APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
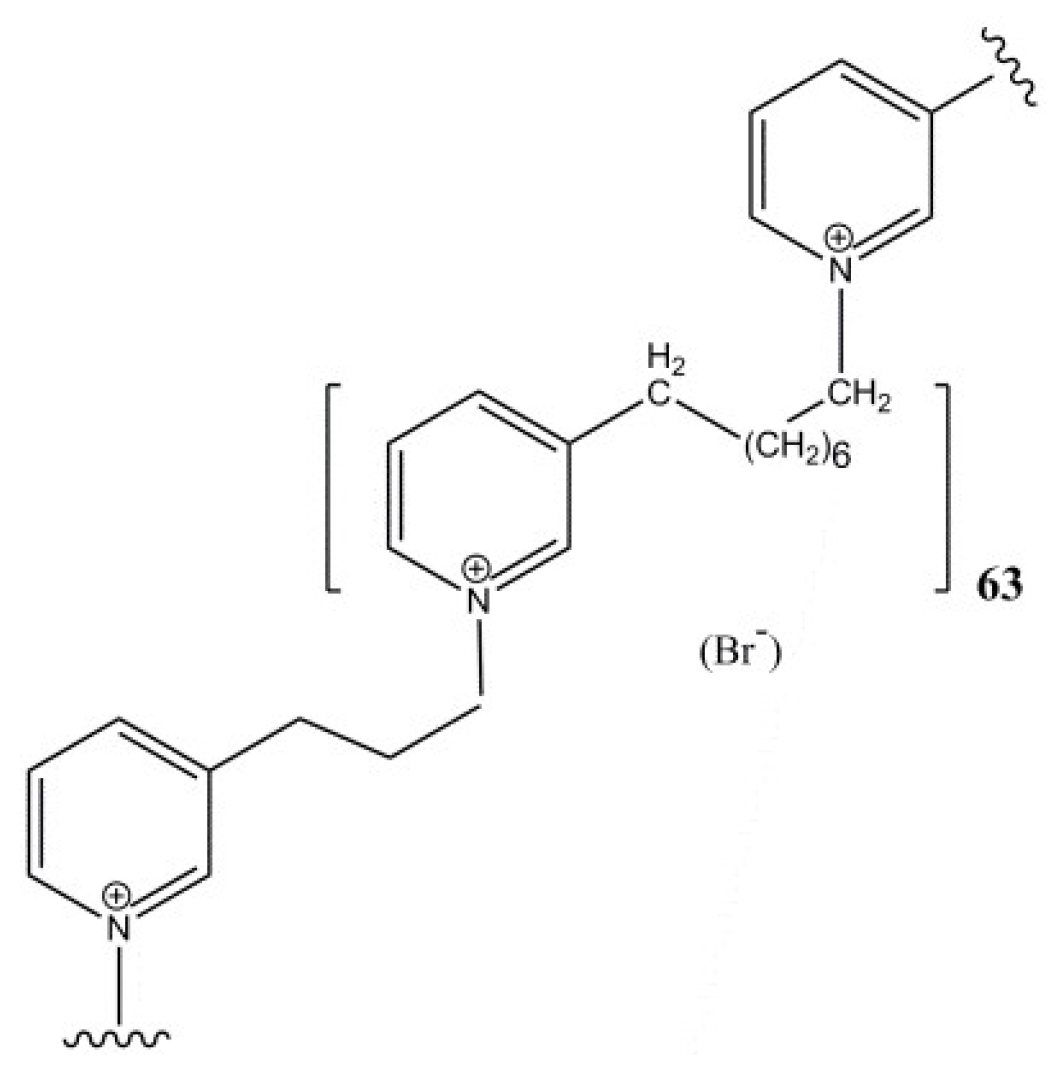
