Cytotoxic Sesterterpenes from Thai Marine Sponge Hyrtios erectus

Abstract
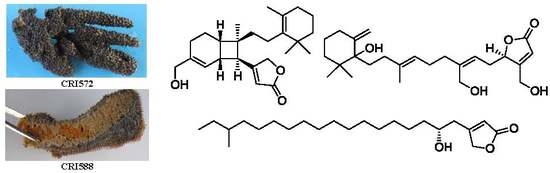
1. Introduction
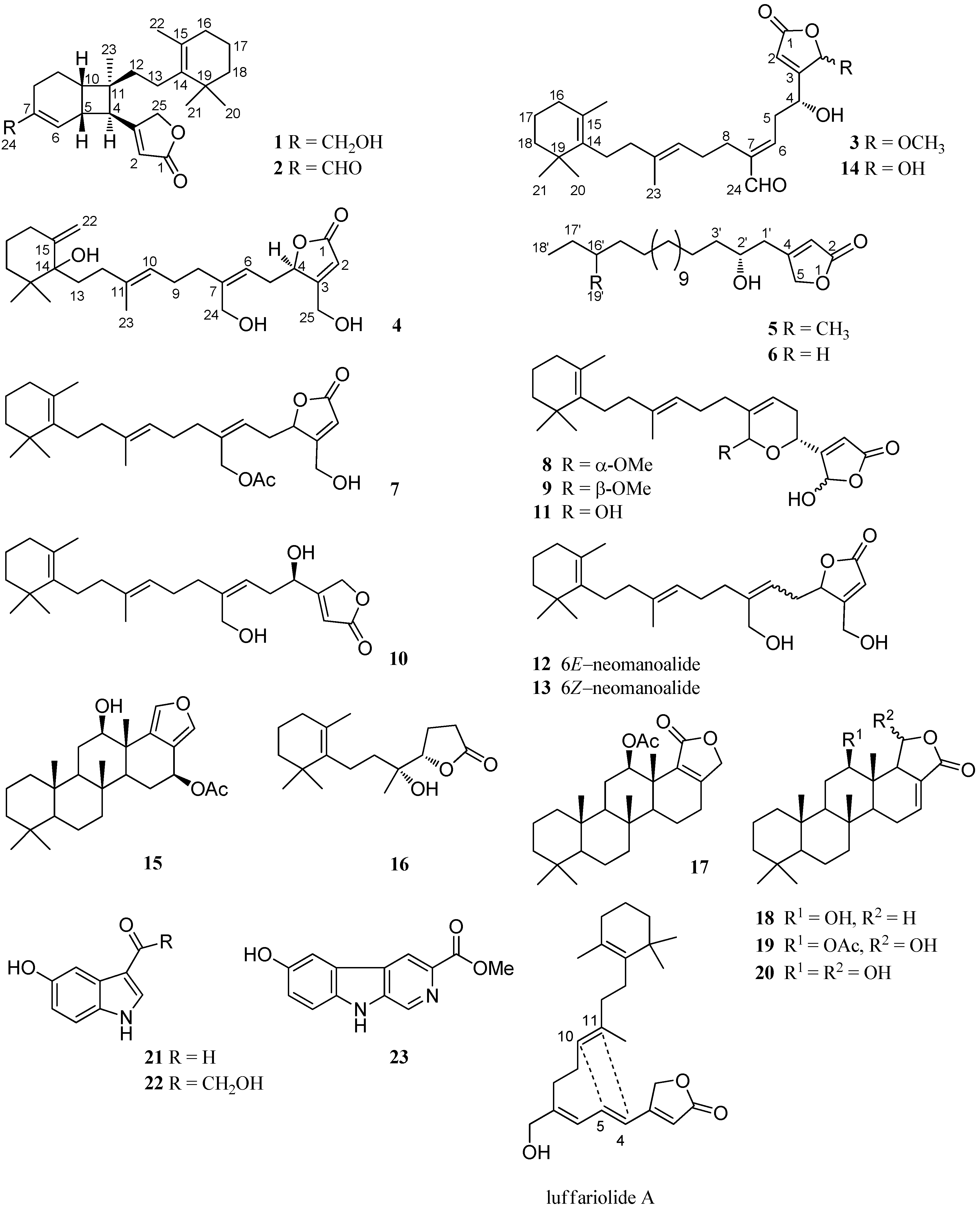
2. Results
3. Experimental Section
3.1. General Experimental Procedures
3.2. Sponge Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bugni, S.T.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Kaweetripob, W.; Mahidol, C.; Wongbundit, S.; Tuntiwachwuttikul, P.; Ruchirawat, S.; Prawat, H. Sesterterpenes and phenolic alkenes from the Thai sponge Hyrtios erectus. Tetrahedron 2018, 74, 316–323. [Google Scholar] [CrossRef]
- Jiao, W.H.; Hong, L.L.; Sun, J.B.; Piao, S.J.; Chen, G.D.; Deng, H.; Wang, S.P.; Yang, F.; Lin, H.W. (±)-Hippolide J—A Pair of Unusual Antifungal Enantiomeric Sesterterpenoids from the Marine Sponge Hippospongia lachne. Eur. J. Org. Chem. 2017, 24, 3421–3426. [Google Scholar] [CrossRef]
- Hawas, U.W.; Abou El-Kassem, L.T.; Abdelfattah, M.S.; Elmallah, M.I.Y.; Eid, M.A.G.; Monier, M.; Nithiyanandam, M. Cytotoxic activity of alkyl benzoate and fatty acids from the red sea sponge Hyrtios erectus. Nat. Prod. Res. 2018, 32, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Elhady, S.S.; Al-Abd, A.M.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. Antiproliferative Scalarane-Based Metabolites from the Red Sea Sponge Hyrtios erectus. Mar. Drugs 2016, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Kato, H.; Rotinsulu, H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Yokosawa, H.; Tsukamoto, S. Variabines A and B: New β-carboline alkaloids from the marine sponge Luffariella variabilis. J. Nat. Med. 2014, 68, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Mahidol, C.; Prawat, H.; Sangpetsiripan, S.; Ruchirawat, S. Bioactive