Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
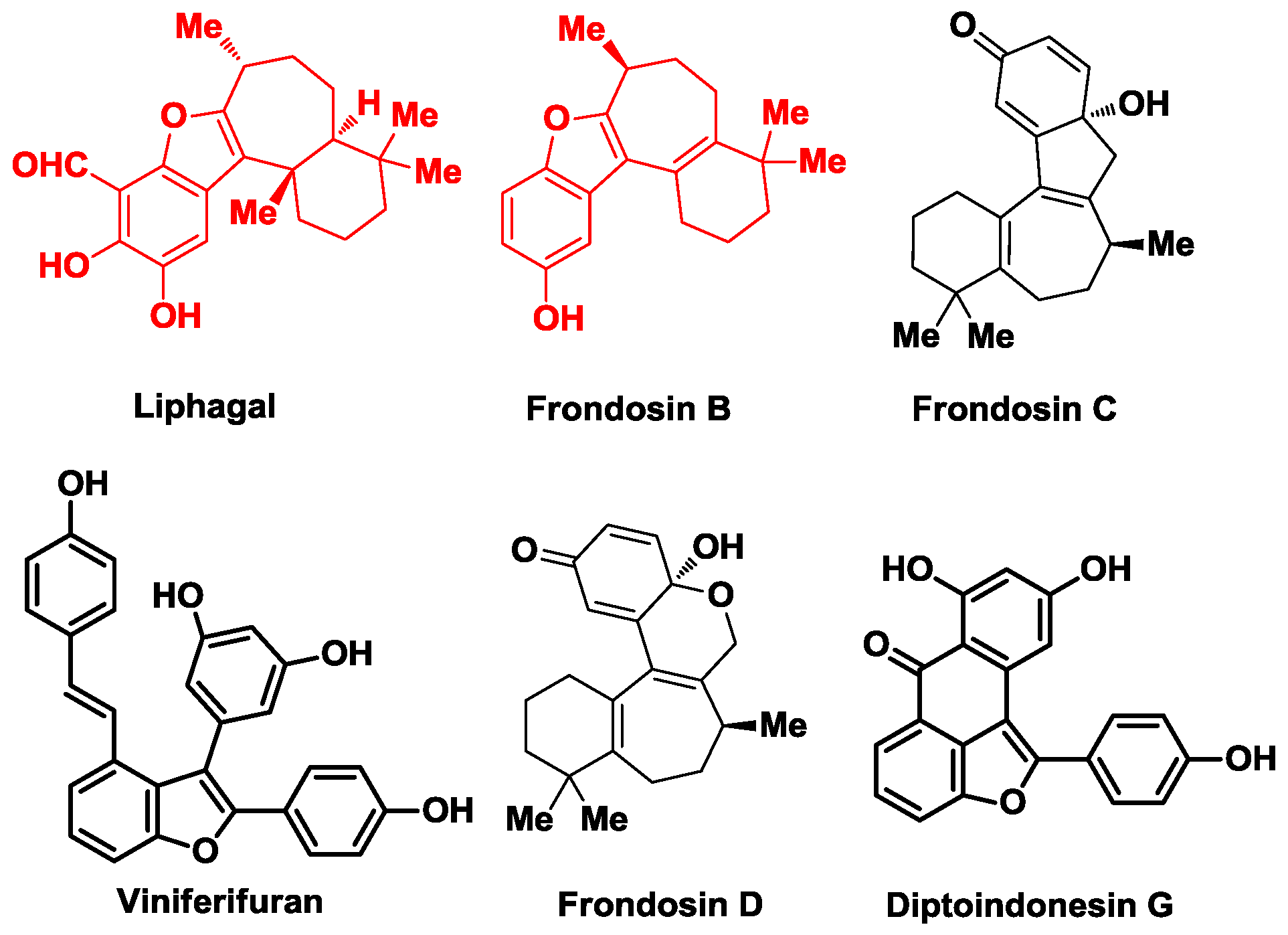
1. Introduction
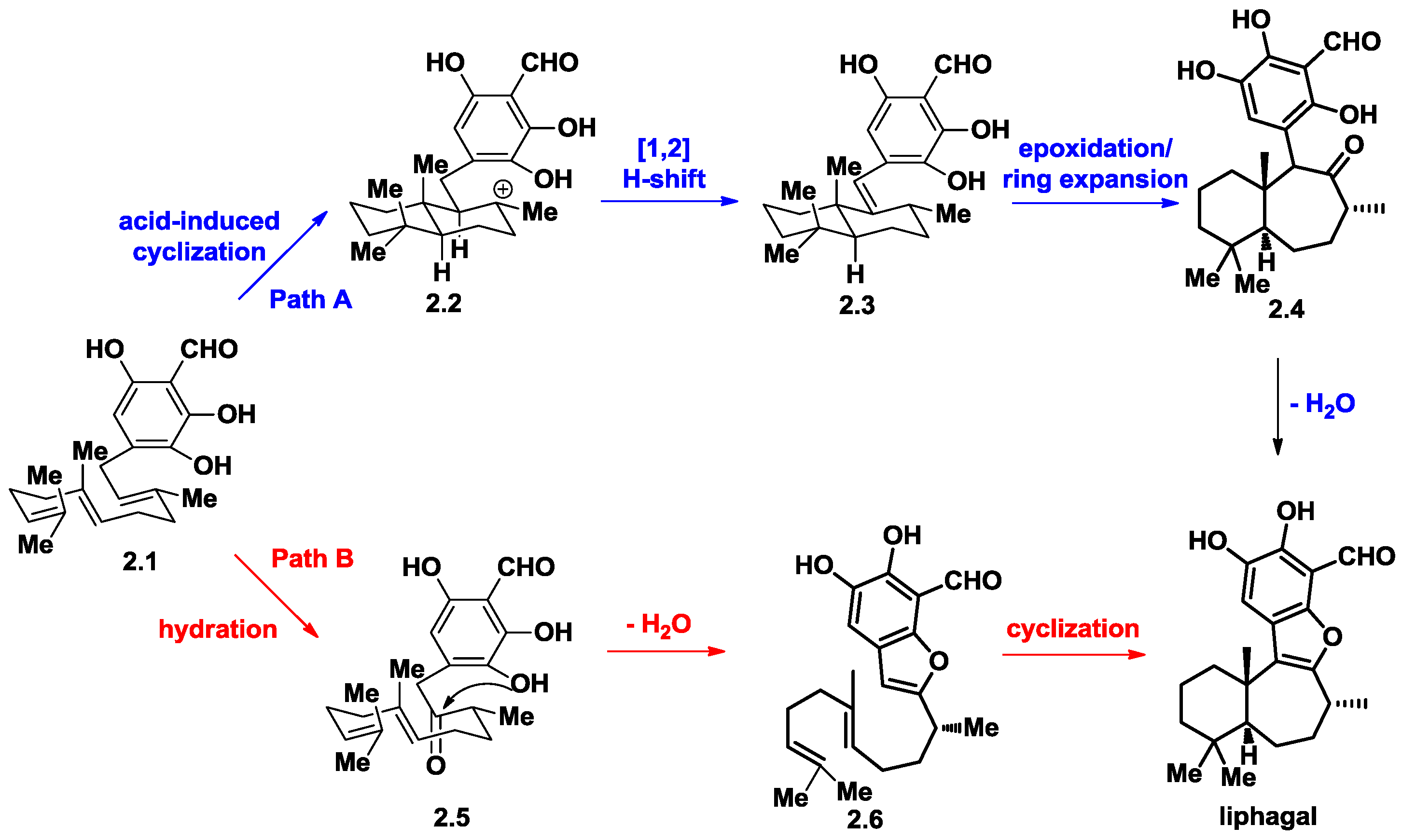
2. The Biosynthetic Proposal for Liphagal
3. Total Synthesis of Liphagal
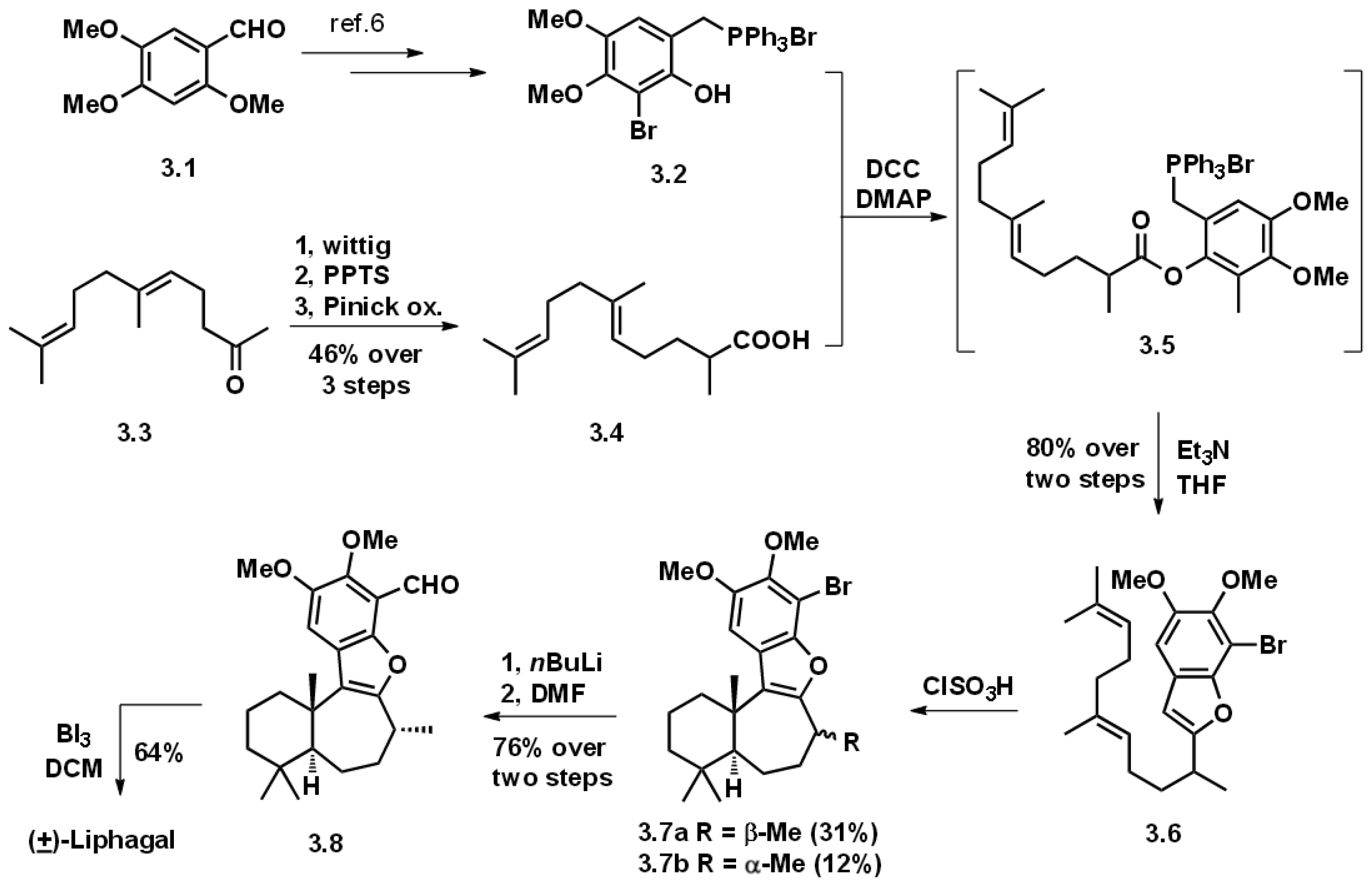
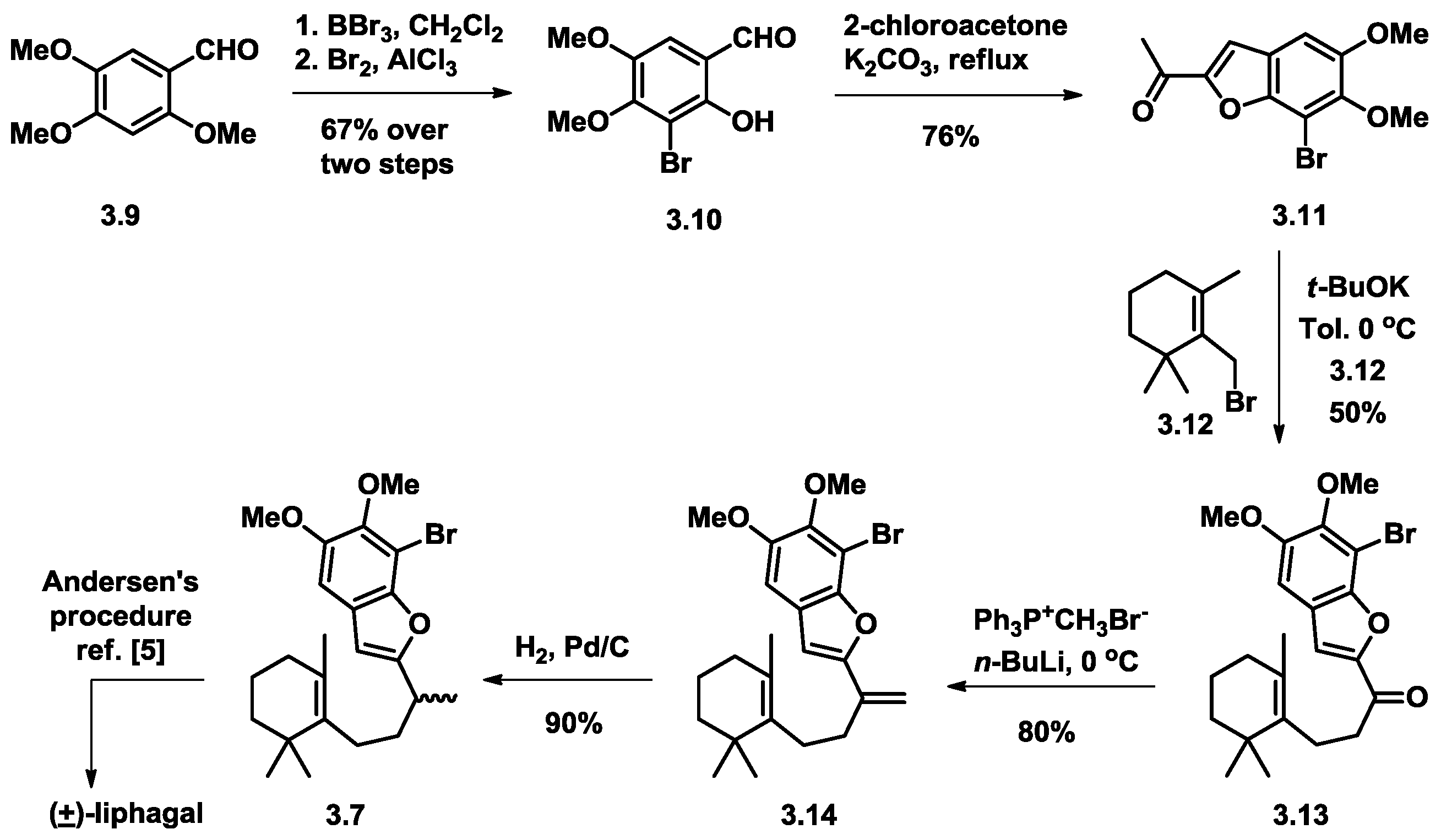
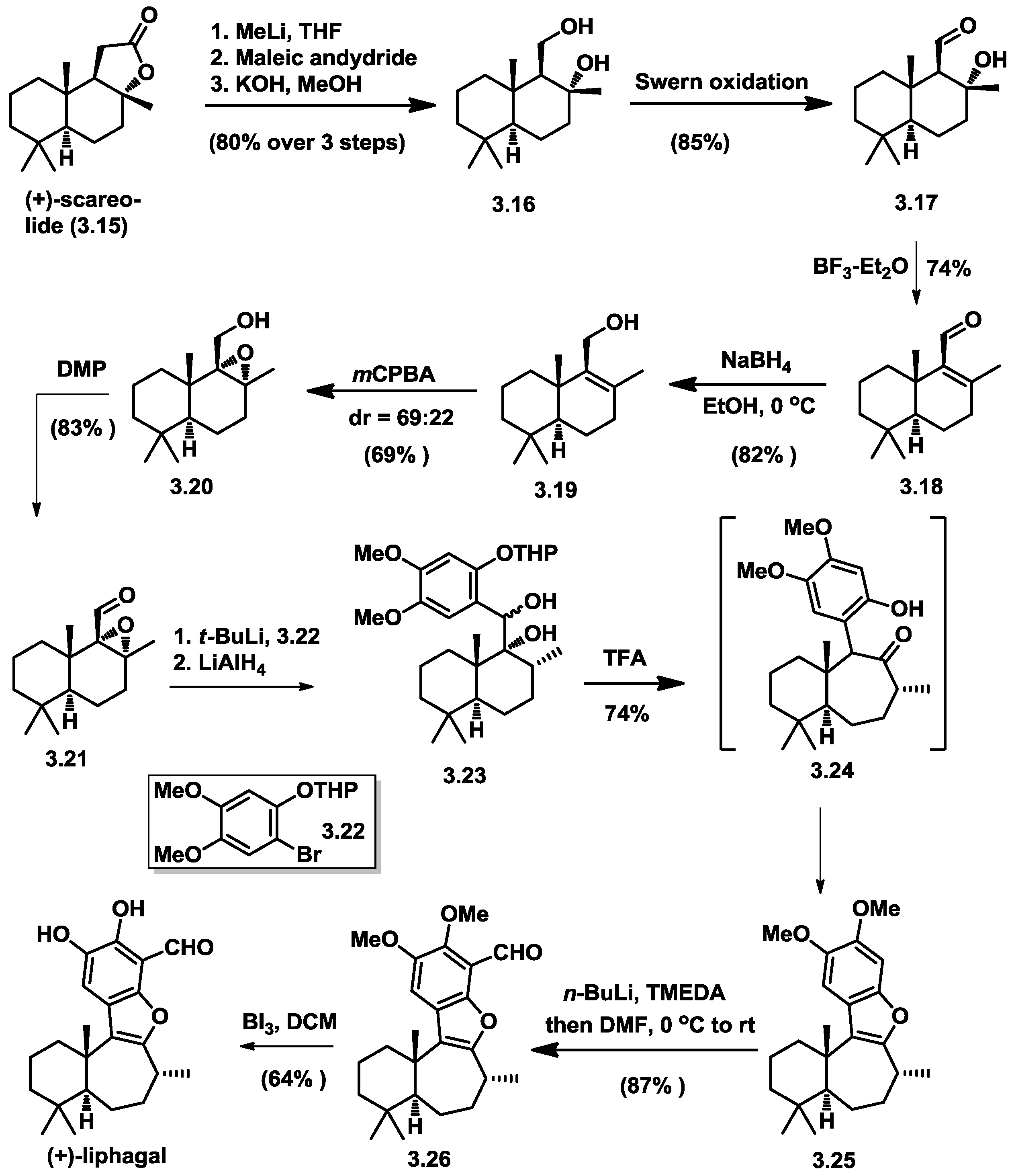
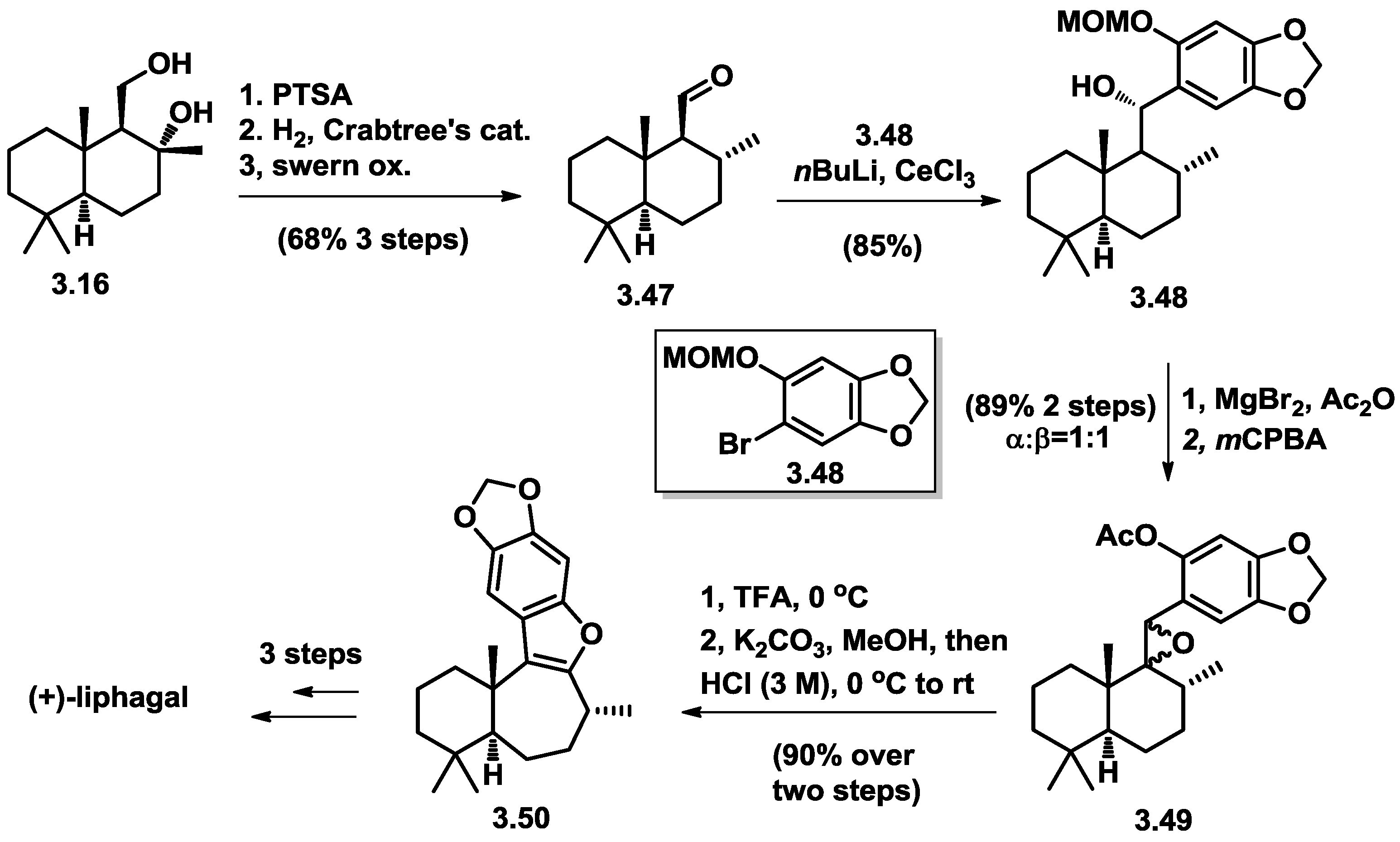
3.1. Biomimetic Total Synthesis of (+)-Liphagal
3.2. Mehta’s Formal Synthesis of (+)-Liphagal
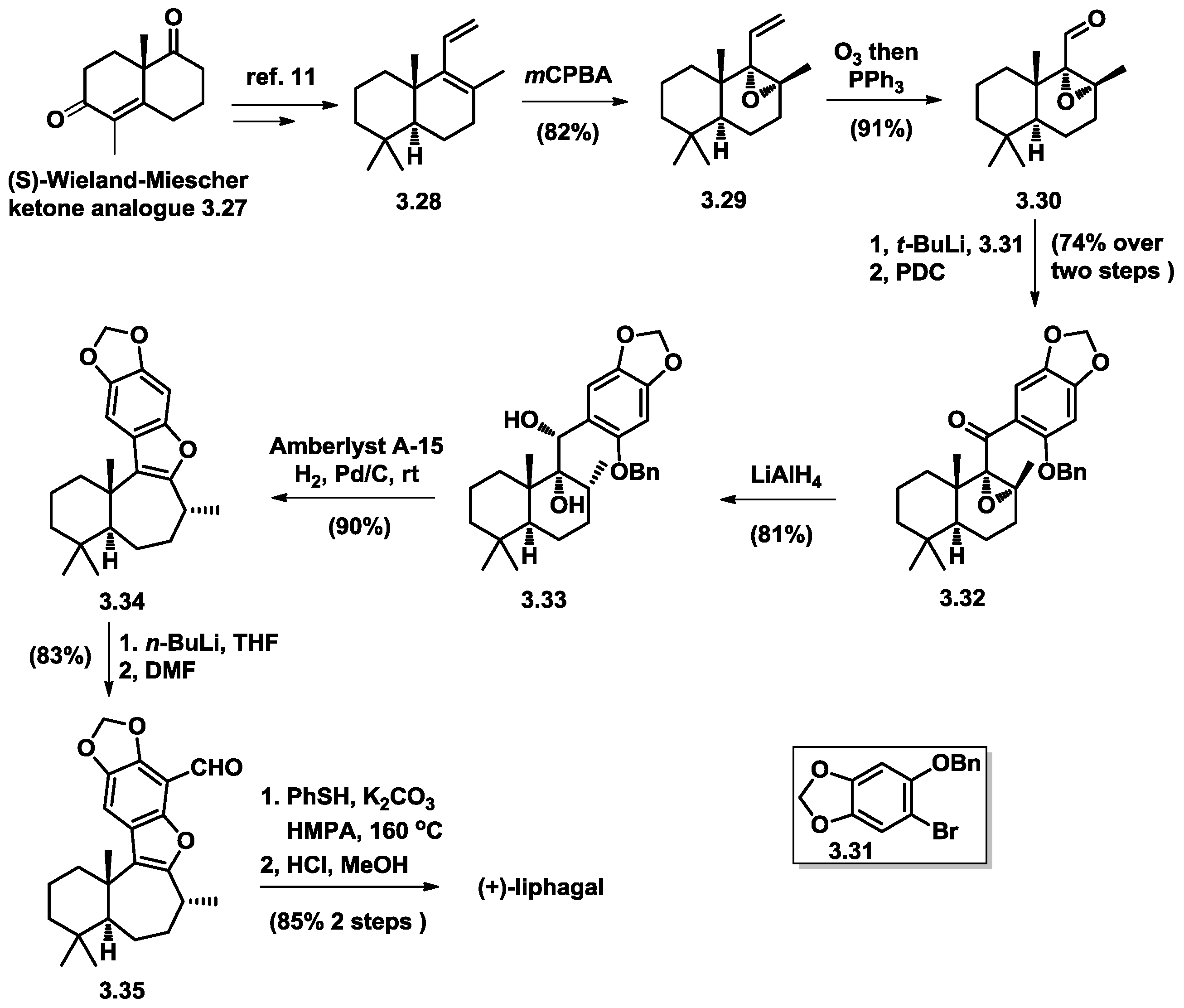
3.3. George’s Total Synthesis of (+)-Liphagal
3.4. Manzaneda’s Total Synthesis of (+)-Liphagal
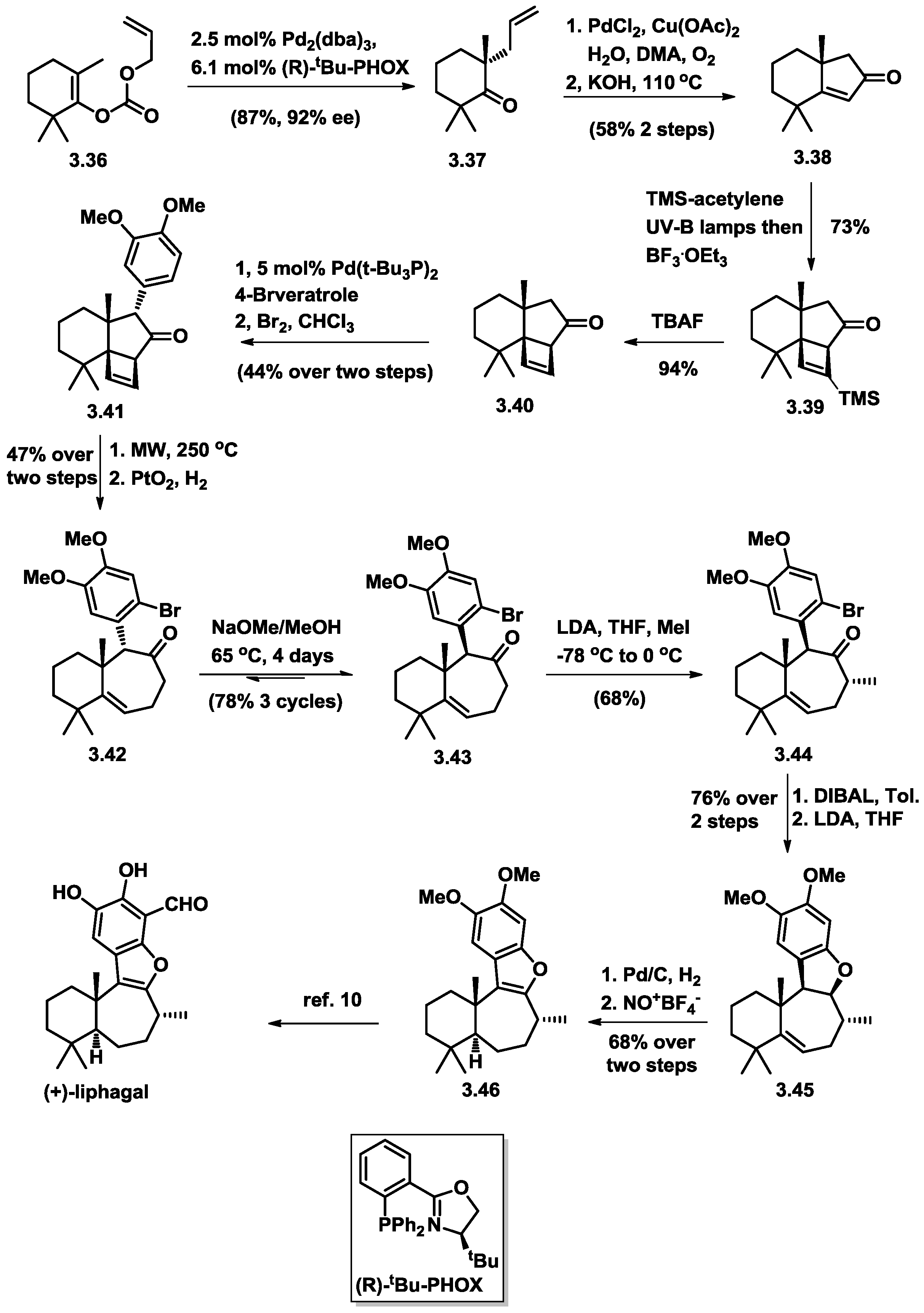
3.5. Stoltz’s Total Synthesis of (+)-Liphagal
3.6. Katol’s Total Synthesis of (+)-Liphagal
3.7. Synthetic Studies towards Liphagal
4. Total Synthesis of Frondosin B
4.1. Danishefsky’s Total Synthesis of (+)-Frondosin B
4.2. Trauner’s Total Synthesis of (−)-Frondosin B
4.3. Ovaska’s Total Synthesis of (−)-Frondosin B
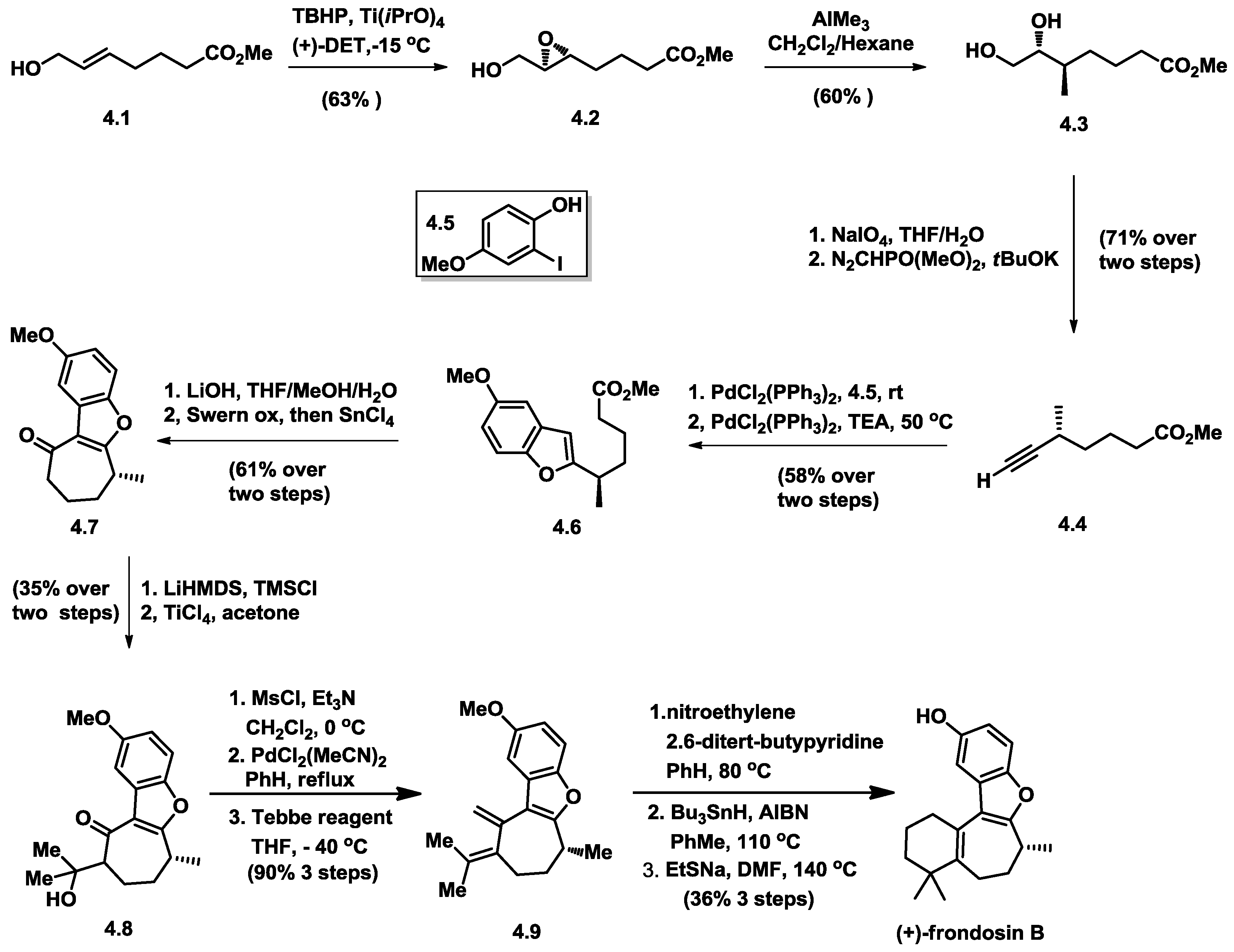
4.4. MacMillan’s Total Synthesis of (+)-Frondosin B
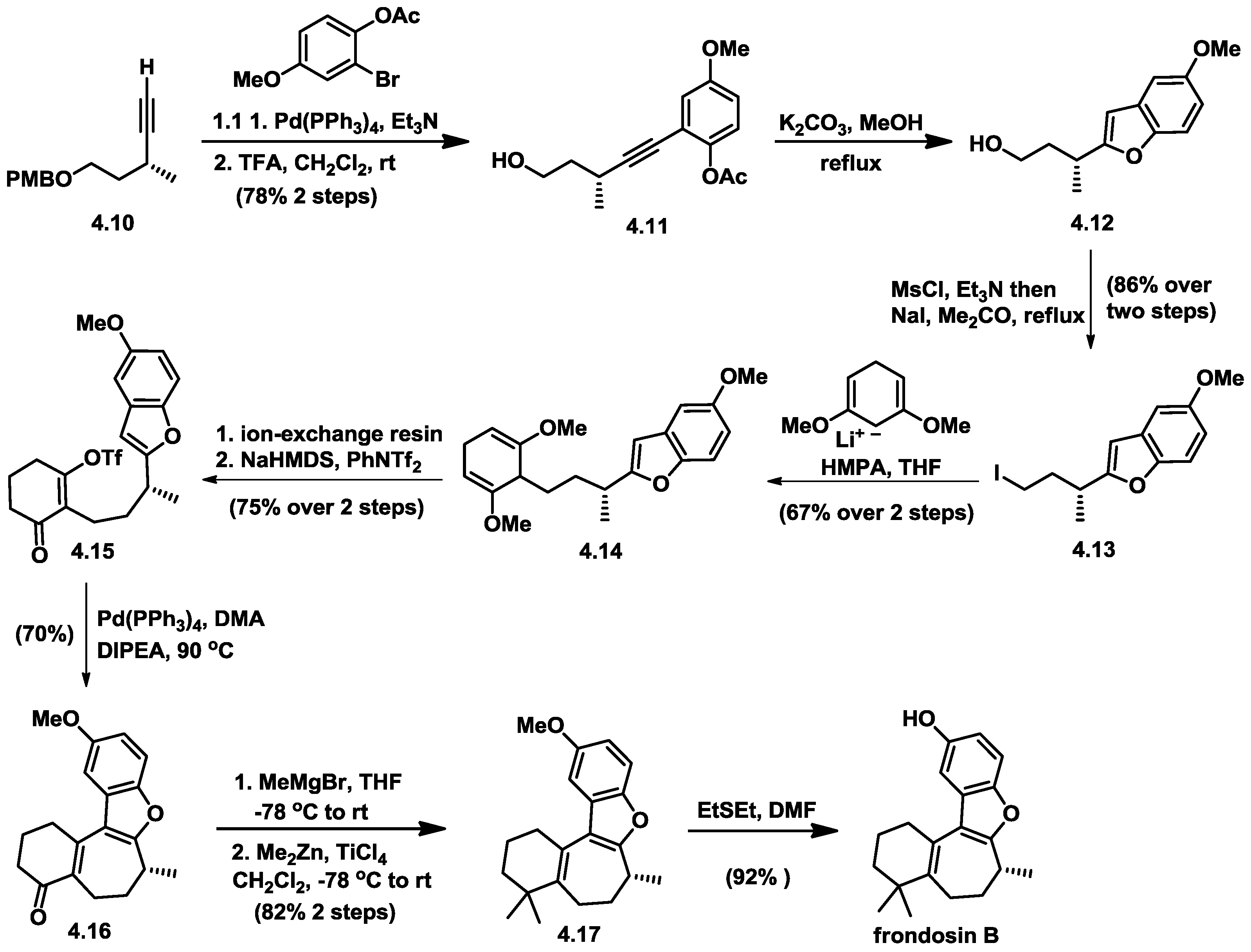
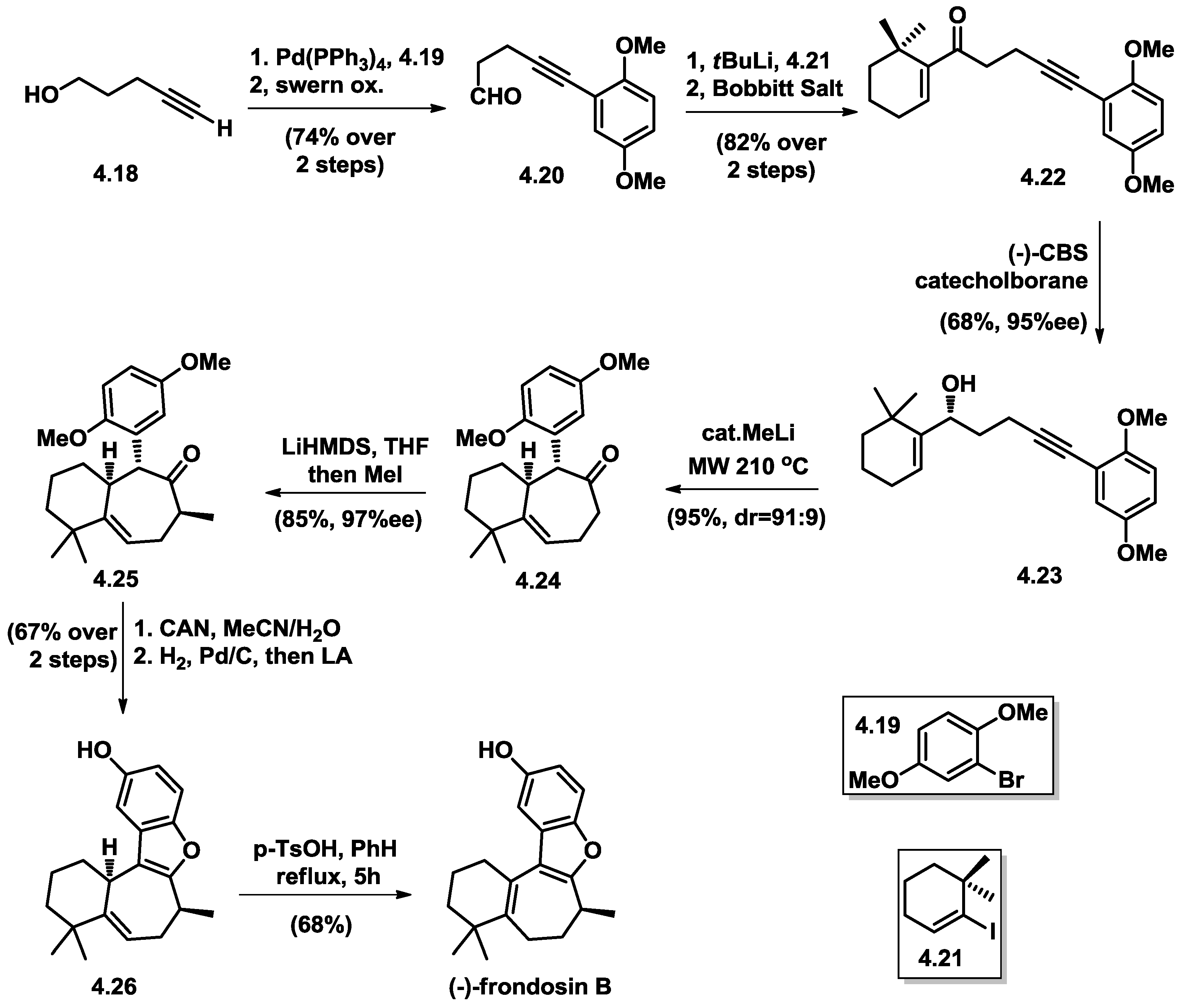
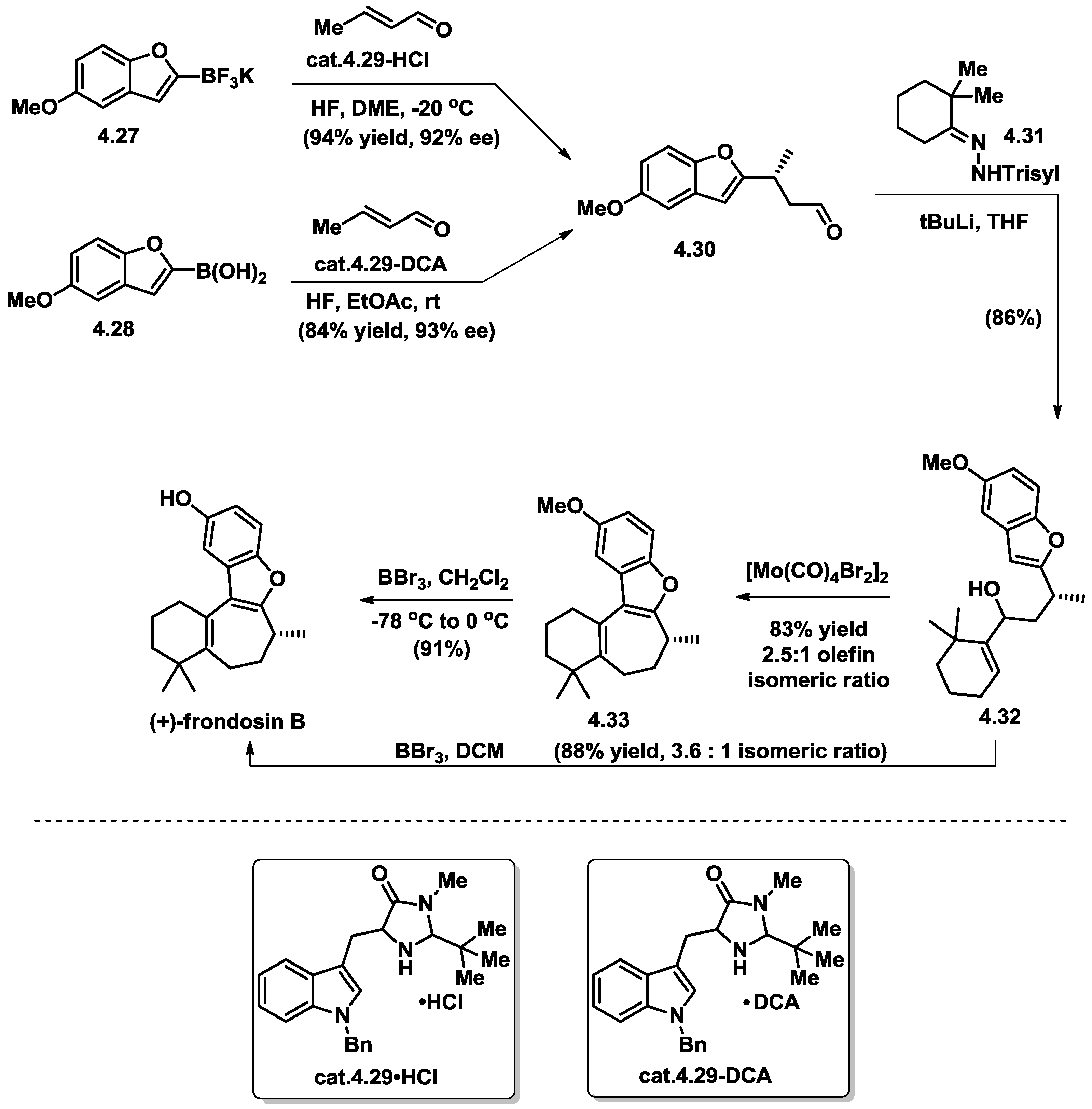
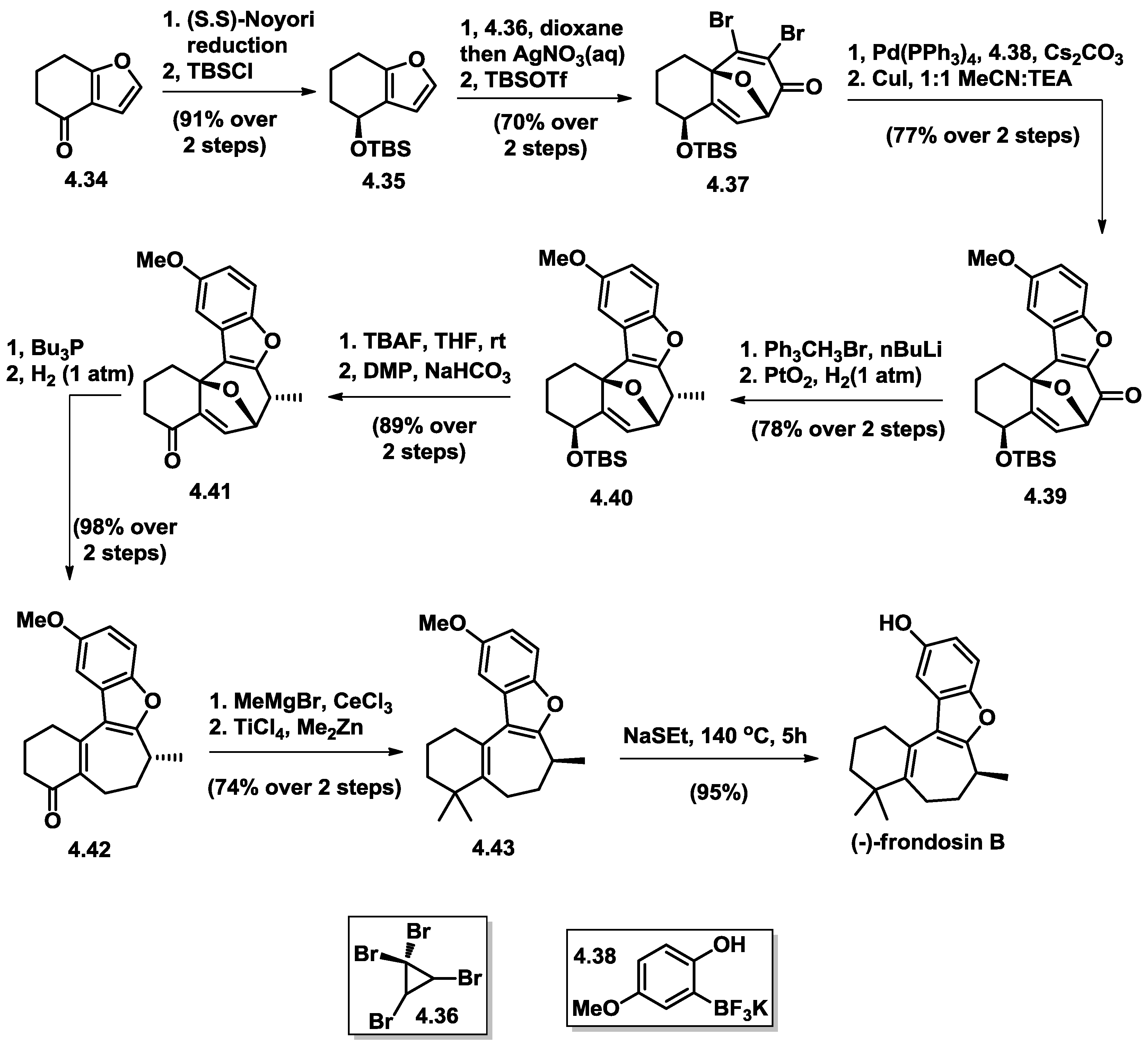
