Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008

 ,
, 
Abstract
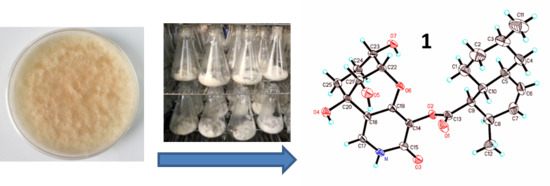
:
1. Introduction
2. Results and discussion
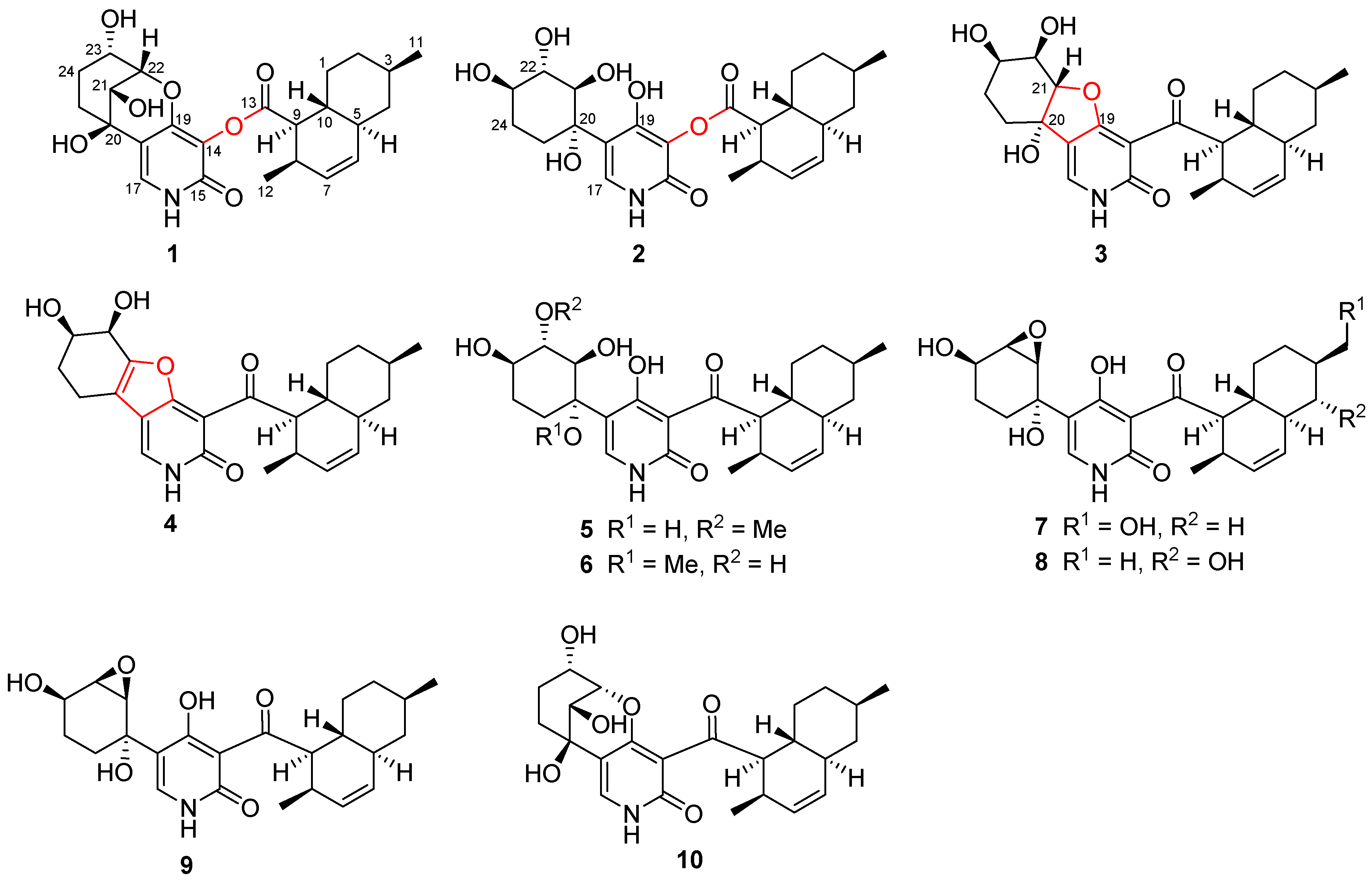
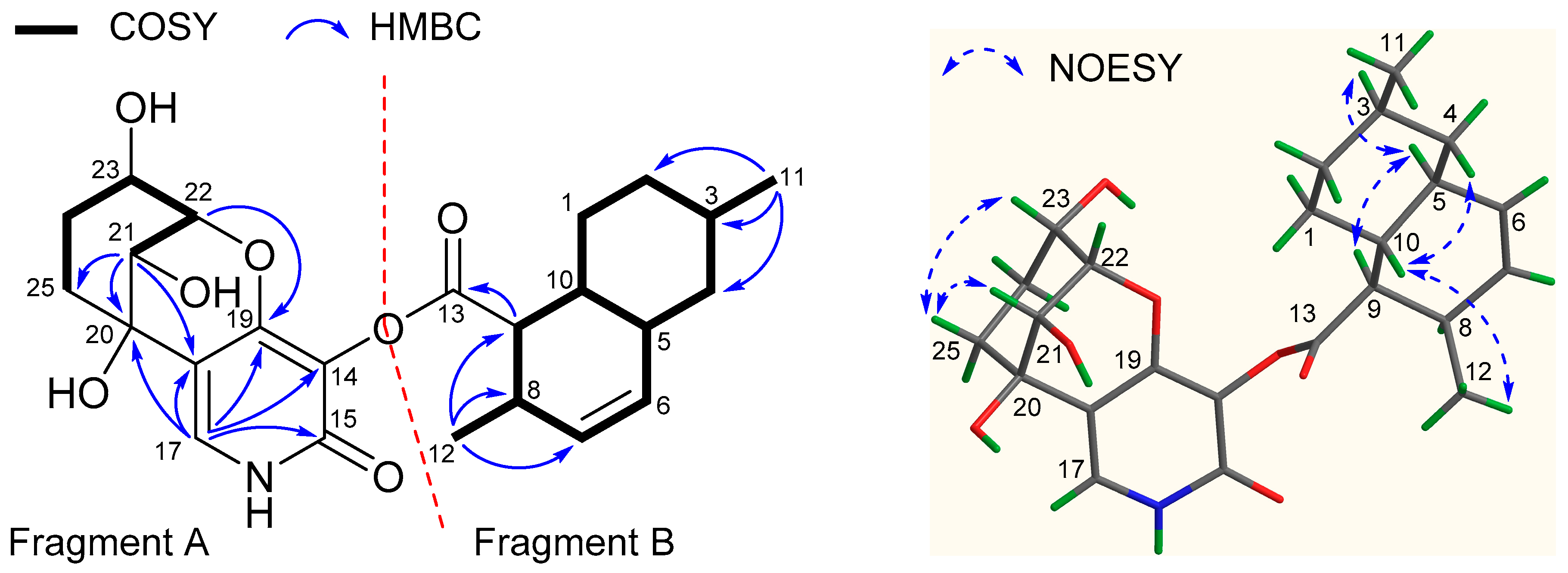
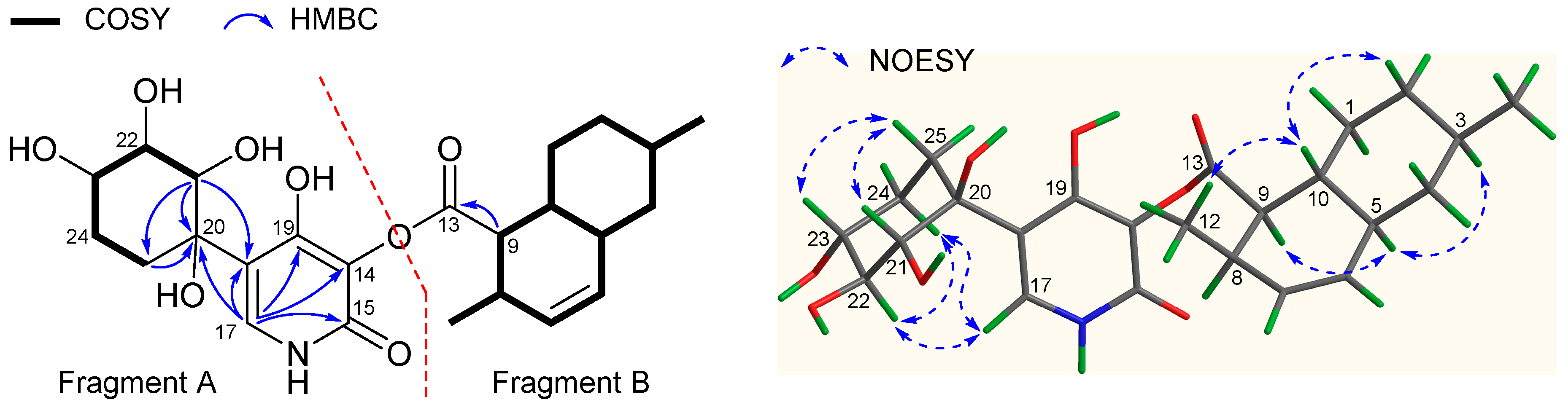
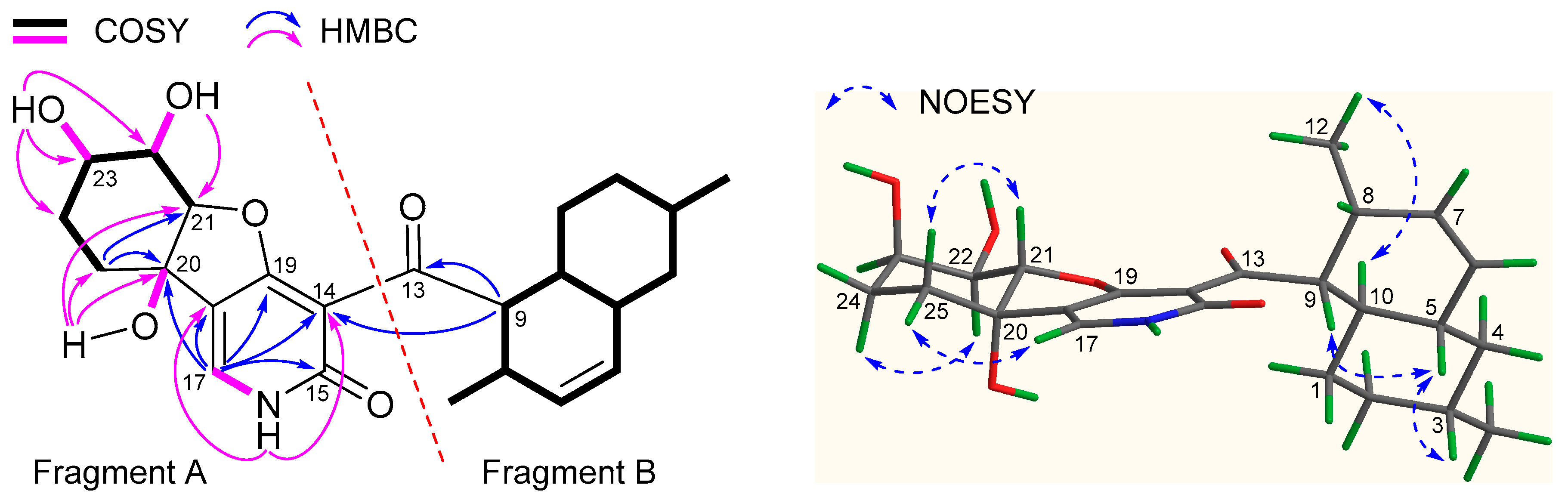
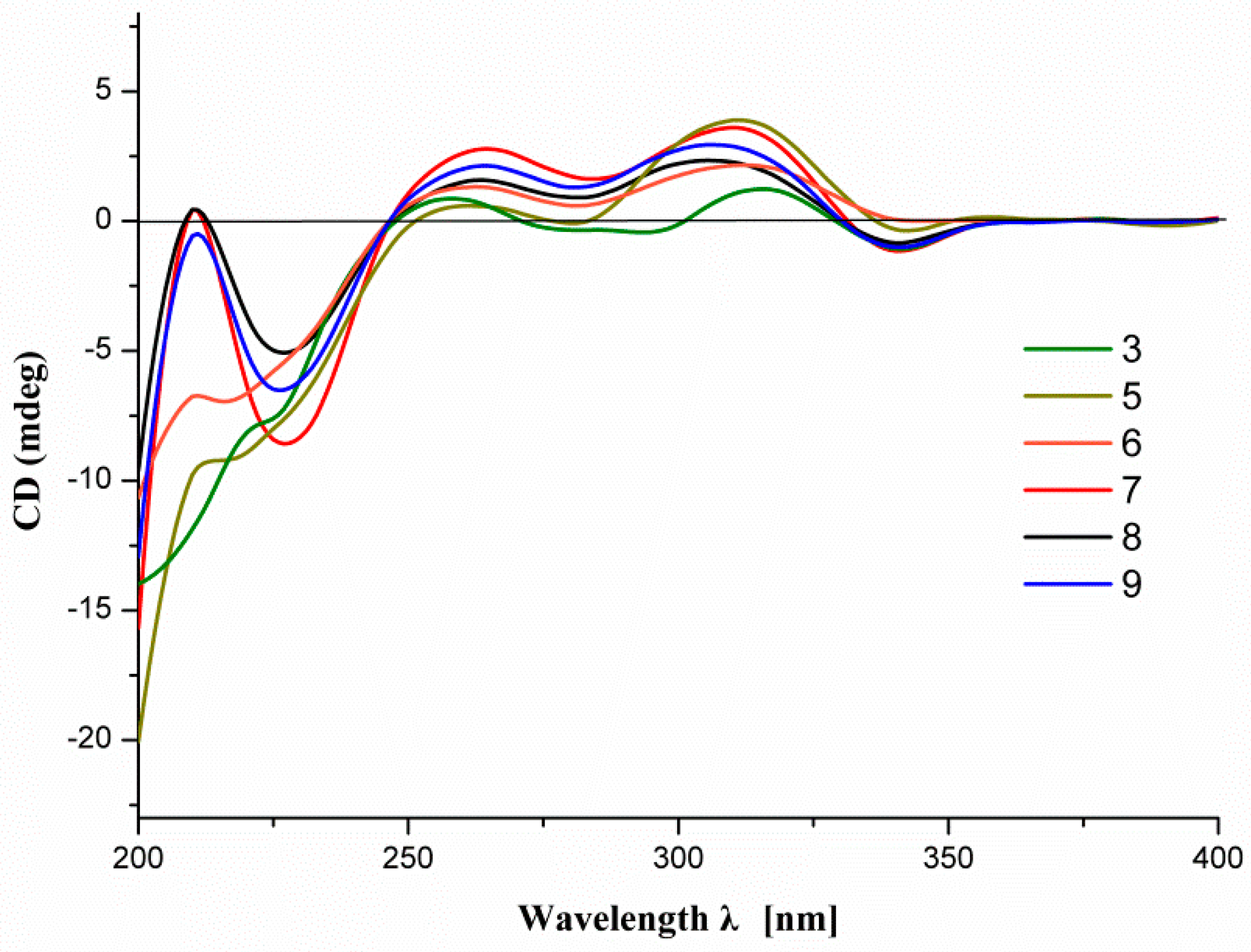
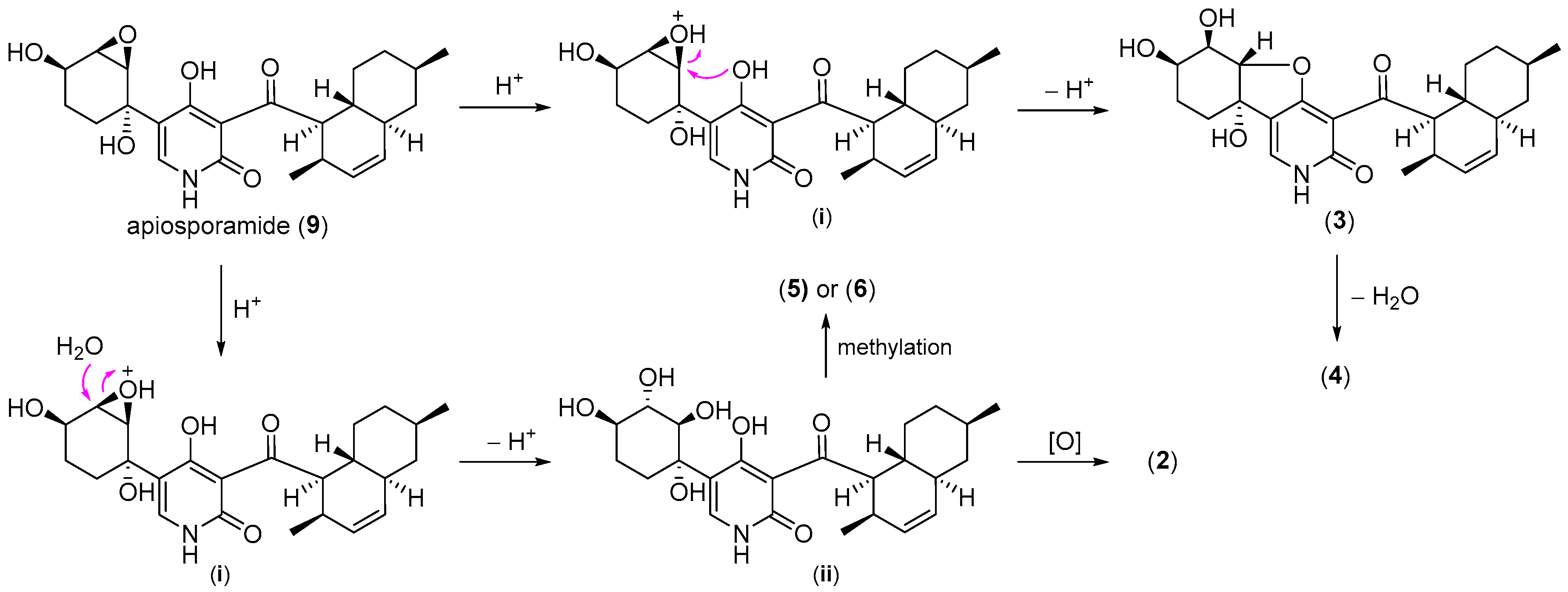
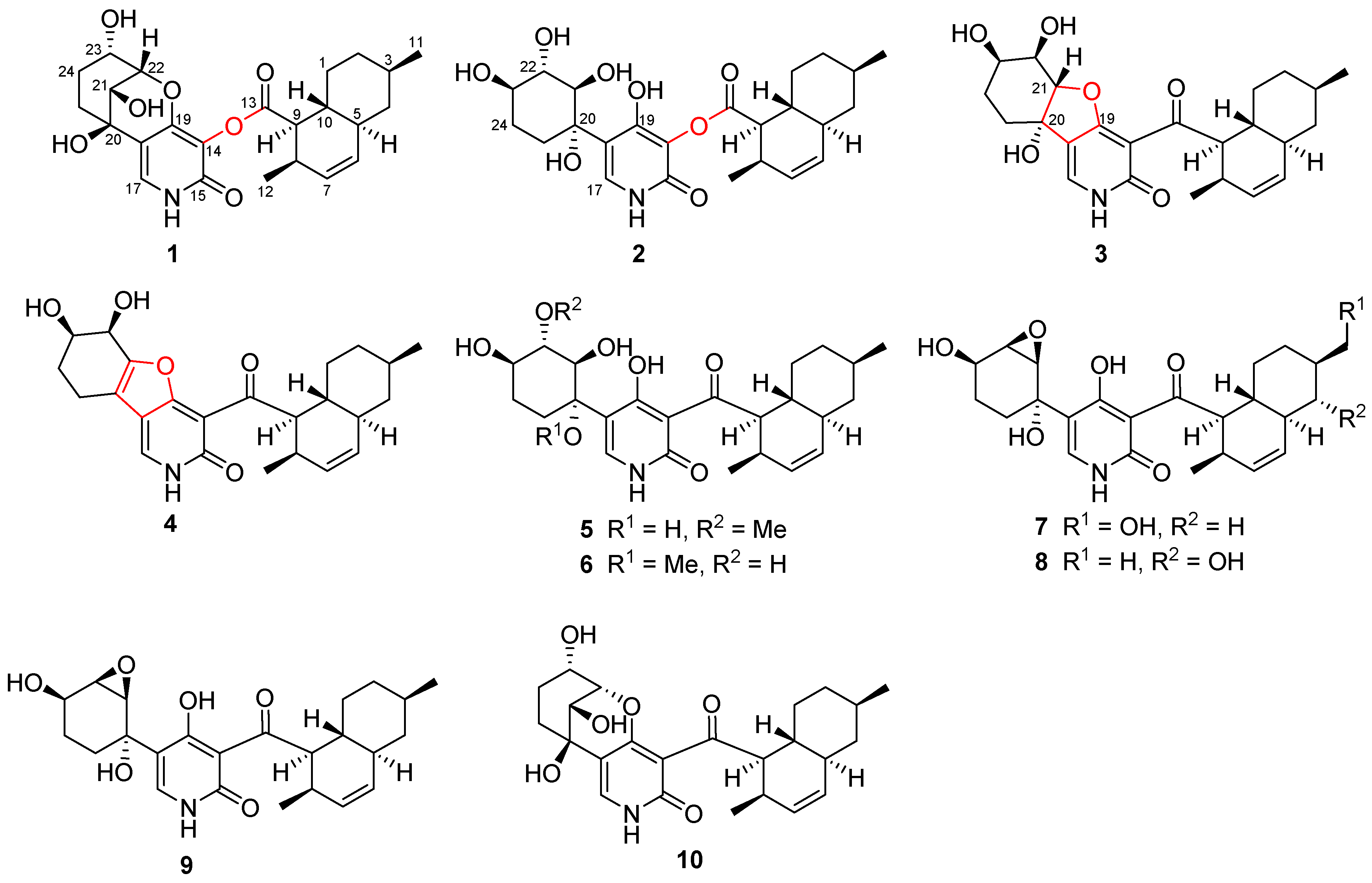
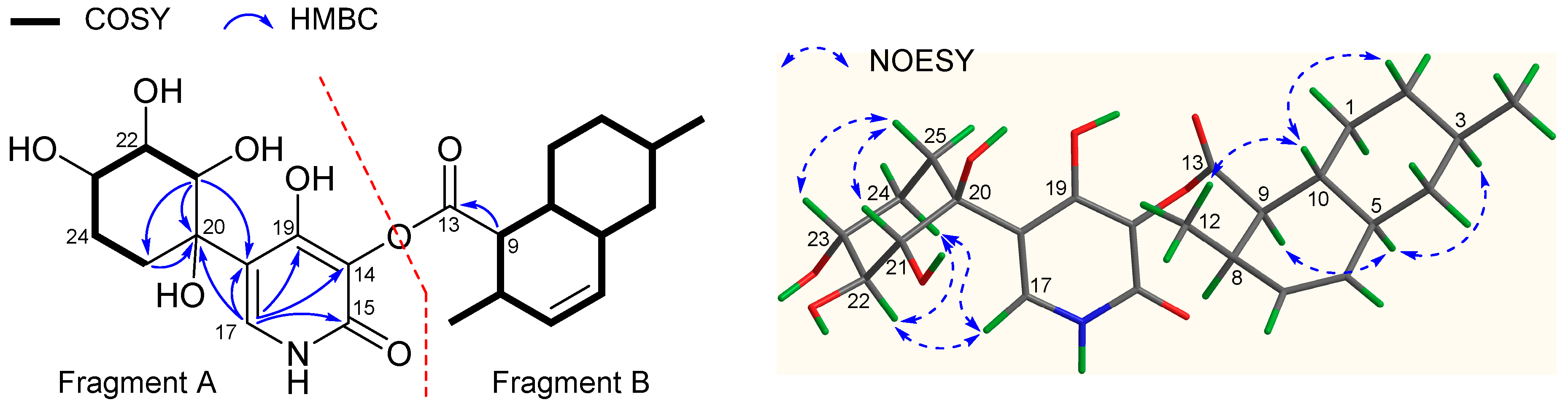
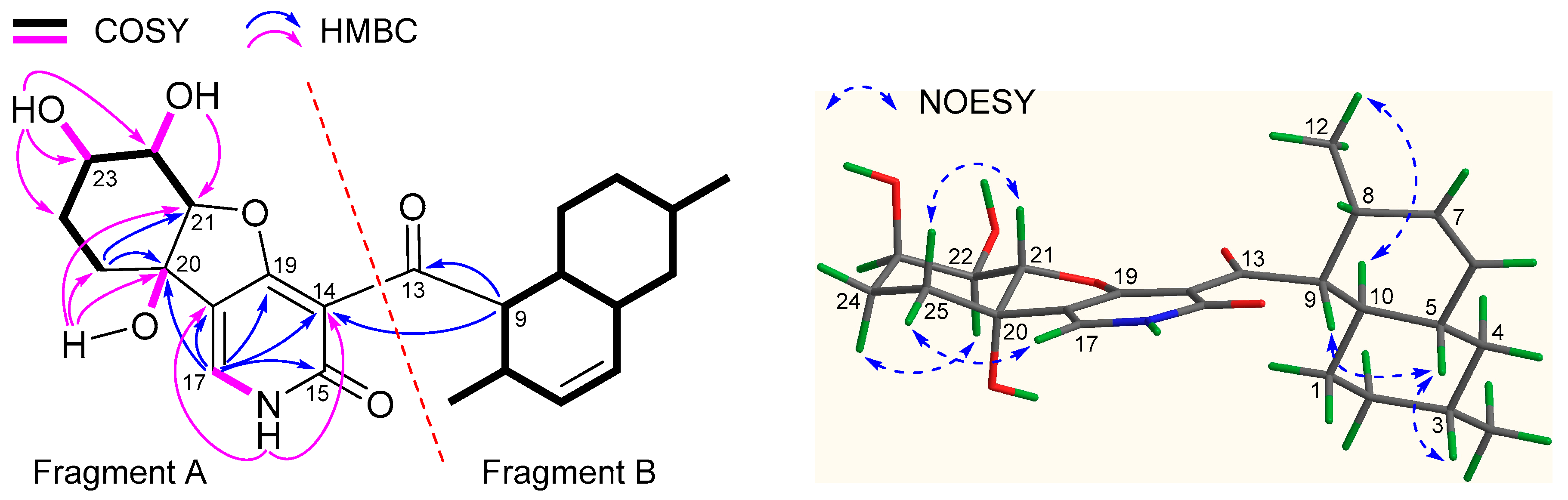
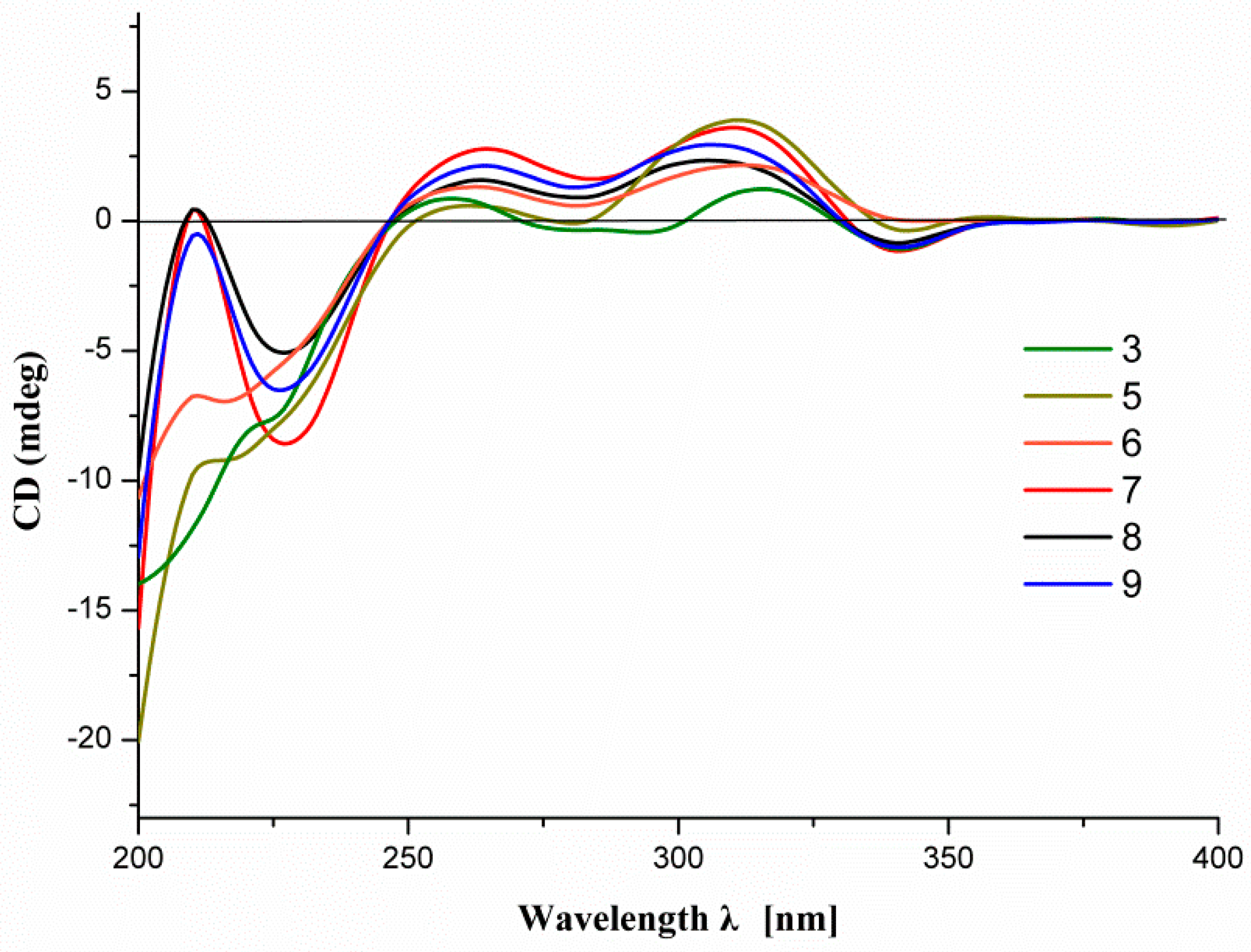
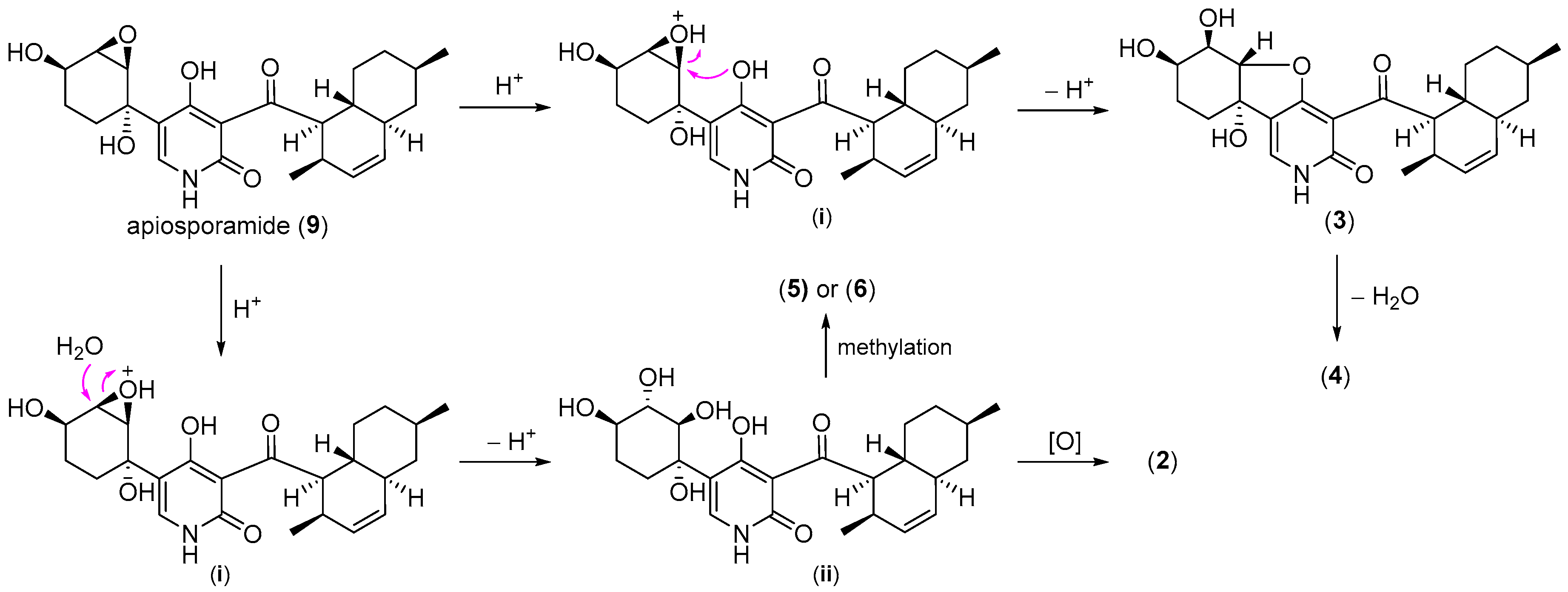
2.1. Structure Elucidation
2.2. Biological Activity
3. Experimental Section
3.1. General Experimental Procedures
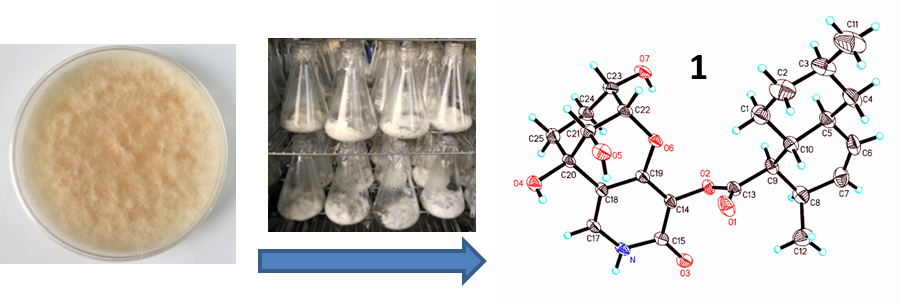
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.4.1. Arthpyrone D (1)
3.4.2. Arthpyrone E (2)
3.4.3. Arthpyrone F (3)
3.4.4. Arthpyrone G (4)
3.4.5. Arthpyrone H (5)
3.4.6. Arthpyrone I (6)
3.4.7. Arthpyrone J (7)
3.4.8. Arthpyrone K (8)
3.5. Chemical Transformation of 10 to 1
3.6. Antimicrobial Assays
3.7. AChE Inhibitory Assay
3.8. Cytotoxic Assay
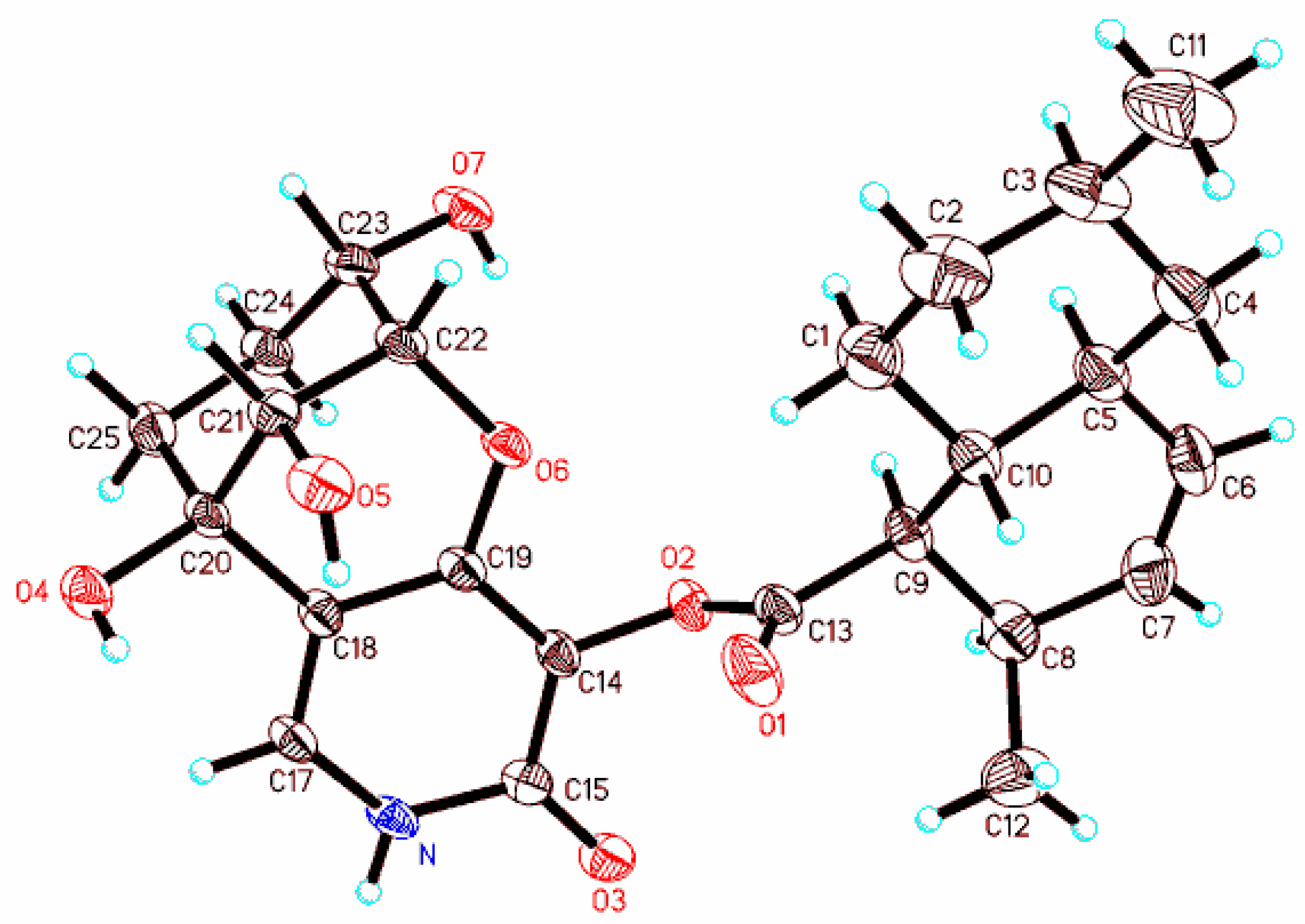
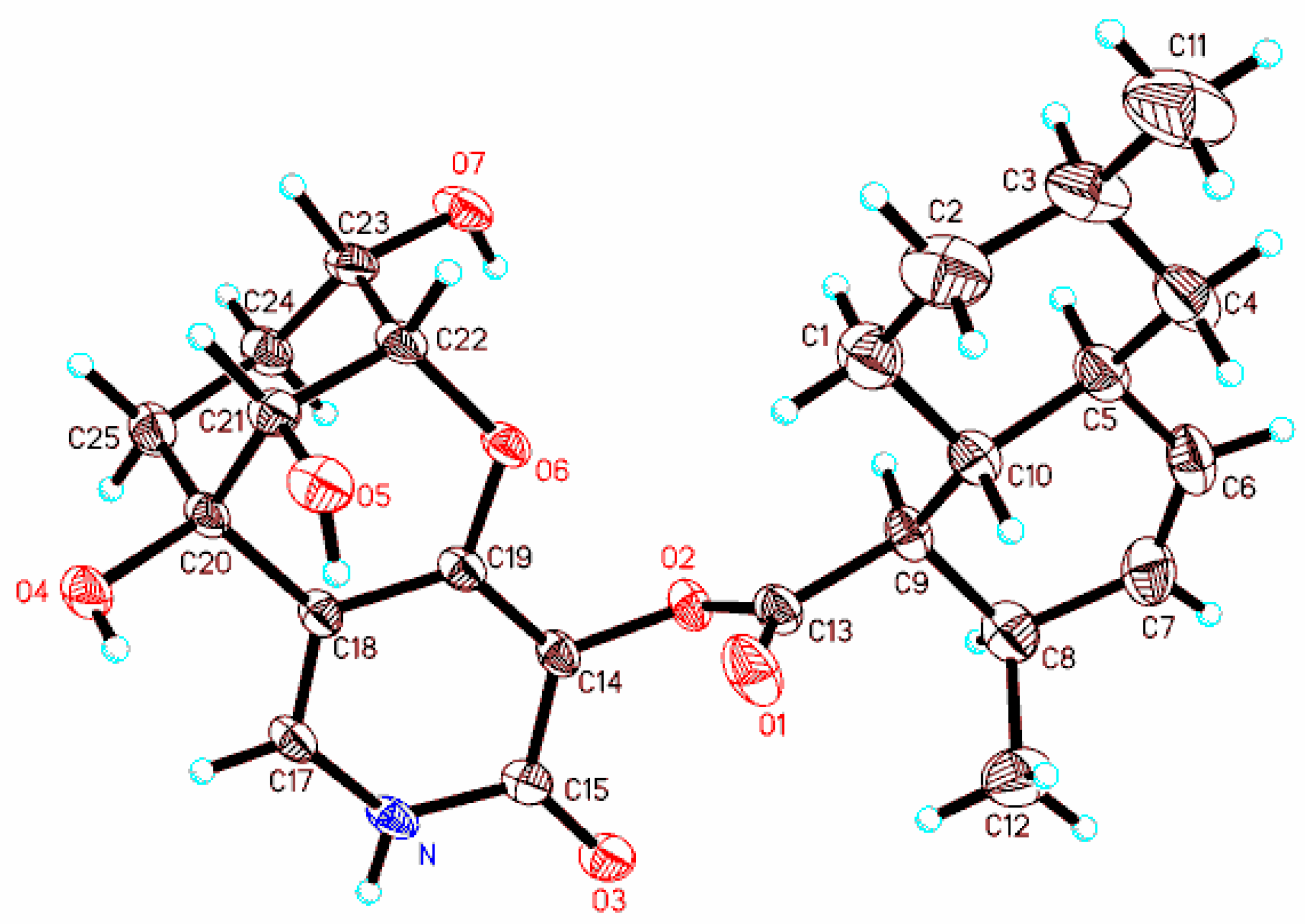
3.9. X-ray Diffraction Analysis of Arthpyrone D (1)
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Choudhary, A.; Naughton, L.M.; Montanchez, I.; Dobson, A.D.W.; Rai, D.K. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Xue, Y.R.; Liu, C.H. A brief review of bioactive metabolites derived from deep-Sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Wei, X.Y.; Qin, X.C.; Lin, X.P.; Zhou, X.F.; Liao, S.R.; Yang, B.; Liu, J.; Tu, Z.C.; Liu, Y.H. Arthpyrones A–C, pyridone alkaloids from a sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Org. Lett. 2015, 17, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Schulz, B.; Wray, V.; Totzke, F.; Kubbutat, M.H.G.; Müller, W.E.G.; Hamacher, A.; Kassack, M.U.; Lin, W.H.; Proksch, P. Arthrinins A–D: Novel diterpenoids and further constituents from the sponge derived fungus Arthrinium sp. Bioorg. Med. Chem. 2011, 19, 4644–4651. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Wang, J.F.; He, W.J.; Lin, X.P.; Zhou, X.J.; Liu, Y.H. One Strain-many compounds method for production of polyketide metabolites using the sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Chem. Nat. Compd. 2017, 53, 373–374. [Google Scholar] [CrossRef]
- Wang, J.F.; Wang, Z.; Ju, Z.R.; Wan, J.T.; Liao, S.R.; Lin, X.P.; Zhang, T.Y.; Zhou, X.F.; Chen, H.; Tu, Z.C.; et al. Cytotoxic cytochalasins from marine-derived fungus Arthrinium arundinis. Planta Med. 2015, 81, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Bloor, S. Arthrinic acid, a novel antifungal polyhydroxyacid from Arthrinium phaeospermum. J. Antibiot. 2008, 61, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Guan, F.F.; Du, S.Y.; Wei, M.Y.; Wang, C.Y.; Shao, C.L. Two polyhydroxy xanthones and their antiviral activity from gorgonian coral-derived fungus Arthrinium sp. Chin. J. Mar. Drugs 2016, 35, 30–34. [Google Scholar]
- Wang, J.F.; Xu, F.Q.; Wang, Z.; Lu, X.; Wan, J.T.; Yang, B.; Zhou, X.F.; Zhang, T.Y.; Tu, Z.C.; Liu, Y.H. A new naphthalene glycoside from the sponge-derived fungus Arthrinium sp. ZSDS1-F3. Nat. Prod. Res. 2014, 28, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Iimura, S.; Tenmyo, O.; Sawada, Y.; Sugawara, M.; Ohkusa, N.; Yamamoto, H.; Kawano, K.; Hu, S.L.; Fukagawa, Y. Terpestacin, a new syncytium formation inhibitor from Arthrinium sp. J. Antibiot. 