.C1 was purified by preparative HPLC with an ODS column eluted with CH3CN/H2O (30:70 to 100:0 over 28 min, then hold 7 min, 9 mL/min) to afford 7 (0.6 mg) and 6 (0.6 mg) at tR 13.5 min, 14.5 min, respectively. Fr.C2 was purified by preparative HPLC with an ODS column eluted with CH3CN/H2O (30:70 to 100:0 over 28 min, then hold 7 min, 9 mL/min) to afford 8 (0.7 mg), 9 (0.6 mg), and 5 (0.6 mg) at tR 15.5 min, 16.5 min, and 26 min, respectively. Fr.C3 was purified by preparative HPLC with an ODS column eluted with CH3CN/H2O (30:70 to 100:0 over 28 min, then hold 7 min, 9 mL/min) to afford 2 (8 mg) and 3 (7 mg) at tR 32 min, 32.5 min, respectively. Fr.C6 was purified by preparative HPLC eluted with CH3CN/H2O (30:70 to 100:0 over 28 min, then held for 7 min, 9 mL/min) to afford 4 (8 mg) at tR 25 min.
3.4. Spectral Data
Urdamycin N1 (1). Dark red powder; [α + 24 (c 0.05, CDCl3); UV (CDCl3) λmax (log ε) 240 (4.06), 285 (4.27), 411 (3.69); IR νmax 3391, 2932, 1705, 1667, 1632, 1582, 1492, 1364, 1279, 1061, 1011, 754 cm−1; 1H NMR (500 MHz, CDCl3/CD3OD) and 13C NMR (125 MHz, CDCl3/CD3OD) data, Table 1; (-)HR-ESI-MS m/z 725.2834 ([M − H]−, calcd for C38H45O14, 725.2815).
Urdamycin N2 (2). Dark green powder; [α + 235 (c 0.06, CDCl3); UV (CDCl3) λmax (log ε) 241 (4.62), 263 (4.55), 324 (4.71), 434 (4.17), IR νmax 3379, 2926, 1631, 1504, 1435, 1300, 1061, 1011, 758 cm−1; 1H NMR (700 MHz, DMSO-d6) and 13C NMR (176 MHz, DMSO-d6) data, Table 1; (-)HR-ESI-MS m/z 707.2708 ([M − H]−, calcd for C38H43O13, 707.2709).
Urdamycin N3 (3). Dark green powder; [α − 430 (c 0.04, CDCl3); UV (CDCl3) λmax (log ε) 240 (4.48), 263 (4.39), 324 (4.55), 433 (4.02), IR νmax 3360, 2920, 1630, 1506, 1435, 1229, 1090, 1057, 772 cm−1; 1H NMR (700 MHz, DMSO-d6/CD3OD) and 13C NMR (176 MHz, DMSO-d6/CD3OD) data, Table 1; (-)HR-ESI-MS m/z 463.1409 ([M − H]−, calcd for C26H23O8, 463.1398).
Urdamycin N4 (4). Dark green powder; [α + 125 (c 0.06, CDCl3); UV (CDCl3) λmax (log ε) 207 (3.92), 240 (4.16), 263 (4.05), 323 (4.18), 434 (3.67), IR νmax 3397, 2930, 1628, 1437, 1298, 1096, 1061, 1013, 852, 786 cm−1; 1H NMR (500 MHz, DMSO-d6/CD3OD) and 13C NMR (125MHz, DMSO-d6/CD3OD) data, Table 2; (-)HR-ESI-MS m/z 693.2554 ([M − H]−, calcd for C37H41O13, 693.2553).
Table 2.
The 1H and 13C NMR data of compounds 4–6 (δ in ppm, J in Hz).
Urdamycin N5 (5). Dark green powder; [α + 250 (c 0.01, CDCl3); UV (CDCl3) λmax (log ε) 240 (4.98), 322 (4.85), 437 (4.46), IR νmax 3392, 2924, 1626, 1435, 1267, 1061, 1013, 773 cm−1; 1H NMR (700 MHz, DMSO-d6/CD3OD) and 13C NMR (176 MHz, DMSO-d6/CD3OD) data, Table 2; (-)HR-ESI-MS m/z 677.2606 ([M − H]−, calcd for C37H41O12, 677.2604).