Scalaranes from the Thai Sponge Hyrtios gumminae. J. Nat. Prod. 2009, 72, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Konig, G.; Wrignt, A.D.; Sticher, O. Four new antibacterial sesterterpenes from a marine sponge of the genus Luffariella. J. Nat. Prod. 1992, 55, 174–178. [Google Scholar] [CrossRef]
- Namikoshi, M.; Suzuki, S.; Meguro, S.; Nagai, H.; Koike, Y.; Kitazawa, A.; Kobayashi, H.; Oda, T.; Yamada, J. Manoalide derivatives from a marine sponge Luffariella sp. collected in Palau. Fish. Sci. 2004, 70, 152–158. [Google Scholar] [CrossRef]
- Pailee, P.; Mahidol, C.; Ruchirawat, S.; Prachyawarakorn, V. Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity. Mar. Drugs 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Okamoto, T.; Hayashi, K.; Yokoyama, N.; Sasaki, T.; Kitagawa, I. Marine Natural Products. XXXII. Absolute Configurations of C-4 of the Manoalide Family, Biologically Active Sesterterpenes from the Marine Sponge Hyrtions erecta. Chem. Pharm. Bull. 1994, 42, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Shigemori, H.; Ishibashi, M.; Mizobe, F.; Kawashima, A.; Nakaike, S.; Kobayashi, J. New Sesterterpenes with Nerve Growth Factor Synthesis-Stimulating Activity from the Okinawan Marine Sponge Hyrtios sp. Chem. Pharm. Bull. 1993, 41, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.A.; Elkhayat, E.S.; Ebel, R.; Edrada, R.; Prokschc, P. Indole alkaloid from the Red Sea sponge Hyrtios erectus. Arkivoc 2007, XV, 225–231. [Google Scholar]
