Abstract
Five new compounds named asperpenes A-C (1–3), 12,13-dedihydroversiol (4), and methyl 6-oxo-3,6-dihydro-2H-pyran-4-carboxylate (5), along with 10 known compounds (6–15), were isolated from the fermentation broth of Aspergillus sp. SCS-KFD66 associated with a bivalve mollusk, Sanguinolaria chinensis, collected from Haikou Bay, China. The structures of the compounds, including the absolute configurations of their stereogenic carbons, were unambiguously determined by spectroscopic data, single-crystal X-ray diffraction analysis, and electronic circular dichroism (ECD) spectral analysis, along with quantum ECD calculations. The growth inhibitory activity of the compounds against four pathogenic bacterial (Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 6538, Listeria monocytogenes ATCC 1911, and Bacillus subtilis ATCC 6633), their enzyme inhibitory activities against acetylcholinesterase and α-glucosidase, and their DPPH radical scavenging activity were evaluated.
1. Introduction
In the past few decades, natural products have occupied a very important position in modern drug research and development, providing more efficient means for human health care, nutrition, medical care, and other aspects [1]. From 1940 to 2014, 175 new anticancer drugs were approved worldwide, 75% of which came from natural products or their derivatives [2]. Therefore, the study of natural products is of great significance for drug development. Because of the special environmental conditions, marine fungi have been proven to be a rich source of various types of compounds with complex structures and remarkable activities, thereby attracting the attention of for which many natural product chemists turned their attention to them [3,4].
Our previous research on secondary metabolites from marine animal-derived fungi have led to the isolation and identification of a series of structurally new and biologically active natural products, including new quorum-sensing inhibitors from Penicillium sp. SCS-KFD08, chlorinated meroterpenoids with anti-H1N1 activity from Penicillium sp. SCS-KFD09, and helvolic acid derivatives with potent antibacterial activity from Aspergillus fumigatus HNMF0047 [5,6,7,8,9]. In the course of our ongoing research, Aspergillus sp. SCS-KFD66 was isolated and identified from a bivalve mollusk, Sanguinolaria chinensis, from Haikou Bay, Hainan province, in China. The chemical investigation on the EtOAc extract of the fungal fermentation broth led to the isolation and purification of five new compounds, named asperpenes A-C (1-3), 12,13-dedihydroversiol (4), and methyl 6-oxo-3, 6-dihydro-2H-pyran-4-carboxylate (5), as well as 10 known compounds, i.e., versiol (6) [10], (E)-4-oxonon-2-enoic acid (7) [11], ergosta-5, 7,22-triene-3β-ol (8) [12], β-sitosterol (9) [13], (22E)-5α,8α-epidioxyergosta-6,22-dien-3β-ol (10) [14], 15α-hydroxy-(22E,24R)-ergosta-3,5,8(14),22-tetraen-7-one (11) [15], volemolide (12) [16], oxaline (13) [17], fumitremorgin B (14) [18], and helvolic acid (15) [19] (Figure 1). Herein, the structure and bioactivities of these compounds are reported.
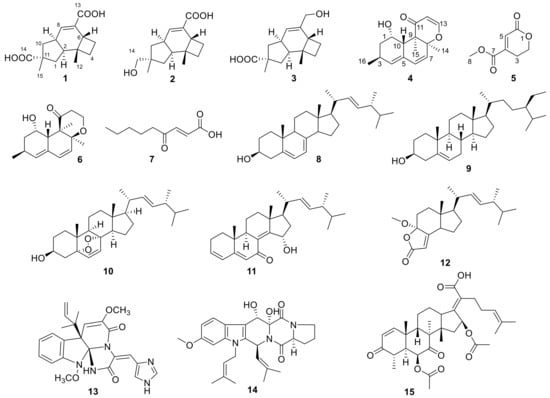
Figure 1.
Structures of compounds 1–15.