1993, 46, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Ondeyka, J.G.; Ball, R.G.; Garcia, M.L.; Dombrowski, A.W.; Sabnis, G.; Kaczorowski, G.J.; Zink, D.L.; Bills, G.F.; Goetz, M.A.; Schmalhofer, W.A.; et al. A carotane sesquiterpene as a potent modulator of the Maxi-K channel from Arthrinium phaesospermum. Bioorg. Med. Chem. Lett. 1995, 5, 733–734. [Google Scholar] [CrossRef]
- Wei, M.Y.; Xu, R.F.; Du, S.Y.; Wang, C.Y.; Xu, T.Y.; Shao, C.L. A new griseofulvin derivative from the marine-derived Arthrinium sp. fungus and its biological activity. Chem. Nat. Compd. 2016, 52, 1011–1014. [Google Scholar] [CrossRef]
- Williams, D.R.; Kammler, D.C.; Donnell, A.F.; Goundry, W.R.F. Total synthesis of (+)-apiosporamide: Assignment of relative and absolute configuration. Angew. Chem. Int. Ed. 2005, 44, 6715–6718. [Google Scholar] [CrossRef] [PubMed]
- Alfatafta, A.A.; Gloer, J.B.; Scott, J.A.; Malloch, D. Apiosporamide, a new antifungal agent from the coprophilous fungus Apiospora montagnei. J. Nat. Prod. 1994, 57, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Burns, A.M.; Liu, M.X.; Faeth, S.H.; Gunatilaka, A.A.L. Search for cell motility and angiogenesis inhibitors with potential anticancer activity: Beauvericin and other constituents of two endophytic strains of Fusarium oxysporum. J. Nat. Prod. 2007, 70, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Tsuchinari, M.; Shimanuki, K.; Hiramatsu, F.; Murayama, T.; Koseki, T.; Shiono, Y. Fusapyridons A and B, novel pyridone alkaloids from an endophytic fungus, Fusarium sp. YG-45. Z. Naturforsch. B 2007, 62, 1203–1207. [Google Scholar] [CrossRef]
- Wang, Q.X.; Li, S.F.; Zhao, F.; Dai, H.Q.; Bao, L.; Ding, R.; Gao, H.; Zhang, L.X.; Wen, H.A.; Liu, H.W. Chemical constituents from endophytic fungus Fusarium oxysporum. Fitoterapia 2011, 82, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Zhang, J.; Rodrigues, M.C.; Ding, D.J.; Longo, J.P.F.