- Tsuda, M.; Shigemori, H.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Luffariolides AE, new cytotoxic sesterterpenes from the Okinawan marine sponge Luffariella sp. J. Org. Chem. 1992, 57, 3503–3507. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Miura, S.; van Soest, R.W.M.; Ohta, T. Three New Cytotoxic Sesterterpenes from a Marine Sponge Spongia sp. J. Nat. Prod. 2003, 66, 438–440. [Google Scholar] [CrossRef] [PubMed]
- De Silva, E.D.; Scheuer, P.J. Three new sesterterpenoid antibiotics from the marine sponge Luffariella variabilis (Polejaff). Tetrahedron Lett. 1981, 22, 3147–3150. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Prachyawarakorn, V.; Tuntiwachwuttikul, P.; Ruchirawat, S. Sesquiterpene isocyanides, isothiocyanates, thiocyanates, and formamides from the Thai sponge Halichondria sp. Tetrahedron 2016, 72, 4222–4229. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Wittayalai, S.; Ruchirawat, S. Iodo-sesquiterpene hydroquinone and brominated indole alkaloids from the Thai sponge Smenospongia sp. Tetrahedron 2012, 68, 6881–6886. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Wittayalai, S.; Intachote, P.; Kanchanapoom, T.; Ruchirawat, S. Nitrogenous sesquiterpenes from the Thai marine sponge Halichondria sp. Tetrahedron 2011, 67, 5651–5655. [Google Scholar] [CrossRef]
- Bergquist, P.R.; Cambie, R.C.; Kernan, M.R. Scalarane sesterterpenes from Collospongia auris, a new thorectid sponge. Biochem. Syst. Ecol. 1990, 18, 349–357. [Google Scholar] [CrossRef]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Jefford, C.W.; Jaggi, D.; Bernardinelli, G.; Boukouvalas, J. The synthesis of (±)-cavernosine. Tetrahedron Lett. 1987, 28, 4041–4044. [Google Scholar] [CrossRef]
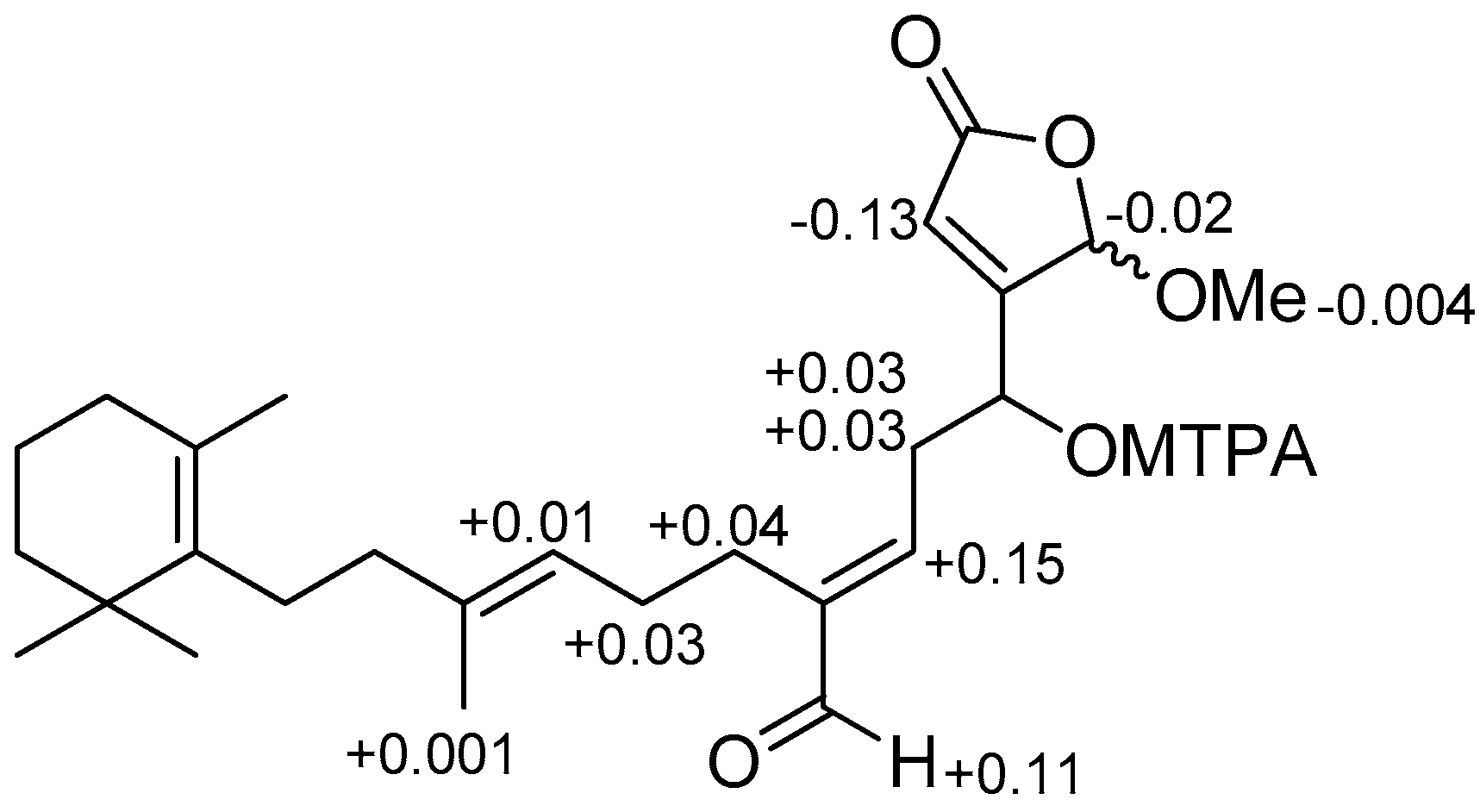