2. Results and Discussion
Compound 1 was obtained as a colorless crystal, and its molecular formula C15H20O4 was established from the HRESIMS m/z 263.1283 [M − H]−. The IR absorptions at 3422, 1695, and 1622 cm−1 revealed the presence of a hydroxyl and a conjugated carboxylic group, respectively, which was further confirmed by a characteristic UV λmax at 221 nm. The 1H and 13C NMR spectra (Supplementary Materials, Figures S1 and S2) in combination with the HSQC spectra (Supplementary Materials, Figure S4) revealed the presence of two methyls, four sp3 methylenes, three sp3 methines, two sp3 non-protonated carbons, one tri-substituted double bond, and two carboxylic groups. These data were closely related to those of russujaponol H [20], suggesting that 1 was also an illudoid sesquiterpene. The COSY correlations (Supplementary Materials, Figure S5) revealed the connectivities from in CH-1–CH-2–CH-9–CH2-10, CH-8–CH-9, and CH2-4–CH2-5–CH-6. These structure fragments were assembled into a whole structure on the basis of the HMBC correlations (Supplementary Materials, Figure S6) from H3-12 (δH 1.25) to C-2 (δC 47.0), C-3 (δC 49.2), C-4 (δC 26.6), and C-6 (δC 37.2), from H3-15 (δH 1.18) to C-1 (δC 39.8), C-10 (δC 44.5), C-11 (δC 39.0), and C-14 (δC 182.4), from H-6 (δH 2.54) to C-5 (δC 30.7) and C-7 (δC 133.9), and from H-8 (δH 6.82) to C-13 (δC 170.3) (Figure 2). ROESY correlations from H-6/H-1β (δH 0.96)/H3-15 and H-8 (δH 6.82)/H-10β (δH 1.59) (Figure 3) suggested that H-6 and H3-15 were on the same face of the ring system, and H-2 (δH 1.98) and H-9 (δH 2.92) were on the opposite face of the molecule (Figure 3). To support the above deduction and determine the absolute configuration of 1, a single-crystal X-ray diffraction pattern was obtained using the anomalous scattering of Cu Kα radiation (Figure 4), allowing an explicit assignment of the absolute structure as 2S, 3R, 6R, 9S, and 11R. This was further corroborated by electronic circular dichroism (ECD) quantum chemical calculations in Gaussian 03 [7]. The experimental and calculated ECD spectra for (2S, 3R, 6R, 9S, 11R)-1 showed good agreement (Figure 5). Thus, 1 was elucidated and named asperpene A.
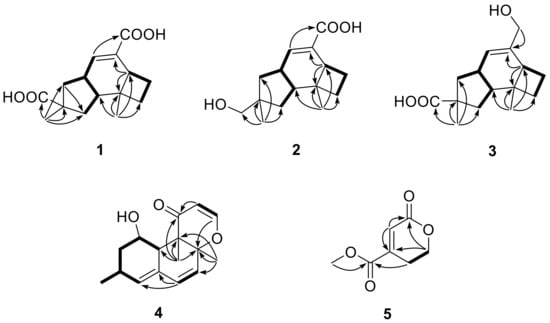
Figure 2.
Key COSY (▬) and HMBC (→) correlations of 1–5.
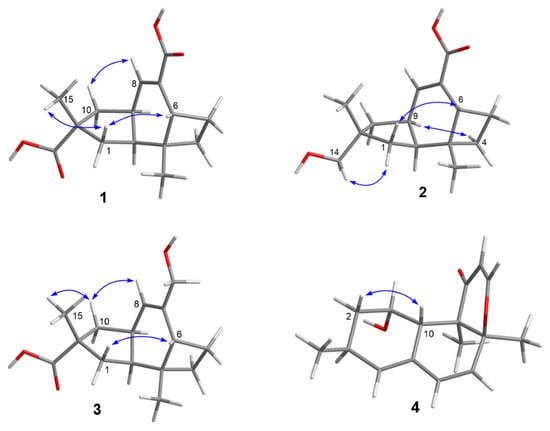
Figure 3.
Key ROESY correlations of 1–4.
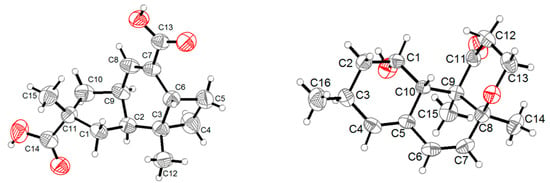
Figure 4.
ORTEP diagrams of 1 and 6.