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.S. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Jiang, L.L.; Li, B.; Wang, G.B.; Wang, J.S.; Fu, Y.H. Oxymatrine suppresses the growth and invasion of MG63 cells by up-regulating PTEN and promoting its nuclear translocation. Oncotarget 2017, 8, 65100–65110. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; Liu, C.C.; Li, L.; Zhou, H.; Liu, L.; Bao, L.; Chen, Q.; Song, F.H.; Zhang, L.X.; Li, E.W.; et al. Decalin-containing tetramic acids and 4-Hydroxy-2-pyridones with antimicrobial and cytotoxic activity from the fungus Coniochaeta cephalothecoides collected in Tibetan Plateau (Medog). J. Org. Chem. 2017, 82, 11474–11486. [Google Scholar] [CrossRef] [PubMed]
- Haga, A.; Tamoto, H.; Ishino, M.; Kimura, E.; Sugita, T.; Kinoshita, K.; Takahashi, K.; Shiro, M.; Koyama, K. Pyridone alkaloids from a marine-derived fungus, Stagonosporopsis cucurbitacearum, and their activities against azole-resistant Candida albicans. J. Nat. Prod. 2013, 76, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Bremmer, M.L.; Brown, D.L.; D’Antuono, J. Total synthesis of (+−)-ilicicolin H. J. Org. Chem. 1985, 50, 2807–2809. [Google Scholar] [CrossRef]
- Shibazaki, M.; Taniguchi, M.; Yokoi, T.; Nagai, K.; Watanabe, M.; Suzuki, K.; Yamamoto, T. YM-215343, a novel antifungal compound from Phoma sp. QN04621. J. Antibiot. 2004, 57, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Ikeda, M.; Yamamoto, K.; Yamazaki, M. Structure of fischerin, a new toxic metabolite from an ascomycete, Neosartorya fischeri var. fischeri. J. Nat. Prod. 1993, 56, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Coval, S.J.; Clardy, J. A cholesteryl ester transfer protein inhibitor from an insect-associated fungus. J. Antibiot. 