- Ohtani, I.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Absolute configurations of marine diterpenes possessing a xenicane skeleton. An application of an advanced Mosher’s method. Tetrahedron Lett. 1989, 30, 3147–3150. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semi-automated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Doyle, A.; Griffiths, J.B. Mammalian Cell Culture: Essential Techniques; John Wiley & Sons: Chichester, UK, 1997. [Google Scholar]

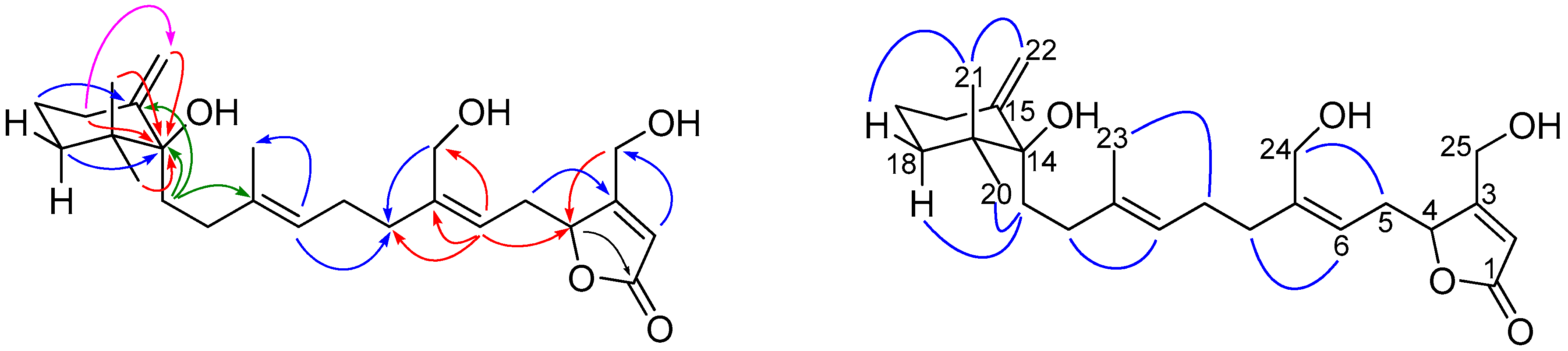
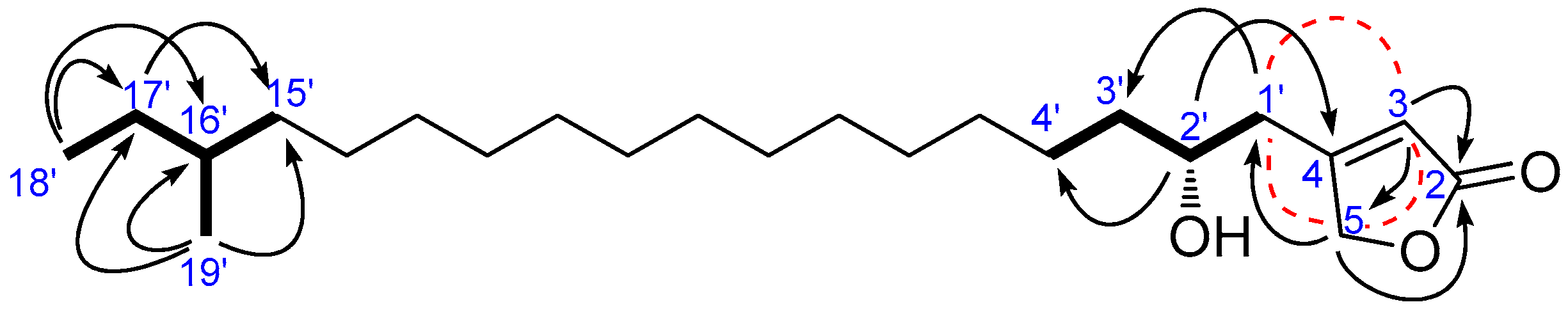
 and
and  ), HMBC (
), HMBC ( ), and NOESY (
), and NOESY ( ) correlations of 1.
) correlations of 1.

 ), and NOESY (
), and NOESY ( ) correlations of 4.
) correlations of 4.
 ), long rang COSY (
), long rang COSY ( ), and HMBC (
), and HMBC ( ) correlations of 5.
) correlations of 5.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| a δC (Type) | a δH, Mult. (J in Hz) | b δH, Mult. (J in Hz) | a δC (Type) | a δH, Mult. (J in Hz) | b δH, Mult. (J in Hz) | |
| 1 | 174.0 (C) | - | - | 173.3 (C) | - | - |
| 2 | 115.3 (CH) | 5.86, brd (1.5) | 5.66, q (1.6) | 116.0 (CH) | 5.93, brd (1.6) | 5.46, brs |
| 3 | 169.7 (C) | - | - | 168.1 (C) | - | - |
| 4 | 51.0 (CH) | 2.74, d (10.0) | 2.24, d (10.1) | 50.8 (CH) | 2.85, d (9.9) | 1.93, d (10.5) |
| 5 | 33.3 (CH) | 2.85, brt (9.0) | 2.60–2.66, m | 34.5 (CH) | 3.14, brt (8.8) | 2.40, brt (10.5) |
| 6 | 122.1 (CH) | 5.67, brd (0.9) | 5.48, brd (1.4) | 149.1 (CH) | 6.74, t (3.0) | 5.85, brt (3.1) |
| 7 | 140.9 (C) | - | - | 143.7 (C) | - | - |
| 8 | 24.0 (CH2) | 1.90–2.00, o 2.09, dt (16.4, 3.4) | 1.76, brt (13.0) 1.90–1.98, o | 19.4 (CH2) | 1.86–1.93, o 2.65, dt (16.8, 3.6) | 1.51–1.62, o 2.60, dt (13.3, 3.1) |
| 9 | 22.8 (CH2) | 1.65, qd (12.2, 4.2) 1.82–2.00, o | 1.42, qd (12.3, 4.1) 1.57–1.67, m | 22.3 (CH2) | 1.50–1.58, o 1.88–1.99, o | 0.88–0.96, m 1.27–1.37, 0 |