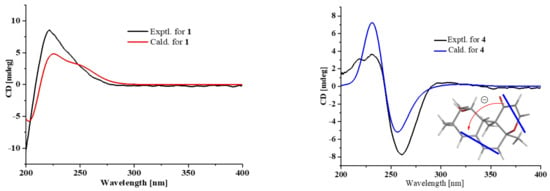
Figure 5.
Comparison of measured and calculated ECD spectra for 1 and 4 and ECD exciton chirality model for 4.
Compound 2 was obtained as a colorless powder, whose molecular formula was established as C15H22O3 by HRESIMS m/z 273.1455 [M + Na]+. Comparison of the 1H and 13C NMR data (Supplementary Materials, Figures S9 and S10) of 2 with those of 1 revealed the presence of a hydroxymethyl group (δC/H 72.1/3.3, C-14) and a carboxylic group in 2 instead of two carboxyl groups as in 1. The above data, together with HMBC correlations from H3-15 (δH 0.96) to the hydroxymethyl carbon (δC 72.1), indicated that the carboxylic group in 1 was replaced by a hydroxymethyl in 2. In the ROESY spectrum (Figure 3), correlations from H2-14 (δH 3.31, 3.33)/H-1α (δH 1.56), H-6 (δH 1.91)/H-1β (δH 0.85), and H-9 (δH 2.88)/H-4α (δH 1.86) suggested that 2 shared the same configuration at the stereogenic C-2, C-3, C-6, C-9, and C-11. Thus, the structure of 2 was established and named as asperpene B.
Compound 3 possessed the same molecular formula as 2, as determined by HRESIMS data. The 1H and 13C NMR data of 3 (Supplementary Materials, Figures S17 and S18) were also quite similar to those of 2. However, in the HMBC spectrum of 3 (Figure 2), correlations from H3-15 (δH 1.26) to the carbonyl (δC 201.5) and from H2-13 (δH 4.01, 4.03) to C-7 (δC 139.5) and C-8 (δC 125.5) suggested the positions of the carboxylic acid and the hydroxymethyl groups at C-11 and C-7, respectively, which is resulted different from those of 2. ROESY correlations (Figure 3) of H-10β (δH 1.57)/H-8 (δH 5.49) and H3-15/H-1β (δH 1.09)/H-6 (δH 1.99) suggested that 3 had the same configurations of its stereogenic carbons as 2.
Compound 4 was obtained as a yellow oil, and its molecular formula was determined as C16H20O3 on the basis of the HRESIMS data, implying seven degrees of unsaturation. The 1H and 13C NMR data (Supplementary Materials, Figures S25 and S26) of 4 indicated the presence of three methyls, one methylene, eight methines (including five olefinic and one oxygenated), and four non-protonated carbons (including one ketone carbonyl, one olefinic, and one oxygenated sp3). These data were quite similar to those of versiol (6) [10], suggesting that they were structurally related, and the only difference was that one disubstituted double bond in 4 was saturated to two vicinal methylenes in 6, as supported by HMBC correlations from H-13 (δH 7.22) to C-8 (δC 85.9) and from H-12 (δH 5.46) to C-11 (δC 198.8). ROESY correlation between H-2β (δH 1.19) and H-10 (δH 2.69) suggested their cofacial relationship, while the absence of ROESY correlations from H3-14 (δH 1.46) and H3-15 (δH 1.17) to H-10 suggested that H3-14 and H3-15 were on the opposite face with respect to H-10. Considering that versiol (6) and 4 were biosynthetically related and a relatively large amount of versiol (6) was isolated, we crystalized versiol (6) successfully and subjected it to a single-crystal X-ray diffraction experiment (Figure 4), finally allowing an explicit assignment of the absolute structure of versiol (6) as 1S, 3S, 8S, 9R, and 10S. The absolute configurations of the stereogenic carbons of 4 were also suggested to be 1S, 3S, 8S, 9R, and 10S on the basis of a biosynthetic consideration. The experimental ECD spectrum (Figure 5) of 4 showed characteristic exciton CDs absorption bands at 261 (-0.31) and 230 (+0.14) nm due to the a negative couplet of the α,β-unsaturated carbonyl and the conjugated double-bond moieties, which further confirmed the absolute configuration assignment. Moreover, the experimental and calculated ECD spectra for 4 also matched well (Figure 5).
