Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation


 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Biochemical Characterization of POE
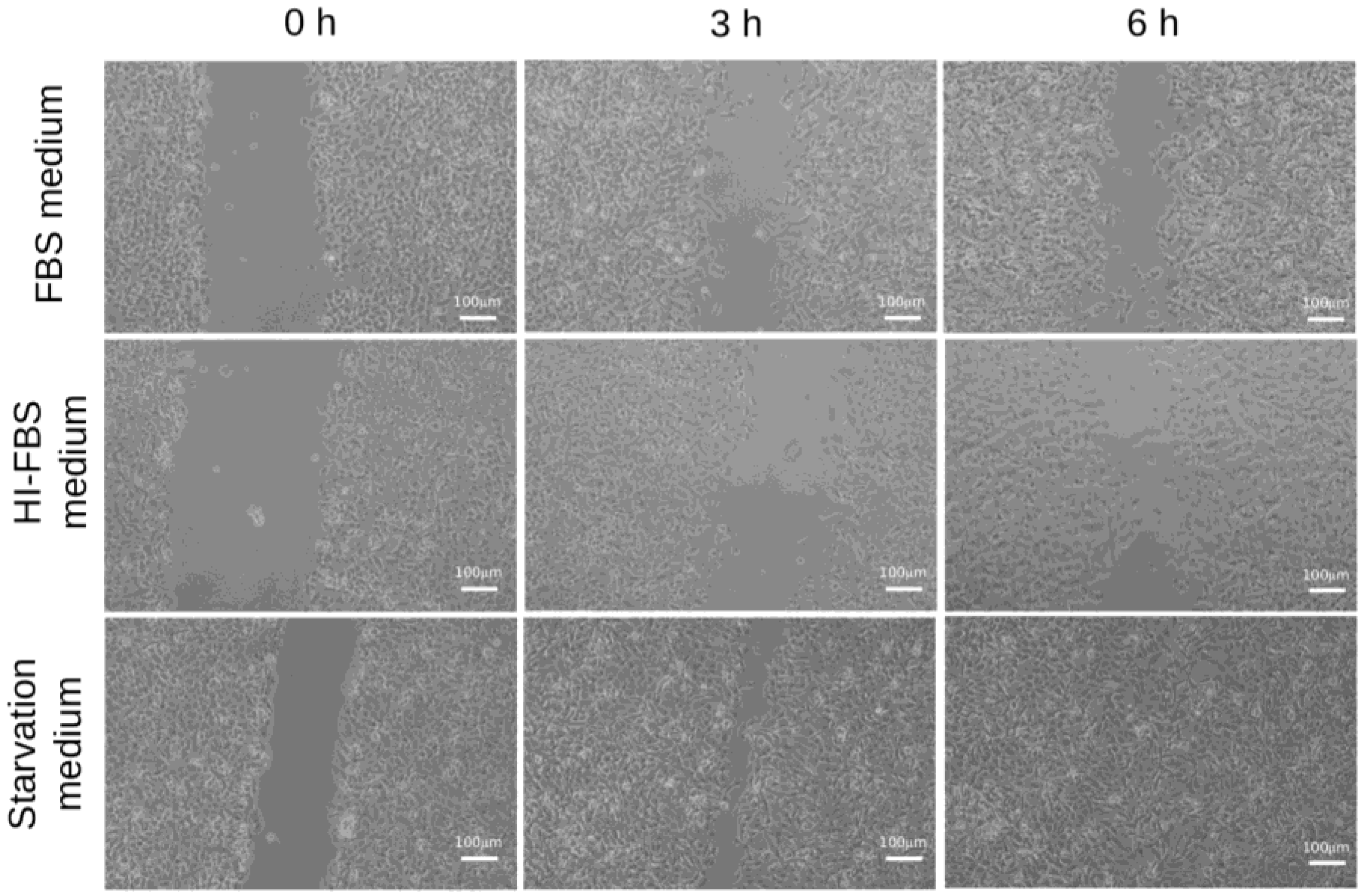
2.2. HT1080 Cell Migration in Heat-Inactivated Fetal Bovine Serum (FBS) Medium
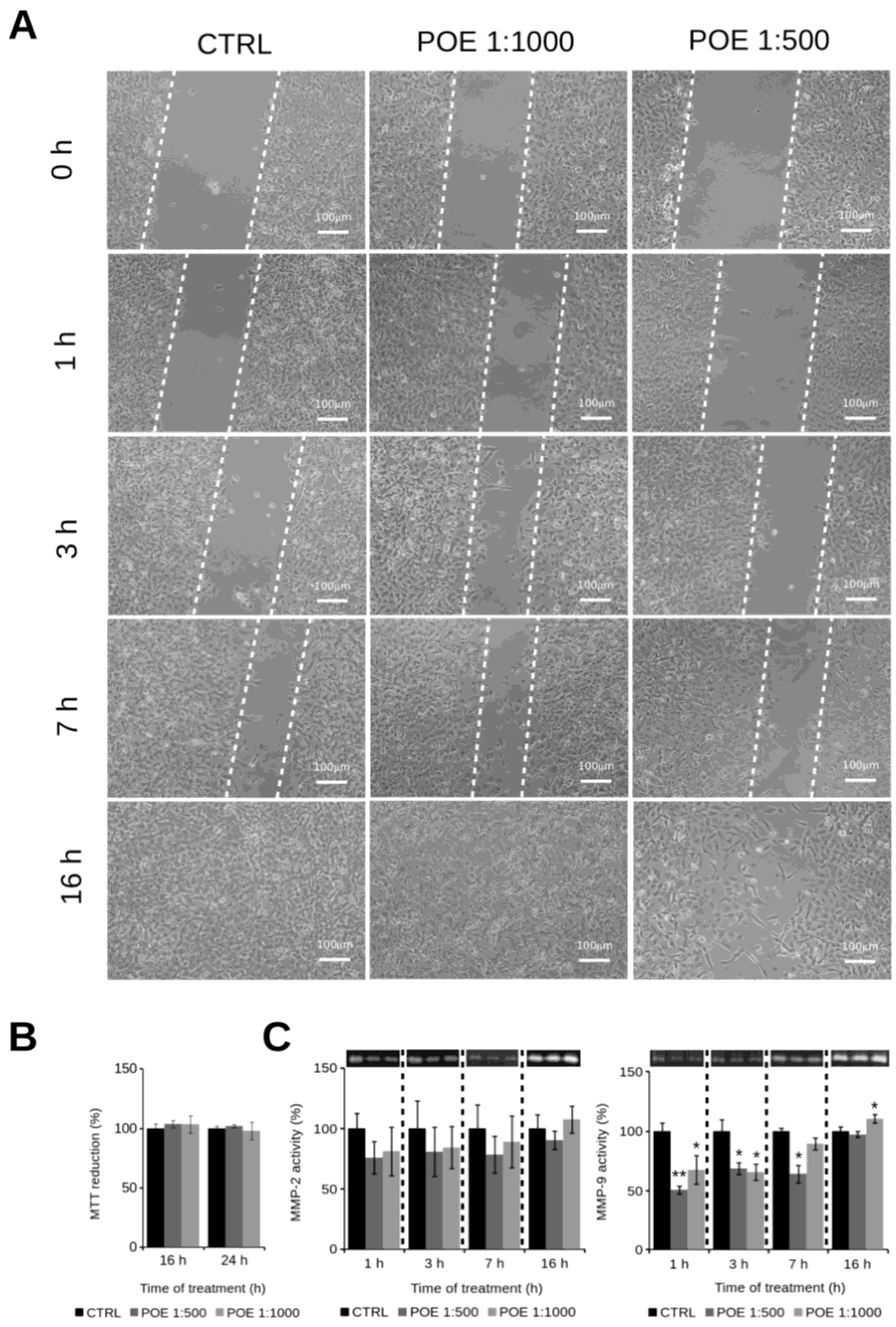
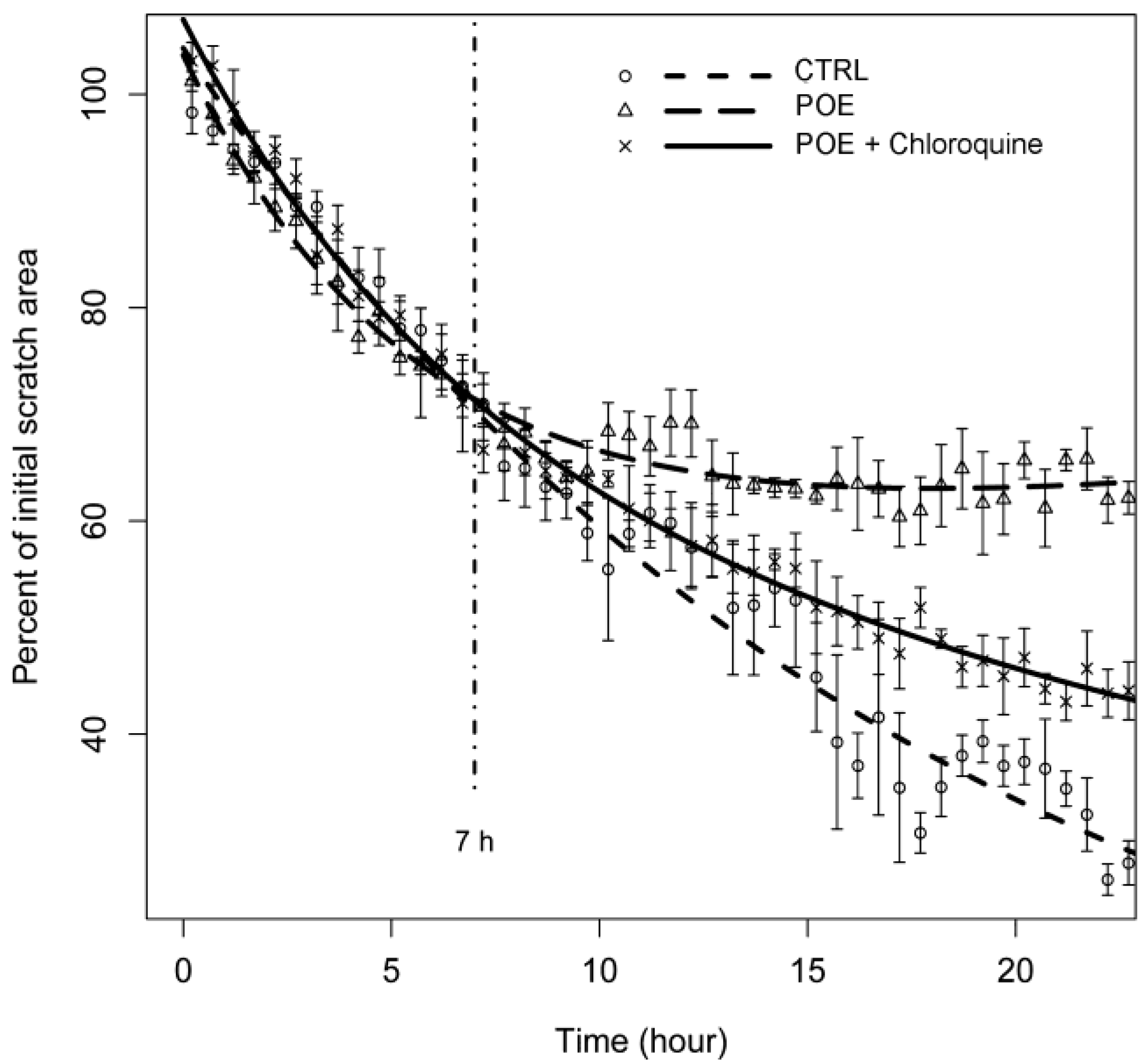
2.3. HT1080 Cell Migration Impairment Following POE Treatment
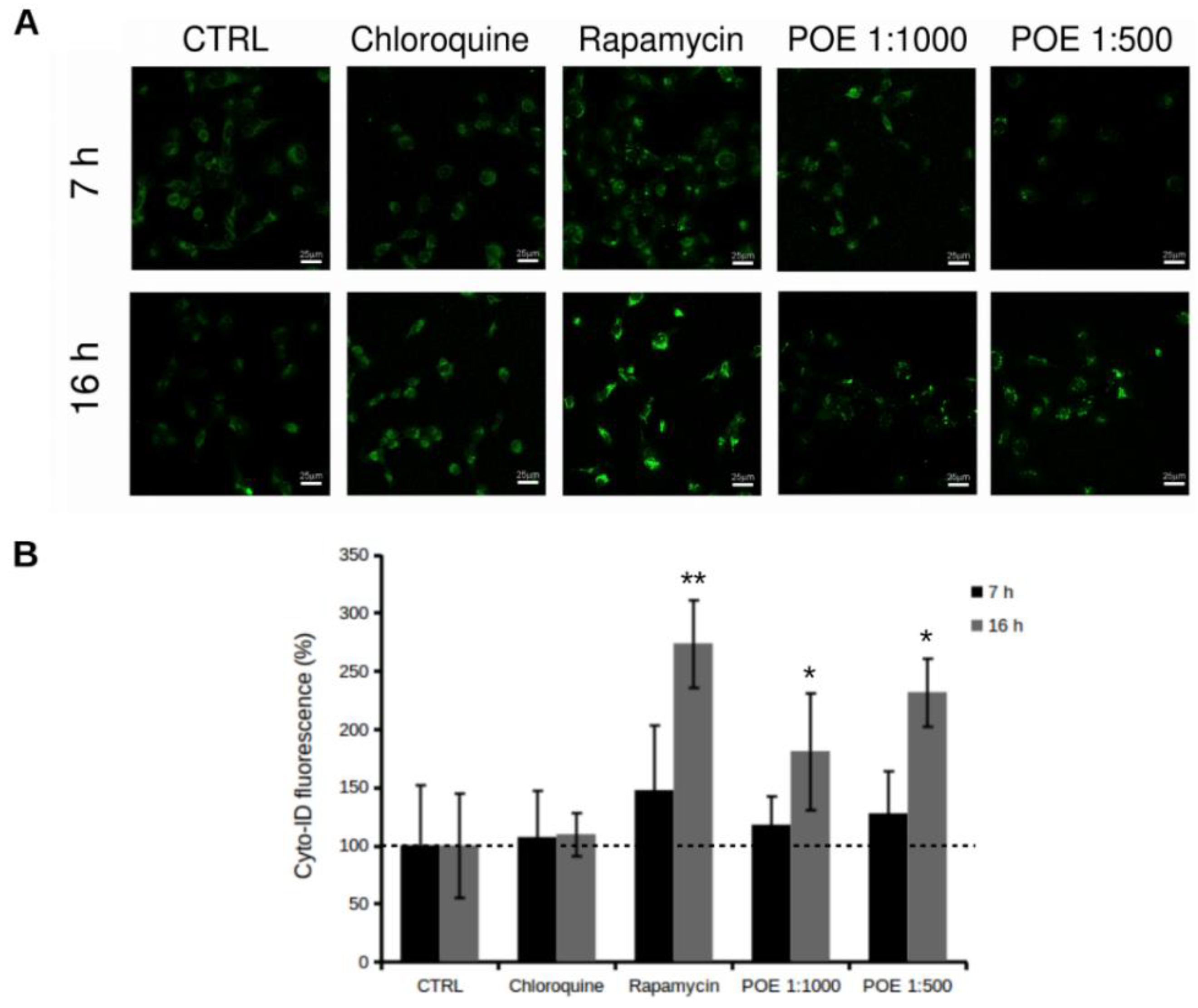
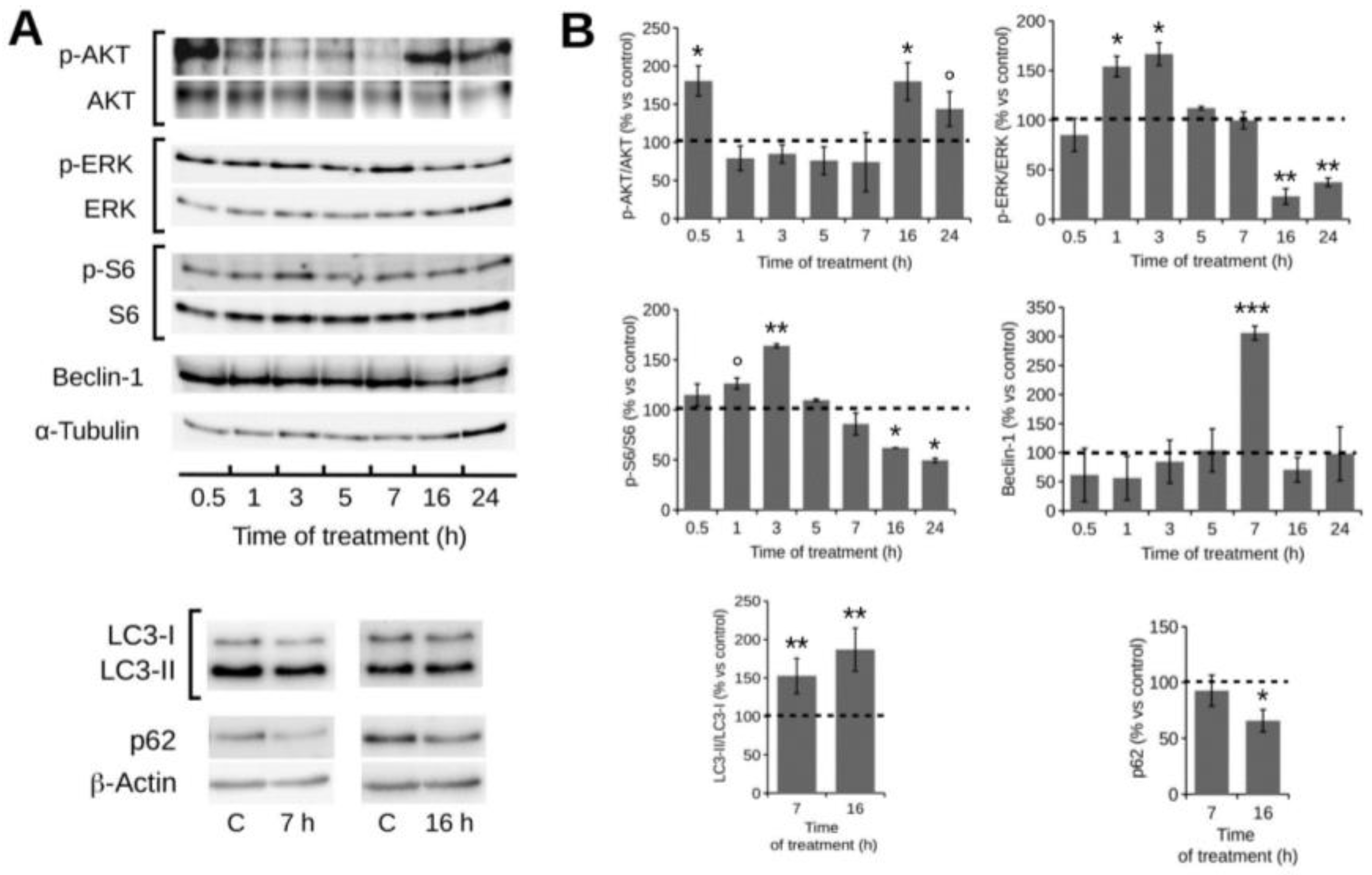
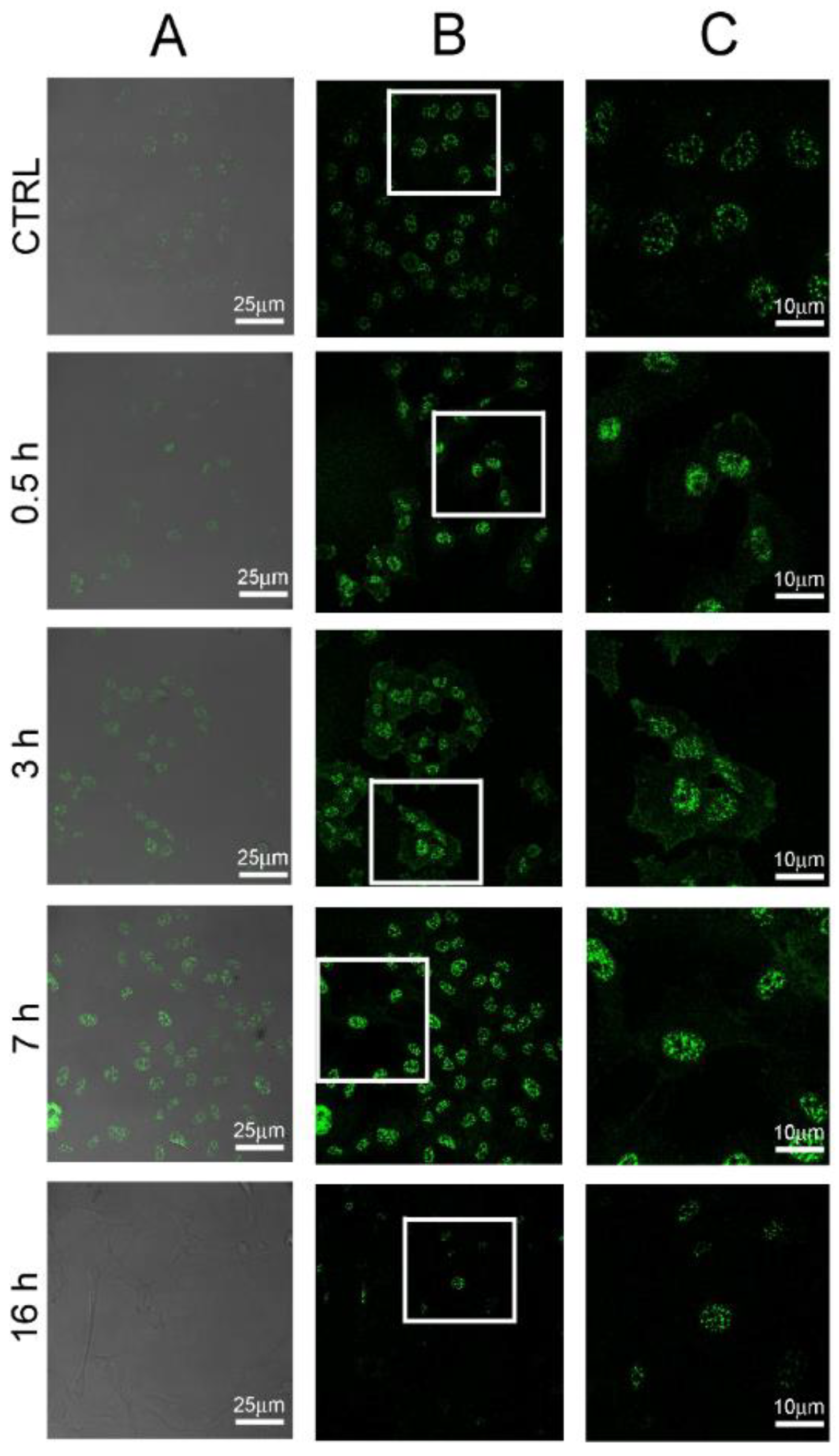
2.4. POE Treatment Induces Autophagy in HT1080 Cells
2.5. Autophagy Modulation by POE Decreases Cell Migration
3. Materials and Methods
3.1. Materials
3.2. Preparation of P. oceanica Extract
3.3. Determination of Total Polyphenol Content
3.4. Determination of Total Carbohydrate Content
3.5. Determination of Radical Scavenging Activity
3.6. Determination of Total Antioxidant Activity
3.7. Cell Line and Culture Conditions
3.8. Cell Viability Assay
3.9. Cell Migration Assay