| 10 | 37.0 (CH) | 2.25, dt (12.2, 6.9) | 2.12, ddd (12.8, 7.9, 6.8) | 38.0 (CH) | 2.37, dt (11.7, 6.7) | 1.82–1.88, o |
| 11 | 43.0 (C) | - | - | 43.7 (C) | - | - |
| 12 | 36.6 (CH2) | 1.08–1.20, m 1.49–1.61, o | 1.28–1.36, m 1.58–1.66, o | 36.6 (CH2) | 1.15–1.18, o 1.55–1.58, o | 1.10–1.17, m 1.35–1.40, o |
| 13 | 22.4 (CH2) | 1.90–2.00, o | 1.94–2.06, o | 22.2 (CH2) | 1.88–1.99, o | 1.23–1.40, o 1.78–1.84, o |
| 14 | 136.3 (C) | - | - | 136.0 (C) | - | - |
| 15 | 127.5 (C) | - | - | 127.7 (C) | - | - |
| 16 | 32.7 (CH2) | 1.89–1.98, o | 1.92–2.06, o | 32.7 (CH2) | 1.85–1.99, o | 1.78–1.89, o |
| 17 | 19.4 (CH2) | 1.53–1.62, o | 1.60–1.68, o | 19.4 (CH2) | 1.52–1.62, o | 1.49–1.58, o |
| 18 | 39.7 (CH2) | 1.40–1.44, m | 1.49–1.52, m | 39.7 (CH2) | 1.40–1.44, m | 1.37–1.42, o |
| 19 | 35.1 (C) | - | - | 35.1 (C) | - | - |
| 20 | 28.71 (CH3) | 0.96 *, s | 1.14, s | 28.74 (CH3) | 0.96, s | 0.99, s |
| 21 | 28.72 (CH3) | 0.97 *, s | 1.11, s | 28.7 (CH3) | 0.98, s | 1.02, s |
| 22 | 19.8 (CH3) | 1.55, s | 1.66, s | 19.8 (CH3) | 1.56, s | 1.52, s |
| 23 | 21.5 (CH3) | 1.13, s | 0.88, s | 21.5 (CH3) | 1.16, s | 0.63, s |
| 24 | 66.8 (CH2) | 4.03, d (13.2) 4.06, d (13.2) | 3.88, s | 192.9 (CH) | 9.45, s | 9.37, s |
| 25 | 73.2 (CH2) | 4.62, dd (17.4, 1.6) 4.65, dd (17.4, 1.0) | 4.02, dd (17.1, 0.8) 4.16, dd (17.1, 1.7) | 72.9 (CH2) | 4.64, dd (17.3, 1.8) 4.68, dd (17.3, 1.1) | 3.85, dd (17.2) 4.00, dd (17.2) |
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δC (Type) | δH, Mult. (J in Hz) | δC (Type) | δH, Mult. (J in Hz) | |
| 1 | 169.2 (C) | - | 172.7 (C) | - |
| 2 | 119.3 (CH) | 6.09, brt (1.3) | 116.0 (CH) | 6.04, brd (1.4) |
| 3 | 166.3 (C) | - | 171.7 (C) | - |