4.5. Wright’s Total Synthesis of (−)-Frondosin B
4.6. Synthetic Studies towards Frondosin B
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reusch, T.B.H. Climate change in the oceans: Evolutionary versus phenotypically plastic responses of marine animals and plants. Evol. Appl. 2014, 7, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Croteau, R.; Kutchan, T.M.; Lewis, N.G. Natural products (secondary metabolites). In Biochemistry and Molecular Biology of Plants; American Society of Plant Physiologists: Rockville, MD, USA, 2004; Chapter 24; pp. 1250–1319. [Google Scholar]
- Newman, D.; Cragg, G.M. Marine Natural Products and Related Compounds in Clinical and Advanced Preclinical Trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef] [PubMed]
- Grothaus, P.G.; Cragg, G.M.; Newman, D. New horizons for Old Drugs and Drug Leads. J. Nat. Prod. 2014, 77, 703–723. [Google Scholar] [CrossRef]
- Marion, F.; Williams, D.E.; Patrick, B.O.; Hollander, I.; Mallon, R.; Kim, S.C.; Roll, D.M.; Feldberg, L.; VanSoest, R.; Andersen, R.J. Liphagal, a Selective Inhibitor of PI3 Kinase α Isolated from the Sponge Aka coralliphaga: Structure Elucidation and Biomimetic Synthesis. Org. Lett. 2006, 8, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.W.; Faulkner, J.; Matsumoto, G.K.; He, C.H.; Clardy, J. Metabolites of the burrowing sponge Siphonodictyon coralliphagum. J. Org. Chem. 1986, 51, 4568–4573. [Google Scholar] [CrossRef]
- Sullivan, B.; Djura, P.; McIntyre, D.; Faulkner, D.J. Antimicrobial constituents of the sponge siphonodictyon coralliphagum. Tetrahedron 1981, 37, 979–982. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Killmer, L.; Offen, P.; Carte, B.; Jurewicz, A.J.; Johnson, R.K. Frondosins, five new sesquiterpene hydroquinone derivatives with novel skeletons from the sponge Dysidea frondosa: Inhibitors of interleukin-8 receptors. Tetrahedron 1997, 53, 5047–5060. [Google Scholar] [CrossRef]
- Mehta, G.; Likhite, N.S.; Kumar, C.S.A. A concise synthesis of the bioactive meroterpenoid natural product (±)-liphagal, a potent PI3K inhibitor. Tetrahedron Lett. 2009, 50, 5260–5262. [Google Scholar] [CrossRef]
- George, J.H.; Baldwin, J.E.; Adlington, R.M. Enantiospecific, Biosynthetically Inspired Formal Total Synthesis of (+)-Liphagal. Org. Lett. 2010, 12, 2394–2397. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Manzaneda, E.; Chahboun, R.; Alvarez, E.; Cano, M.J.; Haidour, A.; Alvarez-Manzaneda, R. Enantioselective Total Synthesis of the Selective PI3 Kinase Inhibitor Liphagal. Org. Lett. 2010, 12, 4450–4453. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Takeuchi, F.; Nozawa, M.; Hoshi, T.; Suzuki, T. The first total synthesis and determination of the absolute configuration of chapecoderin A, B and C. Tetrahedron 2004, 60, 1983–1989. [Google Scholar] [CrossRef]
- Day, J.J.; McFadden, R.M.; Virgil, S.C.; Kolding, H.; Alleva, J.L.; Stoltz, B.M. The Catalytic Enantioselective Total Synthesis of (+)-Liphagal. Angew. Chem. Int. Ed. 2011, 50, 6814–6818. [Google Scholar] [CrossRef] [PubMed]
- Behenna, D.C.; Stoltz, B.M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef] [PubMed]
- McFadden, R.M.; Stoltz, B.M. The Catalytic Enantioselective, Protecting Group-Free Total Synthesis of (+)-Dichroanone. J. Am. Chem. Soc. 2006, 128, 7738–7739. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kamishima, T.; Kikuchi, T.; Narita, K.; Katoh, T. Biogenetically Inspired Total Synthesis of (+)-Liphagal: A Potent and Selective Phosphoinositide 3-Kinase α (PI3Kα) Inhibitor from the Marine Sponge Aka coralliphaga. Eur. J. Org. Chem. 2014, 3443–3450. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Wang, Y.; Wang, W.; Xue, J.; Li, Y. A Novel, Facile Approach to Frondosin B and 5-epi-Liphagal via a New [4 + 3]-Cycloaddition. Org. Lett. 2012, 14, 4528–4530. [Google Scholar] [CrossRef] [PubMed]