3.10. Gelatin Zymography
3.11. Analysis of Autophagic Vacuoles
3.12. Analysis of IGF-1R Localization
3.13. Detection of Autophagy Markers
3.14. Data Analysis and Figure Preparation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gokce, G.; Haznedaroglu, M.Z. Evaluation of antidiabetic, antioxidant and vasoprotective effects of Posidonia oceanica extract. J. Ethnopharmacol. 2007, 115, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Cornara, L.; Pastorino, G.; Borghesi, B.; Salis, A.; Clericuzion, M.; Marchetti, C.; Damonte, G.; Burlando, B. Posidonia oceanica (L.) Delile Ethanolic Extract Modulates Cell Activities with Skin Health Applications. Mar. Drugs 2018, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Barletta, E.; Ramazzotti, M.; Fratianni, F.; Pessani, D.; Degl’Innocenti, D. Hydrophilic extract from Posidonia oceanica inhibits activity and expression of gelatinases and prevents HT1080 human fibrosarcoma cell line invasion. Cell Adhes. Migr. 2015, 9, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenviroment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Tuloup-Minguez, V.; Hamaï, A.; Greffard, A.; Nicolas, V.; Codogno, P.; Botti, J. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle 2013, 12, 3317–3328. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–1719. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Waguri, S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010, 648, 193–214. [Google Scholar] [PubMed]
- Kim, D.; Kim, S.; Koh, H.; Yoon, S.O.; Chung, A.S.; Cho, K.S.; Chung, J. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001, 15, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Mutschelknaus, L.; Azimzadeh, O.; Heider, T.; Winkler, K.; Vetter, M.; Kell, R.; Tapio, S.; Merl-Pham, J.; Huber, S.M.; Edalat, L.; et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci. Rep. 2017, 7, 12423. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sawai, H.; Matsuo, Y.; Ochi, N.; Yasuda, A.; Takahashi, H.; Wakasugu, T.; Funahashi, H.; Sato, M.; Takeyanna, H. IGF-1 