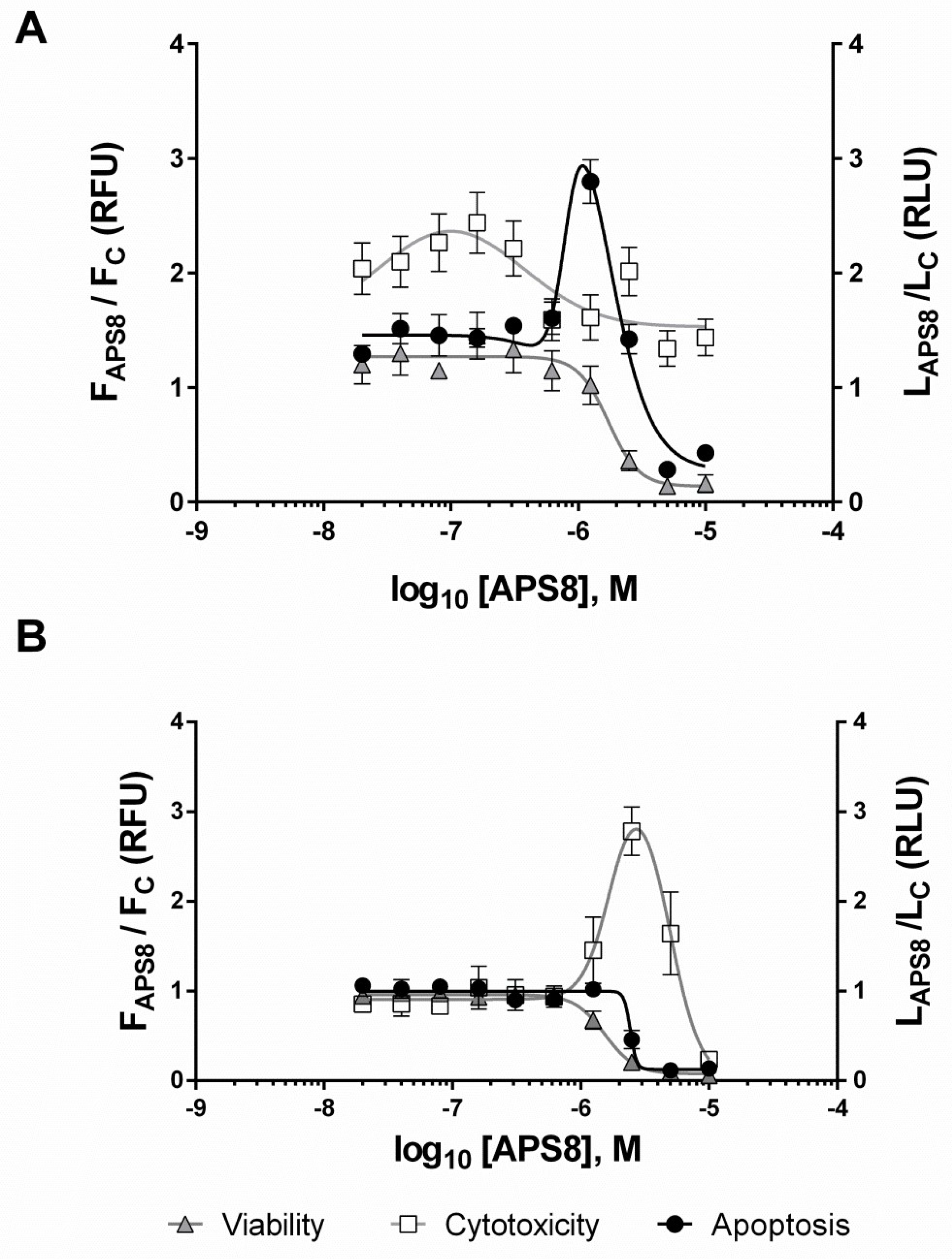
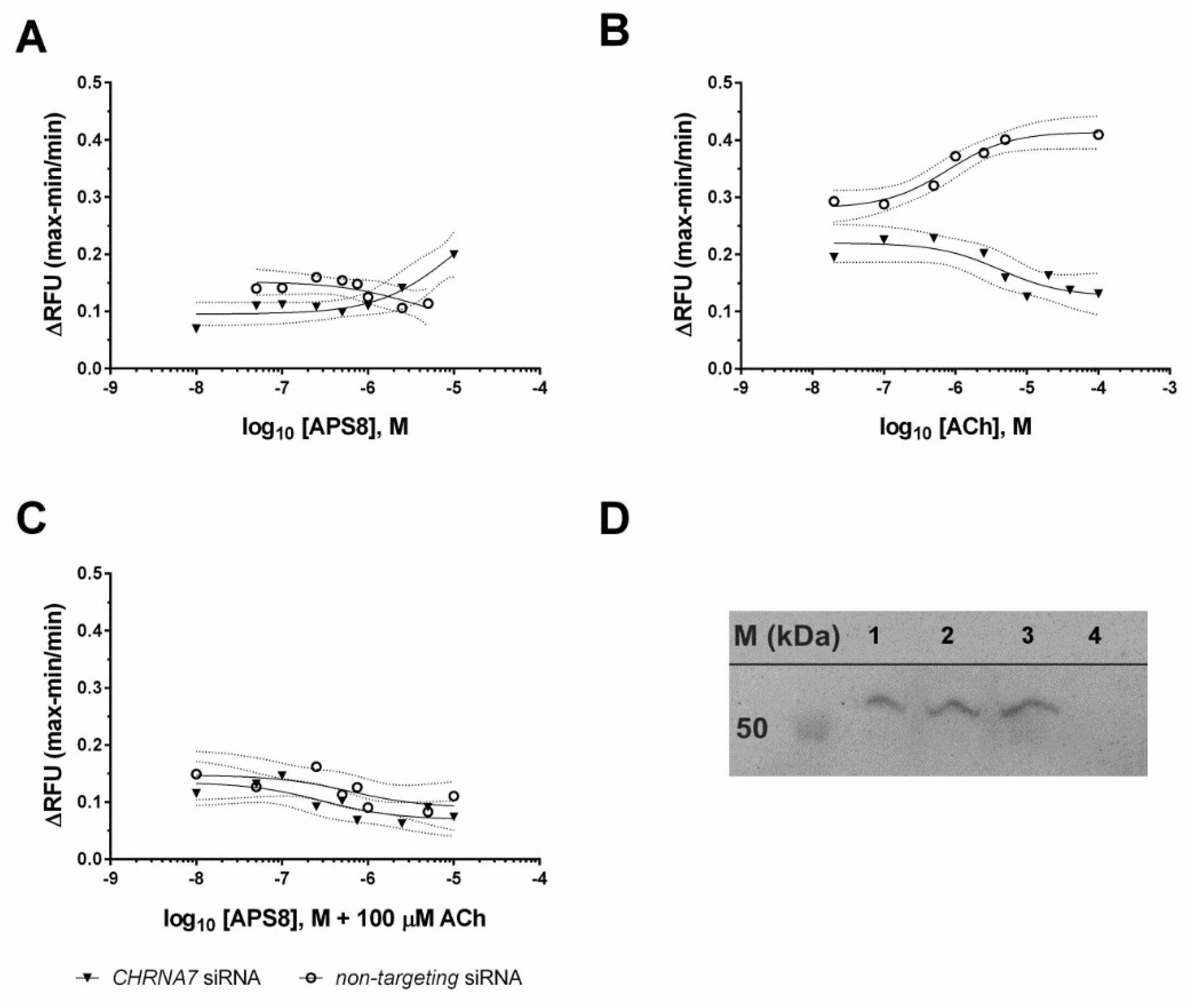