- Laplace, D.R.; Verbraeken, B.; Van Hecke, K.; Winne, J.M. Total Synthesis of (+/−)-Frondosin B and (+/−)-5-epi-Liphagal by Using a Concise (4+3) Cycloaddition Approach. Chem. Eur. J. 2014, 20, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Tian, L.F.; Li, Z.T.; Liu, Q.; Li, C.C.; Yao, X.S.; Yang, Z. InCl3-mediated intramolecular Friedel-Crafts-type cyclization and its application to construct the [6-7-5-6] tetracyclic scaffold of liphagal. Sci. China Chem. 2012, 55, 36–42. [Google Scholar] [CrossRef]
- Peng, J.; Hou, X.; Zhang, S.; Tu, Y. Studies on the Total Synthesis of 8-epi-Liphagal. Acta Chim. Sin. 2012, 70, 2232–2235. [Google Scholar] [CrossRef]
- Wang, J.-L.; Li, H.-J.; Wang, H.-S.; Wu, Y.-C. Regioselective 1,2-Diol Rearrangement by Controlling the Loeading of BF3-Et2O and Its Application to the Synthesis of Related Nor-Sesquiterpene- and Sesquiterpene-Type Marine Natural Products. Org. Lett. 2017, 19, 3811–3814. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.Q.; Seizert, C.A.; Ozumerzifon, T.J.; Allegretti, P.A.; Ferreira, E.M. Platinum-Catalyzed a,b-Unsaturated Carbene Formation in the Formal Syntheses of Frondosin B and Liphagal. Org. Lett. 2017, 19, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Markwell-Heys, A.W.; Kuan, K.K.W.; George, J.H. Total Synthesis and Structure Revision of (-)-Siphonodictyal B and Its Biomimetic Conversion into (+)-Liphagal. Org. Lett. 2015, 17, 4228–4231. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Frontier, A.J.; Danishefsky, S.J. The Total Synthesis of Frondosin B. Angew. Chem. Int. Ed. 2000, 39, 761–764. [Google Scholar] [CrossRef]
- Inoue, M.; Carson, M.W.; Frontier, A.J.; Danishefsky, S.J. Total Synthesis and Determination of the Absolute Configuration of Frondosin B. J. Am. Chem. Soc. 2001, 123, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; Trauner, D. Concise Total Synthesis of (−)-Frondosin B Using a Novel Palladium-Catalyzed Cyclization. Angew. Chem. Int. Ed. 2002, 41, 1569–1572. [Google Scholar] [CrossRef]
- Hughes, C.C.; Trauner, D. Palladium-catalyzed couplings to nucleophilic heteroarenes: The total synthesis of (−)-frondosin B. Tetrahedron 2004, 60, 9675–9686. [Google Scholar] [CrossRef]