mediates PTEN suppression and enhances cell invasion and proliferation via activation of the IGF-1/PI3K/Akt signaling pathway in pancreatic cancer cells. J. Surg. Res. 2010, 160, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 IGF receptor translocates to the nucleus of human tumor cells. Cancer Res. 2011, 70, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- Donglei, Z.; Pnina, B. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene 2003, 22, 974–982. [Google Scholar]
- Karam, A.K.; Santiskulvong, C.; Fekete, M.; Zabih, S.; Eng, C.; Dorigo, O. Cisplatin and PI3kinase Inhibition Decrease Invasion and Migration of Human Ovarian Carcinoma Cells and Regulate Matrix-Metalloproteinase Expression. Cytoskeleton 2010, 67, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, H.; Jia, Y.; Ding, H.; Zhang, L.; Pan, H. Luteolin reduces migration of human cell line via inhibition of the p-IGF-IR/PI3K/AKT/mTOR signaling pathway. Oncol. Lett. 2017, 14, 3545–3551. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2013, 33, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Castellano, L.E.; Almeida, A.M.; Renaut, J.; Argüello, A.; Castro, N. A proteomics study of colostrum and milk from the two major small ruminant dairy breeds from the Canary Islands: A bovine milk comparison perspective. J. Dairy Res. 2016, 83, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Autophagy: Eat thyself, sustain thyself. Nat. Methods 2015, 12, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2015, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J. On the Discovery of TOR as the Target of Rapamycin. PLoS Pathog. 2015, 11, e1005245. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.E.; Marlein, C.; Walker, H.F.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Davies, B.R.; Collins, A.T.; Maitland, N.J. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017, 8, 56698–56713. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.A.; Berhow, M.A.; Singletary, K.W. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006, 27, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef] [PubMed]