| 4 | 66.9 (CH) | 4.73, brdd (6.7, 4.1) | 81.8 (CH) | 5.08, m |
| 5 | 34.7 (CH2) | 2.78, dt (15.7, 7.5) 2.92, ddd (15.7, 7.1, 4.1) | 30.2 (CH2) | 2.57, ddd (15.0, 7.3, 5.5) 2.78, dt (15.0, 7.0) |
| 6 | 147.0 (CH) | 6.54, t (7.1) | 119.9 (CH) | 5.19, t (7.5) |
| 7 | 146.1 (C) | - | 143.0 (C) | - |
| 8 | 24.6 (CH2) | 2.32, t (7.4) | 35.4 (CH2) | 2.10–2.22, m |
| 9 | 26.7 (CH2) | 2.09, q (7.4) | 26.3 (CH2) | 2.07–2.17, m |
| 10 | 122.4 (CH) | 5.13, td (7.3, 1.0) | 123.6 (CH) | 5.07, m |
| 11 | 137.5 (C) | - | 136.6 (C) | |
| 12 | 40.2 (CH2) | 1.95–2.07, m | 33.7 (CH2) | 1.68, td (13.5, 3.5) 1.86–1.96, m |
| 13 | 27.8 (CH2) | 1.97–2.07, m | 31.0 (CH2) | 1.49–1.60, m 1.88, td (11.7, 4.3) |
| 14 | 137.0 (C) | - | 80.4 (C) | - |
| 15 | 127.0 (C) | - | 150.5 (C) | - |
| 16 | 32.8 (CH2) | 1.90, t (6.2) | 34.0 (CH2) | 1.97, td (13.0, 5.7) 2.32, brd (13.0) |
| 17 | 19.5 (CH2) | 1.54–1.59, m | 22.7 (CH2) | 1.43–1.65, m |
| 18 | 39.9 (CH2) | 1.39–1.43, m | 38.0 (CH2) | 1.37, brd (13.7) 1.63, td (13.7, 4.5) |
| 19 | 35.0 (C) | - | 39.7 (C) | - |
| 20 | 28.6 (CH3) | 0.99, s | 24.1 (CH3) | 0.96, s |
| 21 | 28.6 (CH3) | 0.99, s | 22.1 (CH3) | 0.88, s |
| 22 | 19.8 (CH3) | 1.59, s | 108.3 (CH2) | 4.83, brs 4.91, brs |
| 23 | 16.0 (CH3) | 1.61, s | 16.3 (CH3) | 1.60, s |
| 24 | 194.2 (CH) | 9.43, s | 60.3 (CH2) | 4.10, d (12.1) 4.13, d (12.1) |
| 25 | 103.3 (CH) | 5.84, s | 58.6 (CH2) | 4.44, d (16.9) 4.52, d (16.9) |
| OMe-25 | 57.9 (CH3) | 3.65, s | ||
| Position | 5 | 6 | ||||
|---|---|---|---|---|---|---|
| δC (Type) | δH, Mult. (J in Hz) | δC (Type) | δH, Mult. (J in Hz) | |||
| 2 | 173.8 (C) | - | 173.9 (C) | - | ||
| 3 | 117.3 (CH) | 5.93, s | 117.2 (CH) | 5.93, s | ||
| 4 | 167.3 (C) | - | 167.4 (C) | - | ||