1996, 49, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Umeokoli, B.O.; Eze, P.; Heering, C.; Janiak, C.; Mueller, W.E.G.; Orfali, R.S.; Hartmann, R.; Dai, H.F.; Lin, W.H.; et al. Secondary metabolites of the lichen-associated fungus Apiospora montagnei. Tetrahedron Lett. 2017, 58, 1702–1705. [Google Scholar] [CrossRef]
- Wang, J.F.; Liu, Y.H.; Zhou, X.F.; Liao, S.R.; Yang, X.W.; Yang, B.; Lin, X.P.; Liu, J. Faming Zhuanli Shenqing. CN Patent 103948592 B, 2015. (In Chinese). [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Positon | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | |
| 1a | 2.11, m | 2.07, m | 1.93, m | 1.98, m |
| 1b | 1.08, m | 1.07, m | 0.90, m | 0.96, m |
| 2a | 1.78, m | 1.79, m | 1.76, m | 1.75, m |
| 2b | 1.01, m | 1.01, m | 1.05, m | 1.03, m |
| 3 | 1.52, m | 1.54, m | 1.52, m | 1.49, m |
| 4a | 1.74, m | 1.77, m | 1.76, m | 1.73, m |
| 4b | 0.78, q (12.2) | 0.78, q (12.4) | 0.81, q (12.3) | 0.79, q (12.4) |
| 5 | 1.80, m | 1.82, m | 1.83, m | 1.84, m |
| 6 | 5.41, brd (9.9) | 5.41, brd (9.9) | 5.41, brd (9.8) | 5.38, brd (9.8) |
| 7 | 5.61, ddd (9.9, 4.5, 2.8) | 5.62, ddd (9.9, 4.4, 2.8) | 5.61,m | 5.57, ddd (9.8, 4.5, 2.7) |
| 8 | 2.76, m | 2.79, m | 2.84, m | 2.73, m |
| 9 | 2.90, dd (11.7,5.9) | 2.88, dd (11.6, 5.9) | 4.44, dd (11.3, 5.8) | 4.07, dd (11.3,5.7) |
| 10 | 1.39, qd (11.3, 2.5) | 1.38, qd (11.5, 2.3) | 1.57, qd (11.2, 2.0) | 1.54, qd (11.3, 2.6) |
| 11 | 0.93, d (6.6) | 0.93, d (6.6) | 0.94, d (6.5) | 0.92, d (6.5) |
| 12 | 1.11, d (7.0) | 1.08, d (7.1) | 0.83, d (7.2) | 0.76, d (7.1) |
| 17 | 7.38, s | 7.31, s | 7.69, s | 7.96, s |
| 21 | 3.74, d (2.4) | 3.71, d (7.0) | 5.15, d (10.1) | |
| 22 | 4.60, brs | 3.62, dd (7.0, 6.2) | 3.91, dd (10.1, 2.9) | 4.68, d (3.8) |
| 23 | 3.84, ddd (11.6, 5.1, 2.4) | 3.69, m | 4.09, m | 3.95, ddd (10.8, 3.8, 3.2) |