- Oeste, C.L.; Seco, E.; Patton, W.F.; Boya, P.; Pérez-Sala, D. Interactions between autophagic and endo-lysosomal markers in endothelial cells. Histochem. Cell Biol. 2012, 139, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Masuko, T.; Minami, A.; Iwasaki, N.; Majima, T.; Nishimura, S.; Lee, Y.C. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal. Biochem. 2005, 339, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, L.R.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Bravo, L.; Saura-Calixto, F. Antioxidant activity of dietary polyphenols as determined by a modified ferric reducing/antioxidant power assay. J. Agric. Food Chem. 2000, 48, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyphenols | Antioxidant Activity | Radical Scavenging | Carbohydrates | |
|---|---|---|---|---|
| Method | Folin–Ciocalteau | Ferrozine® | DPPH | Phenol/Sulfuric acid |
| Reference control | Gallic acid | Ascorbic acid | Ascorbic acid | Glucose |
| POE | 5.7 ± 0.3 | 1.5 ± 0.3 | 12.8 ± 0.7 | 13 ± 2 |
| Antibody | Target | Dilution | Host | Source |
|---|---|---|---|---|
| SQTSM1/p62 | SQTSM1/p62 protein | 1:1000 | Rabbit | Abcam |
| LC3A/B | Microtubule-associated protein light chain 3 (A/B) | 1:1000 | Rabbit | Cell Signaling |
| P-AKT1 | P-AKT1 serine/threonine kinase (Ser473) | 1:5000 | Rabbit | Abcam |
| AKT1/2 | AKT1/2 serine/threonine kinase | 1:5000 | Rabbit | Abcam |
| p44/42 MAPK(ERK1/2) | Mitogen-activated protein kinases p44/42 (ERK 1/2) | 1:2000 | Mouse | Cell Signaling |
| P-p44/42 MAPK(ERK 1/2) | Mitogen-activated protein kinases p44/42 (ERK 1/2) (Thr202/Thr204) | 1:1000 | Rabbit | Cell Signaling |
| Beclin-1 | Beclin-1 protein | 1:1000 | Rabbit | Cell Signaling |
| S6 | Ribosomial protein S6 | 1:1000 | Rabbit | Cell Signaling |
| P-S6 | Ribosomial protein S6 (Ser235/236) | 1:2000 | Rabbit | Cell Signaling |
| Alpha-Tubulin | Alpha-Tubulin protein | 1:1000 | Mouse | Cell Signaling |
| Actin | Actin protein | 1:1000 | Mouse | Santa Cruz |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leri, M.; Ramazzotti, M.; Vasarri, M.; Peri, S.; Barletta, E.; Pretti, C.; Degl’Innocenti, D. Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation. Mar. Drugs 2018, 16, 137. https://doi.org/10.3390/md16040137
Leri M, Ramazzotti M, Vasarri M, Peri S, Barletta E, Pretti C, Degl’Innocenti D. Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation. Marine Drugs. 2018; 16(4):137. https://doi.org/10.3390/md16040137
Chicago/Turabian StyleLeri, Manuela, Matteo Ramazzotti, Marzia Vasarri, Sara Peri, Emanuela Barletta, Carlo Pretti, and Donatella Degl’Innocenti. 2018. "Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation" Marine Drugs 16, no. 4: 137. https://doi.org/10.3390/md16040137
APA StyleLeri, M., Ramazzotti, M., Vasarri, M., Peri, S., Barletta, E., Pretti, C., & Degl’Innocenti, D. (2018). Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation. Marine Drugs, 16(4), 137. https://doi.org/10.3390/md16040137