| 5 | 73.8 (CH2) | 4.85, s | 73.8 (CH2) | 4.85, s | ||
| 1′ | 36.3 (CH2) | 2.52, dd (14.8, 8.2) 2.64, dd (14.8, 3.2) | 36.5 (CH2) | 2.52, dd (15.2, 8.2) 2.64, dd (15.2, 3.4) | ||
| 2′ | 70.2 (CH) | 3.84–3.90, m | 70.2 (CH) | 3.84–3.89, m | ||
| 3′ | 37.7 (CH2) | 1.49–1.54, m | 37.7 (CH2) | 1.49–1.55, m | ||
| 4′ | 25.5 (CH2) | 1.28–1.38, m 1.39–1.46, m | 25.5 (CH2) | 1.30–1.47, m | ||
 | (CH2)-5′– (CH2)-14′ 30.0, 29.68, 29.64, 29.63, 29.61, 29.57, 29.49, 29.49, 29.48, 27.1 (C-14′) | 1.22–1.50, m | | (CH2)-5′– (CH2)-14′ 29.65, 29.65, 29.65, 29.63, 29.61, 29.61, 29.58, 29.50, 29.50, 29.42 | 1.22–1.38, m | |
| 15′ | 36.6 (CH2) | 1.07–1.17, m 1.23–1.37, m | 29.3 (CH2) | 1.22–1.38, m | ||
| 16′ | 34.4 (CH) | 1.23–1.37, m | 31.9 (CH2) | 1.22–1.31, m | ||
| 17′ | 29.42 (CH2) | 1.22–1.50, m | 22.6 (CH2) | 1.22–1.38, m | ||
| 18′ | 11.3 (CH3) | 0.85, t (7.2) | 14.0 (CH3) | 0.88, t (7.0) | ||
| 19′ | 19.2 (CH3) | 0.84, d (6.0) | - | - | ||
| Compound | MOLT-3 | HepG2 | HeLa | HuCCA-1 | A549 | H69AR | KB | T47D | MDA-MB-231 | MRC-5 |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 15.84 | 27.91 | 20.02 | 24.72 | 41.12 | - a | - a | - a | 40.09 | - a |
| 5 | 31.75 | 107.46 | 28.79 | 65.57 | 57.24 | - a | - a | - a | 89.26 | - a |
| 7 | 40.11 | I | 87.95 | I | I | - a | - a | - a | I | - a |
| 8 | 1.77 | 53.49 | 8.42 | 11.02 | 16.77 | - a | - a | - a | 39.53 | 13.00 |
| 9 | 1.30 | 27.91 | 1.51 | 8.60 | 12.00 | - a | - a | - a | 18.60 | 10.84 |
| 10 | 35.45 | I | 78.71 | I | I | - a | - a | - a | I | - a |
| 11 | 17.38 | I | 87.04 | 84.52 | I | - a | - a | - a | I | - a |
| 12 | 37.81 | I | 79.20 | 119.35 | I | - a | - a | - a | I | - a |
| 13 | 34.10 | 88.73 | 59.30 | 94.65 | 114.30 | - a | - a | - a | 92.04 | - a |