| 24a | 1.86, m | 2.12, m | 2.07, m | 2.03, m |
| 24b | 1.34, m | 1.54, m | 1.72, m | 1.89, m |
| 25a | 1.86, m | 2.43, ddd (14.1, 7.9, 3.7) | 2.88, m | 2.69, m |
| 25b | 1.75, m | 1.67, ddd (14.1, 9.3, 3.8) | 1.51, m | 2.51, m |
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| δc, Type | δc, Type | δc, Type | δc, Type | δc, Type | δc, Type | δc, Type | δc, Type | |
| 1 | 30.7, CH2 | 30.8, CH2 | 31.0, CH2 | 30.8, CH2 | 31.0, CH2 | 31.0, CH2 | 30.4, CH2 | 30.4, CH2 |
| 2 | 36.5, CH2 | 36.5, CH2 | 36.6, CH2 | 36.7, CH2 | 36.6, CH2 | 36.6, CH2 | 30.8, CH2 | 34.6, CH2 |
| 3 | 34.3, CH | 34.3, CH | 34.4, CH | 34.4, CH | 34.4, CH | 34.4, CH | 42.0, CH | 41.7, CH |
| 4 | 43.0, CH2 | 43.0, CH2 | 43.2, CH2 | 43.2, CH2 | 43.2, CH2 | 43.2, CH2 | 37.4, CH2 | 79.6, CH |
| 5 | 43.2, CH | 43.2, CH | 43.2, CH | 43.4, CH | 43.2, CH | 43.2, CH | 42.7, CH | 49.8, CH |
| 6 | 131.6, CH | 131.6, CH | 131.7, CH | 131.7, CH | 131.7, CH | 131.7, CH | 131.6, CH | 127.1, CH |
| 7 | 132.2, CH | 132.2, CH | 132.7, CH | 132.7, CH | 132.7, CH | 132.6, CH | 132.8, CH | 133.3, CH |
| 8 | 33.8, CH | 33.7, CH | 32.4, CH | 32.9, CH | 32.5, CH | 32.5, CH | 32.4, CH | 32.0, CH |
| 9 | 50.3, CH | 50.3, CH | 54.2, CH | 55.5, CH | 54.3, CH | 54.3, CH | 54.2, CH | 54.1, CH |
| 10 | 37.7, CH | 37.7, CH | 37.6, CH | 38.0, CH | 37.6, CH | 37.6, CH | 37.9, CH | 35.9, CH |
| 11 | 22.9, CH3 | 22.9, CH3 | 23.0, CH3 | 23.0, CH3 | 23.0, CH3 | 23.0, CH3 | 68.6, CH2 | 19.3, CH3 |
| 12 | 18.3, CH3 | 18.2, CH3 | 18.3, CH3 | 18.3, CH3 | 18.4, CH3 | 18.4, CH3 | 18.4, CH3 | 18.3, CH3 |
| 13 | 172.9, C | 173.2, C | 212.1, C | 203.8, C | 211.9, C | 211.9, C | 211.8, C | 211.6, C |
| 14 | 123.8, C | 126.4, C | 108.1, C | 110.7, C | 108.9, C | 108.5, C | 108.7, C | 108.8, C |