Compound 5 was isolated as a colorless oil, whose molecular formula was established as C7H8O4 by HRESIMS m/z 179.0316 [M + Na]+. The 1H, 13C, and HSQC NMR spectra (Supplementary Materials, Figures S33, S34, and S36) of 5 showed signals for two ester carbonyls (δC 165.0, 163.6), one tri-substituted double bond (δC/H 126.1/6.77, 145.3), two sp3 methylenes, one of which is oxgenated (δC/H 66.7/4.46), and one methoxyl group (δC/H 53.1/3.87). COSY correlations (Supplementary Materials, Figure S37) of H2-2 (δH 4.46)/H2-3 (δH 2.71) and HMBC correlations (Supplementary Materials, Figure S38) from H2-2 (δH 4.46) and H-5 (δH 6.77) to C-6 (δC 163.6) and C-4 (δC 145.3) and from H2-3 and H3-8 (δH 3.87) to C-7 (δC 165.0) led to the determination of the full structure of 5, as shown in Figure 1.
Compounds 1–15 were tested for their antibacterial activity against Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 6538, Listeria monocytogenes ATCC 1911, and Bacillus subtilis ATCC 6633 by the 96-well microtiter plates method [21]. The results (Table 1) revealed that 7, 8, 12, and 13 showed inhibitory activities against B. subtilis ATCC 6633, with MIC values of 4, 128, 128, and 128 μg/mL, respectively, whereas 7, 8, 14, and 15 showed inhibitory activity against S. aureus ATCC 6538, with MIC values of 16, 128, 128 and 2 μg/mL, respectively; 15 also had inhibitory activity against L. monocytogenes ATCC 1911, with MIC value of 128 μg/mL. None of these compounds showed inhibitory activity against E. coli ATCC 25922.

Table 1.
Antibacterial activities of compounds 7, 8, and 12–15.
Additionally, the DPPH radical scavenging activity and the acetylcholinesterase and α-glucosidase inhibitory activities of all the isolated compounds were evaluated by the DPPH method [22], Ellman colorimetric method [23], and PNPG method [24], respectively. None of these compounds showed inhibitory activities against α-glucosidase and acetylcholinesterase. However, 1, 3, 4, 8, 11, and 15 showed weak DPPH radical scavenging activity, with IC50 values of 1.8, 0.6, 1.1, 0.6, 1.2, and 0.7 mM (ascorbic acid as positive control, IC50 0.04 mM).
3. Experimental Section
3.1. General Experimental Procedure
Optical rotations were measured with a JASCO P-1020 digital polarimeter. The IR spectra were obtained on with a Nicolet Nexus 470 spectrophotometer as KBr discs. The UV spectra were obtained from with a Beckman DU 640 spectrophotometer. ECD data were measured collected on using a JASCO J-715 spectropolarimeter. The NMR spectra were recorded on a Bruker AV-500 spectrometer with TMS as an internal standard. ESIMS, HRESIMS, and HREIMS data were acquired on a Micromass Autospec-Ultima-TOF, API QSTAR Pulsar 1, or Waters Autospec Premier spectrometer. The sea salt was produced by evaporation of seawater collected in Laizhou Bay, Weifang, China (Weifang HaiHua Yu Feng Chemical Factory). Semi-preparative HPLC separation was used octadecyl silane (ODS) columns (YMC-pack ODS-A, 10 × 250 mm, 5 μm, 4 mL/min) and Ph column (YMC-pack Ph, 10 × 250 mm, 5 μm, 4 mL/min) for separation. Thin-layer chromatography (TLC) and column chromatography (CC) were carried out on precoated silica gel GF254 (10–40 μm, Qingdao Marine Chemical Inc., Qingdao, China) and silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), respectively.