Urdamycin N6 (6). Red powder; [α + 325 (c 0.06, CDCl3); UV (CDCl3) λmax (log ε) 219 (4.57), 257 (4.19), 291 (4.04), 325 (3.67), 454 (3.90), IR νmax 3379, 2928, 1614, 1435, 1246, 1086, 752 cm−1; 1H NMR (700 MHz, CDCl3) and 13C NMR (176 MHz, CDCl3) data, Table 2; (-)HR-ESI-MS m/z 495.1667 ([M − H]−, calcd for C27H27O9, 495.1661).
Urdamycin N7 (7). Red powder; [α + 130 (c 0.06, CDCl3); UV (CDCl3) λmax (log ε) 218 (4.50), 259 (4.13), 284 (4.03), 325 (3.64), 451 (3.85), IR νmax 3360, 2918, 1612, 1435, 1238, 1086, 1060, 756 cm−1; 1H NMR (700 MHz, CDCl3) and 13C NMR (176 MHz, CDCl3) data, Table 3; (-)HR-ESI-MS m/z 495.1675 ([M − H]−, calcd for C27H27O9, 495.1661).
Table 3.
1H and 13C-NMR data of compounds 7–9 in CDCl3 (δ in ppm, J in Hz).
Urdamycin N8 (8). Red powder; [α + 75 (c 0.04, CDCl3); UV (CDCl3) λmax (log ε) 240 (4.39), 266 (4.27), 465 (3.91), IR νmax 3379, 2932, 1612, 1435, 1238, 1057, 1009, 748 cm−1; 1H NMR (700 MHz, CDCl3) and 13C NMR (176 MHz, CDCl3) data, Table 3; (-)HR-ESI-MS m/z 739.2984 ([M − H]−, calcd for C39H47O14, 739.2971).
Urdamycin N9 (9). Red powder; [α + 233 (c 0.04, CDCl3); UV (CDCl3) λmax (log ε) 240 (4.31), 265 (4.28), 296 (4.08), 472 (3.94), IR νmax 3395, 2932, 1614, 1435, 1369, 1248, 1088, 1059, 1010.7, 756 cm−1; 1H NMR (700 MHz, CDCl3) and 13C NMR (176 MHz, CDCl3) data, Table 3; (-)HR-ESI-MS m/z 739.2974 ([M − H]−, calcd for C39H47O14, 739.2971).
3.5. Electronic Circular Dichroism (ECD) Calculation
Monte Carlo conformational searches were carried out by means of the Spartan’s 10 software (Wavefunction, Inc., Irvine, CA, USA) using Merck Molecular Force Field (MMFF). The conformers with Boltzmann-population of over 5% were chosen for electronic circular dichroism (ECD) calculations, and then the conformers were initially optimized at B3LYP/6-31+g (d, p) level in methanol using the conductor-like polarizable continuum model (CPCM). The theoretical calculation of ECD was conducted in methanol using Time-dependent Density functional theory (TD-DFT) at the B3LYP/6-311+g (d, p) level for all conformers of compounds 6 and 7. Rotatory strengths for a total of 10 excited states were calculated. ECD spectra were generated using the program SpecDis 1.6 (University of Würzburg, Würzburg, Germany) and GraphPad Prism 5 (University of California San Diego, San Diego, CA, USA) from dipole-length rotational strengths by applying Gaussian band shapes with sigma = 0.3 eV.
4. Conclusions
The early report of urdamycin derivatives, urdamycins A–F, were isolated from Streptomyces fradiae Tu 2717 by Drautz in 1986. Since then, many urdamycin congeners were discovered. Structurally, urdamycins have different aglycone parts, whereas the sugar moieties are always the same [10]. Most of them were decorated with a trisaccharide chain composed of β-olivose-(1→4)-α-rhodinose-(1→3)-β-olivose via a C-C linkage. Diverse biological activities of these angucycline antibiotics were evaluated, and the most important were their cytotoxicities against the tumor cell lines. At the same time, this potent cytotoxicity also limited their use in the clinic. In this study, nine new angucycline glycosides, urdamycin N1–N9 (1–9), together with two known congener urdamycins A (10) and B (11) were obtained from a mangrove-derived Streptomyces diastaticus subsp. SCSIO GJ056. Compounds 7 and 8 are the first naturally occurring (5R, 6R) angucycline glycosides. We will further investigate the biological activity of these new angucycline compounds.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/6/185/s1. This section includes 1D, 2D NMR spectra for new compounds 1–9, and computational details of 6 and 7.