| 15 | 158.2, C | 160.7, C | 164.3, C | 163.4, C | 163.7, C | 164.0, C | 163.9, C | 164.0, C |
| 17 | 129.8, CH | 132.2, CH | 141.0, CH | 133.3, CH | 143.2, CH | 144.2, CH | 139.9, CH | 139.9, CH |
| 18 | 115.6, C | 115.7, C | 118.3, C | 115.4, C | 114.3, C | 111.4, C | 116.6, C | 116.7, C |
| 19 | 160.8, C | 161.9, C | 177.2, C | 166.4, C | 179.8, C | 179.5, C | 179.3, C | 179.4, C |
| 20 | 70.6, C | 78.5, C | 76.9, C | 116.6, C | 76.3, C | 82.4, C | 70.4, C | 70.4, C |
| 21 | 70.0, CH | 77.3, CH | 67.7, CH | 156.2, C | 78.7, CH | 75.2, CH | 60.5, CH | 60.5, CH |
| 22 | 85.2, CH | 75.3, CH | 74.1, CH | 65.3, CH | 86.6, CH | 75.2, CH | 57.6, CH | 57.6, CH |
| 23 | 71.6, CH | 72.6, CH | 71.3, CH | 70.8, CH | 72.4, CH | 72.4, CH | 67.1, CH | 67.1, CH |
| 24 | 27.7, CH2 | 27.6, CH2 | 27.1, CH2 | 26.7, CH2 | 28.2, CH2 | 26.1, CH2 | 25.8, CH2 | 25.8, CH2 |
| 25 | 37.6, CH2 | 31.1, CH2 | 31.2, CH2 | 18.4, CH2 | 30.4, CH2 | 23.7, CH2 | 31.6, CH2 | 31.6, CH2 |
| OCH3 | 60.5, CH3 | 51.0, CH3 |
| Positon | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | δH, mult. (J in Hz) | |
| 1a | 1.91, m | 1.91, m | 1.98, m | 1.86, m |
| 1b | 0.87, m | 0.88, m | 0.90, m | 0.90, m |
| 2a | 1.74, m | 1.74, m | 1.81, m | 1.74, m |
| 2b | 1.03, m | 1.03, m | 1.05, m | 1.14, dq (13.1, 3.5) |
| 3 | 1.51, m | 1.50, m | 1.60, m | 1.39, m |
| 4a | 1.74, m | 1.74, m | 1.84, m | 2.73, dd (9.9, 9.9) |
| 4b | 0.79, q (12.3) | 0.79, q (12.4) | 0.82, m | |
| 5 | 1.81, m | 1.81, m | 1.82, m | 1.77, m |
| 6 | 5.39, brd (9.8) | 5.40,brd (9.8) | 5.42, brd (9.8) | 6.00, brd (10.1) |
| 7 | 5.59, ddd (9.8, 4.3, 2.7) | 5.59, ddd (9.8, 4.4, 2.7) | 5.60, ddd (9.8, 4.4, 2.5) | 5.68, ddd (10.1, 4.4, 2.6) |
| 8 | 2.82, m | 2.81, m | 2.83, m | 2.81, m |
| 9 | 4.41, dd (11.3, 5.8) | 4.41, dd (11.1, 5.6) | 4.43, dd (11.3, 5.7) | 4.46, dd (11.2, 5.8) |