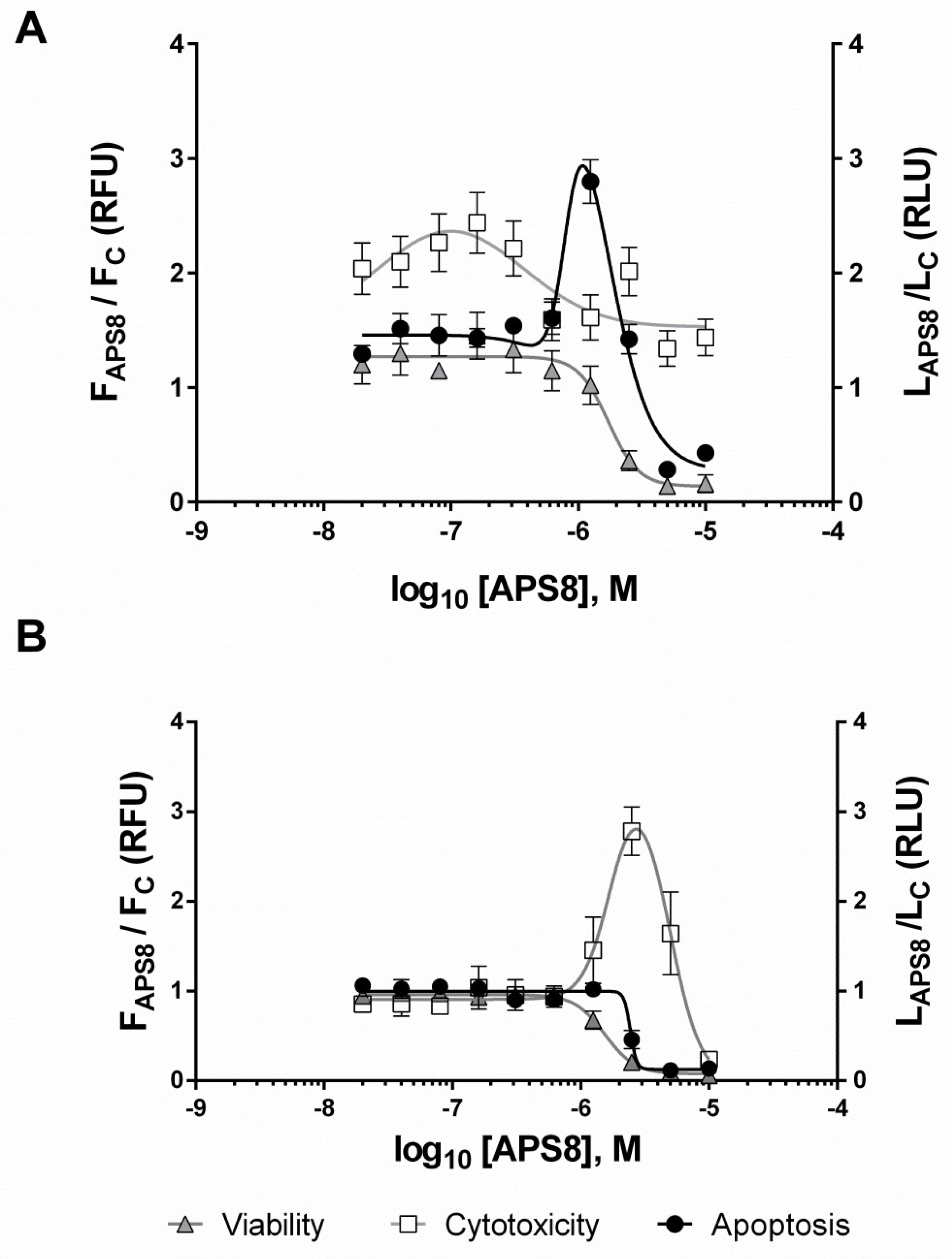