3.2. Fungal Material and Fermentation
The strain SCS-KFD66 was isolated from a bivalve mollusk, Sanguinolaria chinensis, collected from Haikou Bay, Hainan province, in China. After grinding, the sample (1 g) was diluted to 10−2 g/mL with sterile H2O, 100 μL of which was spread on a PDA (200 g potato, 20 g glucose, 20 g agar per liter of sea water collected in Haikou Bay, China) plate containing chloramphenicol (100 μg/mL) as a bacterial inhibitor. Fungal identification was carried out by its examining the morphological characteristics and 18S rRNA gene sequences (GenBank accession No. MK085984, Supporting Information) with of the single coloniesy. A reference culture of Aspergillus sp. SCS-KFD66 is deposited in our laboratory and which maintained at −80 °C. The isolate was cultured on slants of PDA medium at 28 °C for 5 days and then transferred to two hundred 1 L Erlenmeyer flasks containing solid rice medium (80 g rice, 3.96 g sea salt, 120 mL tap water, pH 7.0), used for fermentation. The flasks were incubated under static conditions at room temperature for 30 days.
3.3. Extraction and Isolation
The fermented cultures were extracted with three-fold volumes (3 × 300 mL) of EtOAc, then filtered through a cheesecloth to separate the rice from the mixture. After repeating the procedure three times, the EtOAc extracts were evaporated under a reduced pressure to produce 409.6 g of a crude extract. The extract was fractionated by a silica gel VLC column using different solvents of increasing polarity, from petroleum ether to EtOAc, to yield seven fractions (Frs. 1−7). Fr. 3 (5.3 g) was further purified by HPLC using an octadecyl silane (ODS) silica gel column and eluted with in a MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0) gradient to afford 8 (3.5 mg), 9 (14.0 mg), and three subfractions (Sfrs. 3-1–Fr. 3-3). Sfr. 3-2 (289.7 mg) was subjected to VLC on silica gel and eluted with an EtOAc/petroleum ether stepwise gradient (from 1:10 to 2:1) to afford 4 (3.6 mg). Fr. 4 (8.0 g) was separated into seven subfractions (Sfrs. 4-1–Fr–4-7) by HPLC using an ODS silica gel column with a gradient elution of MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0). Sfr. 4-1 (16.5 mg) was purified by a semipreparative HPLC (YMC-pack ODS-A, 5 μm; 10 × 250 mm; 35% MeCN/H2O; containing 0.1% TFA; 4 mL/min) to afford 7 (tR 18.4 min; 1.7 mg). Fr. 5 (3.7 g) was chromatographed on an ODS silica gel column with a gradient elution of MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0) to yield 14 (3.0 mg), 15 (4.1 mg), 10 (5.2 mg), and four subfractions (Sfrs. 5-1–Fr. 5-4). Sfr. 5-4 (13.3 mg) was purified by a semipreparative HPLC (YMC-pack ODS-A, 5 μm; 10 × 250 mm; 40% MeCN/H2O; containing 0.1% TFA; 4 mL/min) to afford 6 (tR 9.8 min; 6.0 mg). Fr. 6 (4.0 g) was fractionated on an ODS silica gel column with a gradient elution of MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0) to yield six subfractions (Sfrs. 6-1–Fr. 6-6). Sfr. 6-4 (69.9 mg) was subjected to semipreparative HPLC (YMC-pack Ph, 5 μm; 10 × 250 mm; 30% MeCN/H2O; containing 0.1% TFA; 4 mL/min) to afford 1 (tR 15.6 min; 24.5 mg), 2 (tR 18.4 min; 2.5 mg), and 3 (tR 21.5 min; 3.7 mg). Purification of Fr. 7 (28.3 g) by a silica gel VLC column with a stepwise gradient with of MeOH/CHCl3 (from 10:90 to 100:0) gave 13 (78.5 mg) and eight fractions (Sfrs. 7-1–Fr. 7-8). Sfr. 7-1 (119.2 mg) was applied to ODS silica gel with a gradient elution of MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0) to yield 12 (2.0 mg). Sfr. 7-2 (199.4 mg) was purified by ODS silica gel column with a gradient elution with of MeOH/H2O (1:5, 2:3, 3:2, 4:1, 1:0) to give 11 (3.0 mg). Sfr. 7-4 (184.9 mg) was purified by Sephadex LH-20 chromatography and eluted with MeOH to give three subfractions (Sfrs. 7-4-1–Fr. 7-4-3). Sfr. 7-4-1 (149.6 mg) was finally purified by semipreparative HPLC (YMC-pack ODS-A, 5 μm; 10 × 250 mm; 40% MeOH/H2O; 4 mL/min) to obtain 5 (tR 4.4 min; 9.3 mg).