| 10 | 1.55, m | 1.55, m | 1.58, m | 1.71, m |
| 11 | 0.92, d (6.5) | 0.92, d (6.5) | 3.39, dd (6.0, 1.2) | 1.03, d (6.4) |
| 12 | 0.81, d (7.2) | 0.81, d (7.2) | 0.82, d (7.2) | 0.82, d (7.2) |
| 17 | 8.00, s | 7.77, s | 7.57, s | 7.56, s |
| 21 | 3.87, d (7.9) | 4.20, s | 3.63, d (3.6) | 3.63, d (3.8) |
| 22 | 3.35, dd (9.8, 7.9) | 3.65. m | 3.41, t (3.6) | 3.41, dd, (3.8, 2.8) |
| 23 | 3.69, m | 3.73, m | 4.12, m | 4.12, m |
| 24a | 1.91, m | 1.98, m | 1.81, m | 1.81, m |
| 24b | 1.31, m | 1.55, m | 1.34, m | 1.33, m |
| 25a | 2.82, m | 2.59, m | 2.20, m | 2.20, m |
| 25b | 1.48, m | 1.76, m | 1.69, ddd (14.2, 10.2, 2.8) | 1.69, m |
| OCH3 | 3.63, s | 3.18, s |
| Compounds | MS | SA |
|---|---|---|
| 3 | 11.4 | 8.97 |
| 4 | >50 | 42.8 |
| 5 | 19.4 | 8.37 |
| 6 | 35.3 | 14.1 |
| 9 | 2.20 | 1.66 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, J.; Zhai, H.; Zhu, K.; Yu, J.-H.; Zhang, Y.; Wang, Y.; Jiang, C.-S.; Zhang, X.; Zhang, Y.; Zhang, H. Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008. Mar. Drugs 2018, 16, 174. https://doi.org/10.3390/md16050174
Bao J, Zhai H, Zhu K, Yu J-H, Zhang Y, Wang Y, Jiang C-S, Zhang X, Zhang Y, Zhang H. Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008. Marine Drugs. 2018; 16(5):174. https://doi.org/10.3390/md16050174
Chicago/Turabian StyleBao, Jie, Huijuan Zhai, Kongkai Zhu, Jin-Hai Yu, Yuying Zhang, Yinyin Wang, Cheng-Shi Jiang, Xiaoyong Zhang, Yun Zhang, and Hua Zhang. 2018. "Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008" Marine Drugs 16, no. 5: 174. https://doi.org/10.3390/md16050174
APA StyleBao, J., Zhai, H., Zhu, K., Yu, J.-H., Zhang, Y., Wang, Y., Jiang, C.-S., Zhang, X., Zhang, Y., & Zhang, H. (2018). Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008. Marine Drugs, 16(5), 174. https://doi.org/10.3390/md16050174