2.1. Mechanism of APS8 Toxicity
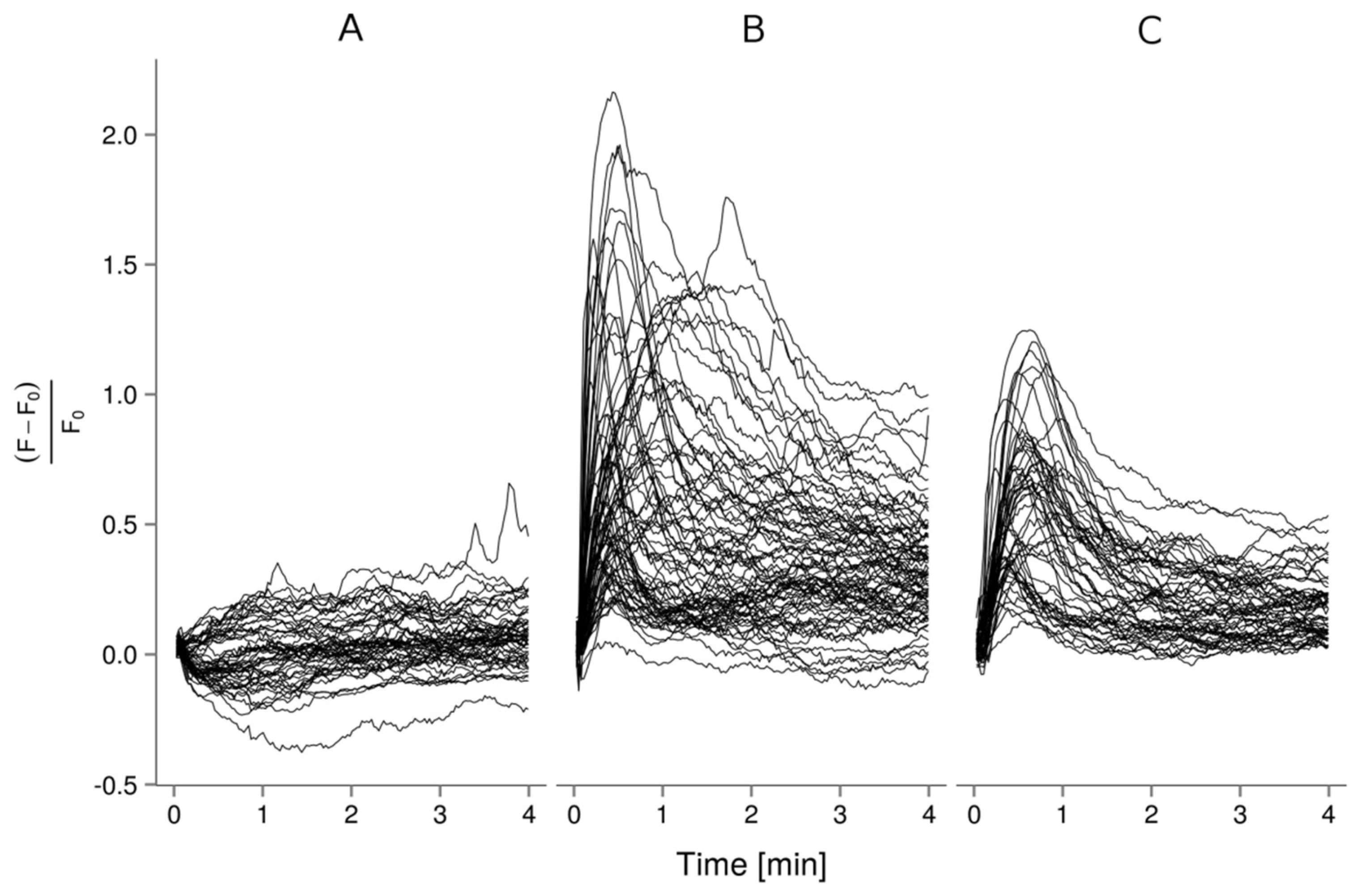