Asperpene A (1): Colorless crystal; mp 194–195 °C; +8 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 203 (3.54) nm; ECD (MeOH) λmax 221 (+0.34) nm; IR (KBr) νmax (cm−1): 3422, 2930, 2851, 1695, 1626, 1453, 1390, 1254, 1121. 1H NMR data, Table 2; 13C NMR data, Table 3; HRESIMS m/z 263.1283 [M − H]− (calcd for C15H19O4, 263.1289).
Table 2.
1H NMR data (500 MHz, δ in ppm, J in Hz) of 1–5.
Table 3.
13C NMR data (125 MHz, δ in ppm) of 1–5.
Asperpene B (2): White powders; −6 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 213 (3.14) nm, 215 (3.14) nm; ECD (MeOH) λmax 295 (-0.06), 255 (+0.02), 229 (-0.08) nm; IR (KBr) νmax (cm−1): 3446, 2932, 2867, 1692, 1638, 1455, 1393, 1255, 1049. 1H NMR data, Table 2; 13C NMR data, Table 3; HRESIMS m/z 273.1455 [M + Na]+ (calcd for C15H23O3Na, 273.1461).
Asperpene C (3): White powders; +5 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 202 (3.34) nm; IR (KBr) νmax (cm−1): 3415, 2959, 1708, 1456, 1184, 1122. 1H NMR data, Table 2; 13C NMR data, Table 3; HRESIMS m/z 273.1457 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
12,13-Dedihydroversiol (4): Yellow oil; −8 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 260 (3.46) nm, 234 (3.58) nm, 219 (3.66) nm; ECD (MeOH) λmax 310 (+0.02), 261 (-0.31), 230 (+0.14) nm; IR (KBr) νmax (cm−1): 3445, 2930, 1723, 1655, 1605, 1454, 1385, 1266, 1108. 1H NMR data, Table 2; 13C NMR data, Table 3; HRESIMS m/z 283.1297 [M + Na]+ (calcd for C16H20O3Na, 283.1305).
Methyl 6-oxo-3, 6-dihydro-2H-pyran-4-carboxylate (5): Colorless oil; UV (MeOH) λmax (log ε): 218 (3.45); IR (KBr) νmax (cm−1): 2927, 1726, 1641, 1441, 1260, 1219, 1084. 1H NMR data, Table 2; 13C NMR data, Table 3; HRESIMS m/z 179.0316 [M + Na]+ (calcd for C7H8O4Na, 179.0315).
X-ray Crystal Data for 1 and 6: Colorless crystals of 1 and 6 were obtained in the mixed solvent of MeOH and H2O. Crystal data of 1 and 6 were obtained on a Bruker D8 QUEST diffractometer (Bruker) with graphite monochromated Cu Kα radiation (λ = 1.54178 Å). Crystallographic data for 1 and 6 have been deposited with in the Cambridge Crystallographic Data Center as supplementary publication numbers CCDC 1875828 and 1875827. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data for 1. Orthorhombic, C15H20O4; space group P 21 21 21 with a = 7.2684(6) Å, b = 9.6858(7) Å, c = 19.5962(15) Å, V = 1379.58(18) Å3, Z = 4, Dcalcd = 1.273 g/cm3, μ = 0.747 mm−1, and F(000) = 568. T = 296(2) K. R1 = 0.0583 (I > 2σ(I)), wR2 = 0.1409 (all data), S = 0.978. Absolute structure parameter: 0.0 (4). The structures were solved using ShelXS. The structural solutions were found by direct methods and refined using the ShelXL package by least-squares minimization. The final structures were examined using the Addsym subroutine of PLATON to assure that no additional symmetry could be applied to the models. All non-hydrogen atoms were refined with anisotropic thermal factors.
Crystal data for 6. Orthorhombic, C16H22O3; space group P 21 21 21 with a = 6.0879(2) Å, b = 9.1456(3) Å, c = 25.1933(9) Å, V = 1402.70(8) Å3, Z = 4, Dcalcd = 1.237 Mg/m3, μ = 0.674 mm−1, and F(000) = 564. T = 296(2) K. R1 = 0.0339 (I > 2σ(I)), wR2 = 0.0793 (all data), S = 1.060. Absolute structure parameter: 0.08(12). The structures were solved using ShelXS. The structural solutions were found by direct methods and refined using the ShelXL package by least-squares minimization. The final structures were examined using the Addsym subroutine of PLATON to assure that no additional symmetry could be applied to the models. All non-hydrogen atoms were refined with anisotropic thermal factors.