Author Contributions
C.G. performed the experiments and wrote the draft manuscript. Y.L. performed the ECD calculations. Z.Z. and S.Z. contributed to the isolation and identification of compounds. Y.H. provided the strain. H.H. revised the manuscript. Y.-C.G. and J.J. supervised the whole work, and edited the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the National Natural Science Foundation of China [grant numbers 41476133, 81425022, and U1501223], the Program of Chinese Academy of Sciences [grant number XDA11030403], and the Natural Science Foundation of Guangdong Province [grant number 2016A030312014].
Acknowledgments
We thank Syngenta Ph.D. Fellowship Awarded to Chun Gui. We are grateful to Zhihui Xiao, Aijun Sun, Yun Zhang and Chuanyun Li in the analytical facility at SCSIO for recording spectroscopic data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rohr, J.; Thiericke, R. Angucycline group antibiotics. Nat. Prod. Rep. 1992, 9, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, H.; Chen, Q.; Luo, M.; Sun, A.; Song, Y.; Ma, J.; Ju, J. Identification of the grincamycin gene cluster unveils divergent roles for GcnQ in different hosts, tailoring the L-rhodinose moiety. Org. Lett. 2013, 15, 3254–3257. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C.; Luzhetskyy, A.; Rebets, Y.; Bechthold, A. Type II polyketide synthases: Gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007, 24, 162–190. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and antibacterial angucycline- and prodigiosin-analogues from the deep-sea derived Streptomyces sp. SCSIO 11594. Mar. Drugs 2015, 13, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Yu, J.; Ling, H.; Song, Y.; Yuan, J.; Ju, J.; Tao, Y.; Huang, H. Grincamycins I–K, cytotoxic angucycline glycosides derived from marine-derived actinomycete Streptomyces lusitanus SCSIO LR32. Planta Med. 2018, 84, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Duan, Y.; Cui, Z.; Wang, Z.; Li, Z.; Zhang, Y.; Ju, J.; Huang, H. Cytotoxic rearranged angucycline glycosides from deep sea-derived Streptomyces lusitanus SCSIO LR32. J. Antibiot. 2017, 70, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.; Zeeck, A. Metabolic products of microorganisms. 240. Urdamycins, new angucycline antibiotics from Streptomyces fradiae. II. Structural studies of urdamycins B to F. J. Antibiot. 1987, 40, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Lu, X.; Ke, A.; Zheng, Z.; Lin, J.; Hao, W.; Zhu, J.; Fan, Y.; Ding, Y.; Jiang, Q.; et al. Three novel members of angucycline group from Streptomyces sp. N05WA963. J. Antibiot. 2011, 64, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Drautz, H.; Zähner, H.; Rohr, J.; Zeeck, A. Metabolic products of microorganisms. 234 Urdamycins, new angucycline antibiotics from Streptomyces fradiae. J. Antibiot. 1986, 39, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Zeeck, A.; Rohr, J.; Sheldrick, G.M.; Jones, P.G.; Paulus, E.F. Structure of a new antibiotic and cytotoxic indicator substance, urdamycin A. J. Chem. Res. 1986, 104–105. [Google Scholar]
- Pérez, M.; Schleissner, C.; Rodríguez, P.; Zúñiga, P.; Benedit, G.; Sánchez-Sancho, F.; de la Calle, F. PM070747, a new cytotoxic angucyclinone from the marine-derived Saccharopolyspora taberi PEM-06-F23-019B. J. Antibiot. 2009, 62, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.T.; Carney, J.R.; Gould, S.J. Cloning and heterologous expression of the entire gene clusters for PD 116740 from Streptomyces strain WP 4669 and tetrangulol and tetrangomycin from Streptomyces rimosus NRRL 3016. J. Bacteriol. 1997, 179, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Ohmori, K.; Suzuki, K. Stereochemical relay via axially chiral styrenes: Asymmetric synthesis of the antibiotic TAN-1085. Angew. Chem. Int. Ed. Engl. 2009, 48, 5633–5637. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Tanaka, Y.; Ohmori, K.; Suzuki, K. Synthesis and stereochemical assignment of angucycline antibiotic, PD-116740. Chem. Lett. 2008, 37, 470–471. [Google Scholar] [CrossRef]
- Ohmori, K.; Mori, K.; Ishikawa, Y.; Tsuruta, H.; Kuwahara, S.; Harada, N.; Suzuki, K. Concise total synthesis and structure assignment of TAN-1085. Angew. Chem. Int. Ed. Engl. 2004, 43, 3167–3171. [Google Scholar] [CrossRef] [PubMed]
- Hauser, F.M.; Dorsch, W.A.; Mal, D. Total synthesis of (±)-O-methyl PD 116740. Org. Lett. 2002, 4, 2237–2239. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).