| 14 | 6.68 | 101.75 | 21.80 | 47.38 | 58.13 | - a | - a | - a | 84.13 | 17.48 |
| 15 | - a | - a | 63.08 | 49.07 | - a | 51.40 | 58.41 | 28.04 | 14.02 | - a |
| 16 | 124.32 | I | 77.54 | I | I | - a | - a | - a | I | - a |
| 17 | - a | - a | 63.08 | 60.75 | - a | 107.48 | 63.08 | 42.06 | 60.75 | - a |
| 18 | - a | - a | 23.32 | 41.45 | - a | 56.99 | 6.99 | 5.18 | 5.96 | - a |
| 19 | 13.83 | 75.45 | 65.32 | 60.81 | 60.81 | - a | - a | - a | - a | - a |
| 20 | 7.64 | 27.36 | - a | 87.06 | 73.38 | - a | - a | - a | - a | 70.97 |
| 23 | 77.98 | I | I | I | I | - a | - a | - a | - a | - a |
| Doxorubicin | - a | 0.36 ± 0.05 | 0.19 ± 0.00 | 0.40 ± 0.03 | 0.35 ± 0.02 | 1.64 ± 0.15 | 1.64 ± 0.17 | 3.16 ± 0.10 | ||
| Etoposide | 0.04 ± 0.01 | - a | - a | - a | - a | - a | - a | - a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaweetripob, W.; Mahidol, C.; Tuntiwachwuttikul, P.; Ruchirawat, S.; Prawat, H. Cytotoxic Sesterterpenes from Thai Marine Sponge Hyrtios erectus. Mar. Drugs 2018, 16, 474. https://doi.org/10.3390/md16120474
Kaweetripob W, Mahidol C, Tuntiwachwuttikul P, Ruchirawat S, Prawat H. Cytotoxic Sesterterpenes from Thai Marine Sponge Hyrtios erectus. Marine Drugs. 2018; 16(12):474. https://doi.org/10.3390/md16120474
Chicago/Turabian StyleKaweetripob, Wirongrong, Chulabhorn Mahidol, Pittaya Tuntiwachwuttikul, Somsak Ruchirawat, and Hunsa Prawat. 2018. "Cytotoxic Sesterterpenes from Thai Marine Sponge Hyrtios erectus" Marine Drugs 16, no. 12: 474. https://doi.org/10.3390/md16120474
APA StyleKaweetripob, W., Mahidol, C., Tuntiwachwuttikul, P., Ruchirawat, S., & Prawat, H. (2018). Cytotoxic Sesterterpenes from Thai Marine Sponge Hyrtios erectus. Marine Drugs, 16(12), 474. https://doi.org/10.3390/md16120474