4. Conclusions
In conclusion, five new compounds (1–5) and 10 known compounds (6–15) were isolated from the fermentation broth of Aspergillus sp. SCS-KFD66 which was isolated from a bivalve mollusk, S.anguinolaria chinensis, collected from Haikou Bay, China. The structures of the isolated compounds were unambiguously determined by spectroscopic data, single-crystal X-ray diffraction analysis, and comparison of the calculated and experimental ECD spectra. Compounds 7, 8, 12, and 13 showed antibacterial activity against Bacillus subtilis, with MIC values of 4, 128, 128, and 128 μg/mL. Compounds 7, 8, 14, and 15 exhibited antibacterial activity against S.taphylococcus aureus, with MIC values of 16, 128, 128, and 2 μg/mL, while 15 also showed inhibitory activity against L.isteria monocytogenes, with MIC value of 128 μg/mL. Compounds 1, 3, 4, 8, 11, and 15 showed a weak DPPH radical scavenging activity, with IC50 values of 1.8, 0.6, 1.1, 0.6, 1.2, and 0.7 mM (ascorbic acid as positive control, IC50 0.04 mM).
Supplementary Materials
The following are available online in http://www.mdpi.com/1660-3397/16/12/468/s1, Figures S1–S39: HRESIMS, IR, and 2D NMR spectra of the new compounds 1–5, the 18S rRNA gene sequence of Aspergillus sp. SCS-KFD66, and the quantum calculation details are supplied.
Author Contributions
C.A. contributed to fungal isolation and fermentation and compounds purification. F.K. was responsible for the structural elucidation, chemical computation, and preparation of the paper. Q.M. and L.Z. contributed to the bioassays. Q.X. identified the fungal strain. J.Y. collected the NMR data. H.D. revised the paper. Z.Y. and Y.Z. designed the work and revised the paper.
Funding
This work was supported by the Natural Science Foundation of Hainan Province (417256), Natural Science Foundation of China (41606088, 81741157), Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, Hainan Normal University (HNSD201705), Financial Fund of the Ministry of Agriculture and Rural Affairs, P. R. of China (NFZX2018), and Central Public-Interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (17CXTD-15, 1630052016008).
Acknowledgments
We wish to thank Junfeng Wang (CAS Key Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China) for collection of ECD spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Hamed, I.; Ozogul, F.; Ozogul, Y.; Regenstein, J.M. Marine bioactive compounds and their health benefits: A review. Compr. Rev. Food Sci. Food Saf. 2015, 14, 446–465. [Google Scholar] [CrossRef]
- Ren, J.W.; Niu, S.B.; Li, L.; Geng, Z.F.; Liu, X.Z.; Che, Y.X. Identification of oxaphenalenone ketals from the ascomycete fungus Neonectria sp. J. Nat. Prod. 2015, 78, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.D.; Ma, Q.Y.; Huang, S.Z.; Wang, P.; Wang, J.F.; Zhou, L.M.; Yuan, J.Z.; Dai, H.F.; Zhao, Y.X. Chrodrimanins K-N and related meroterpenoids from the fungus Penicillium sp. SCS-KFD09 isolated from a marine worm, Sipunculus nudus. J. Nat. Prod. 2017, 80, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.D.; Zhang, R.S.; Ma, Q.Y.; Xie, Q.Y.; Wang, P.; Chen, P.W.; Zhou, L.M.; Dai, H.F.; Luo, D.Q.; Zhao, Y.X. Chrodrimanins O–S from the fungus Penicillium sp. SCS-KFD09 isolated from a marine worm, Sipunculusnudus. Fitoterapia 2017, 122, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.D.; Huang, X.L.; Ma, Q.Y.; Xie, Q.Y.; Wang, P.; Chen, P.W.; Zhou, L.M.; Yuan, J.Z.; Dai, H.F.; Luo, D.Q.; et al. Helvolic acid derivatives with antibacterial activities against Streptococcus agalactiae from the marine-derived fungus Aspergillus fumigatus HNMF0047. J. Nat. Prod. 2018, 81, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.D.; Zhou, L.M.; Ma, Q.Y.; Huang, S.Z.; Wang, P.; Dai, H.F.; Zhao, Y.X. Metabolites with Gram-negative bacteria quorum sensing inhibitory activity from the marine animal endogenic fungus Penicillium sp. SCS-KFD08. Arch. Pharm. Res. 2017, 40, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.M.; Wang, P.; Liao, G.; Zeng, Y.B.; Cai, C.H.; Kong, F.D.; Guo, Z.K.; Proksch, P.; Dai, H.F.; Mei, W.L. New eudesmane-type sesquiterpenoids from the mangrove-derived endophytic fungus Penicillium sp. J-54. Mar. Drugs. 