- Ovaska, T.V.; Sullivan, J.A.; Ovaska, S.I.; Winegrad, J.B.; Fair, J.D. Asymmetric Synthesis of Seven-Membered Carbocyclic Rings via a Sequential Oxyanionic 5-Exo-Dig Cyclization/Claisen Rearrangement Process. Total Synthesis of (−)-Frondosin B. Org. Lett. 2009, 11, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Reiter, M.; Torssell, S.; Lee, S.; MacMillan, D.W.C. The organocatalytic three-step total synthesis of (+)-frondosin B. Chem. Sci. 2010, 1, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Oblak, E.Z.; VanHeyst, M.D.; Li, J.; Wiemer, A.J.; Wright, D.L. Cyclopropene Cycloadditions with Annulated Furans: Total Synthesis of (+)- and (−)-Frondosin B and (+)-Frondosin A. J. Am. Chem. Soc. 2014, 136, 4309–4315. [Google Scholar] [CrossRef] [PubMed]
- VanHeyst, M.D.; Wright, D.L. The Frondosins: An Unusual Synthetic and Stereochemical Journey. Eur. J. Org. Chem. 2015, 1387–1401. [Google Scholar] [CrossRef]
- Mehta, G.; Likhite, N.S. A total synthesis of (±)-frondosins A and B. Tetrahedron Lett. 2008, 49, 7113–7116. [Google Scholar] [CrossRef]
- Kerr, D.J.; Willis, A.C.; Flynn, B.L. Multicomponent Coupling Approach to (±)-Frondosin B and a Ring-Expanded Analogue. Org. Lett. 2004, 6, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Hu, Y.; Horne, D.B. Total Synthesis of (+)-Frondosin A. Application of the Ru-Catalyzed [5+2] Cycloaddition. J. Am. Chem. Soc. 2007, 129, 11781–11790. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Debien, L. Re-orienting coupling of organocuprates with propargyl electrophiles from SN2′ to SN2 with stereocontrol. Chem. Sci. 2016, 7, 4985–4989. [Google Scholar] [CrossRef] [PubMed]
- Ovaska, T.V. Synthesis of cycloheptanoid natural products via a tandem 5-exo-cyclization/Claisen rearrangement process. Arkivoc 2011, 5, 34–44. [Google Scholar] [CrossRef]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, Y.; Wang, W.; Xu, T. Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B. Mar. Drugs 2018, 16, 115. https://doi.org/10.3390/md16040115
Zong Y, Wang W, Xu T. Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B. Marine Drugs. 2018; 16(4):115. https://doi.org/10.3390/md16040115
Chicago/Turabian StyleZong, Yan, Weijia Wang, and Tao Xu. 2018. "Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B" Marine Drugs 16, no. 4: 115. https://doi.org/10.3390/md16040115
APA StyleZong, Y., Wang, W., & Xu, T. (2018). Total Synthesis of Bioactive Marine Meroterpenoids: The Cases of Liphagal and Frondosin B. Marine Drugs, 16(4), 115. https://doi.org/10.3390/md16040115