2018, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Asahara, M.; Ichinoe, M.; Nakajima, H. Fungal melanin inhibitor and related compounds from Penicillium decumbens. Phytochemistry 2002, 60, 703–708. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G. Synthesis of (E)-4-oxonon-2-enoic acid, a natural antibiotic produced by streptomyces olivaceus. J. Nat. Prod. 1998, 61, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Nagia, M.M.; El-Metwally, M.M.; Shaaban, M.; El-Zalabani, S.M.; Hanna, A.G. Four butyrolactones and diverse bioactive secondary metabolites from terrestrial Aspergillus flavipes MM2: Isolation and structure determination. Org. Med. Chem. Lett. 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zhang, Y.; Wang, X.; Wei, J.; Kang, W. Antithrombotic effect and mechanism of Rubus spp. Blackberry. Food. Funct. 2017, 8, 2000–2012. [Google Scholar] [CrossRef] [PubMed]
- Hybelbauerova, S.; Sejbal, J.; Dracinsky, M.; Hahnova, A.; Koutek, B. Chemical constituents of Stereum subtomentosum and two other birch-associated basidiomycetes: An interspecies comparative study. Chem. Biodivers. 2008, 5, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.L.; Liu, T.; Tian, L.; Pei, Y.H.; Hua, H.M. A new steroid with long cross-conjugation structure from the marine-derived fungus Aspergillus aculeatus. Acta. Pharmacol. Sin. 2014, 49, 68–71. [Google Scholar]
- Kobata, K.; Wada, T.; Hayashi, Y.; Shibata, H. Volemolide, a novel norsterol from the fungus Lactarius volemus. Biosci. Biotech. Bioch. 1994, 58, 1542–1544. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.F.; Kim, D.S.; Choi, H.D.; Son, B.W. Indolyl alkaloid derivatives, Nb-Acetyltryptamine and oxaline from a marine-derived fungus. Arch. Pharm. Res. 2003, 26, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.L.; Ma, Y.M. Isolation and Anti-phytopathogenic Activity of Secondary Metabolites from Alternaria sp. FL25, an Endophytic Fungus in Ficus carica. Chin. J. Appl. Environ. Biol. 2010, 16, 76–78. [Google Scholar] [CrossRef]
- Sawadsitang, S.; Mongkolthanaruk, W.; Suwannasai, N.; Sodngam, S. Antimalarial and cytotoxic constituents of Xylaria cf. cubensis PK108. Nat. Prod. Res. 2015, 29, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Matsumoto, Y.; Hama, H.; Tanaka, M.; Zhai, H.F.; Fukuyama, Y.; Arihara, S.; Hashimoto, T. Russujaponols G-L, illudoid sesquiterpenes, and their neurite outgrowth promoting activity from the fruit body of Russula japonica. Chem. Pharm. Bull. 2009, 57, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.J.; Dai, B.L.; Chen, N.P.; Jin, L.X.; Jiang, F.S.; Ding, Z.S.; Qian, C.D. The anti-Staphylococcus aureus activity of the phenanthrene fraction from fibrous roots of Bletilla striata. BMC. Complem. Altern. Med. 2016, 16, 491. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.I.; Tomas-Barberan, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food. Chem. 2000, 48, 4581–4589. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Feather-stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Jong-Anurakkun, N.; Bhandari, M.R.; Kawabata, J. α-glucosidase inhibitors from Devil tree (Alstonia scholaris). Food. Chem. 2007, 103, 1319–1323. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).