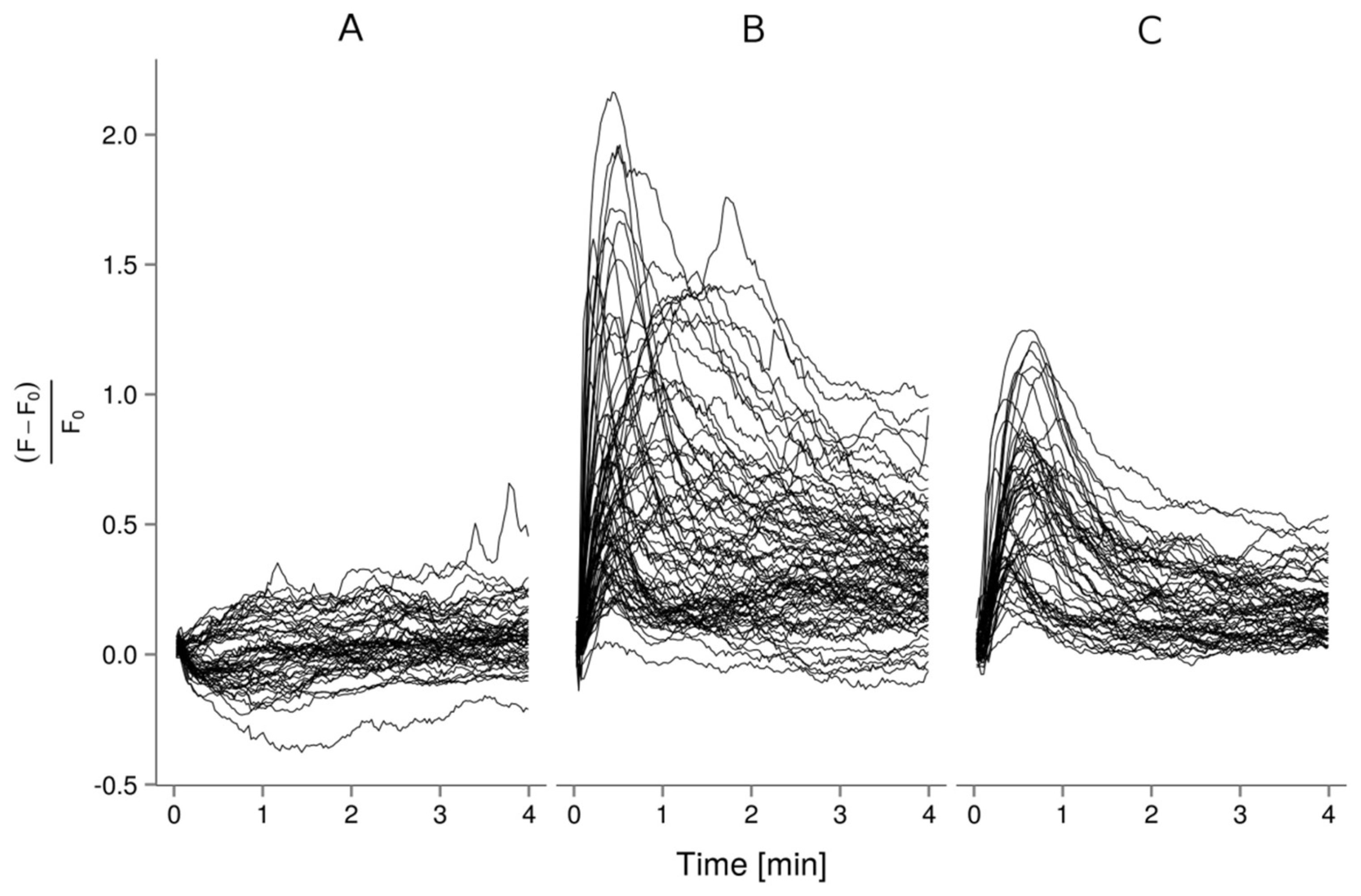
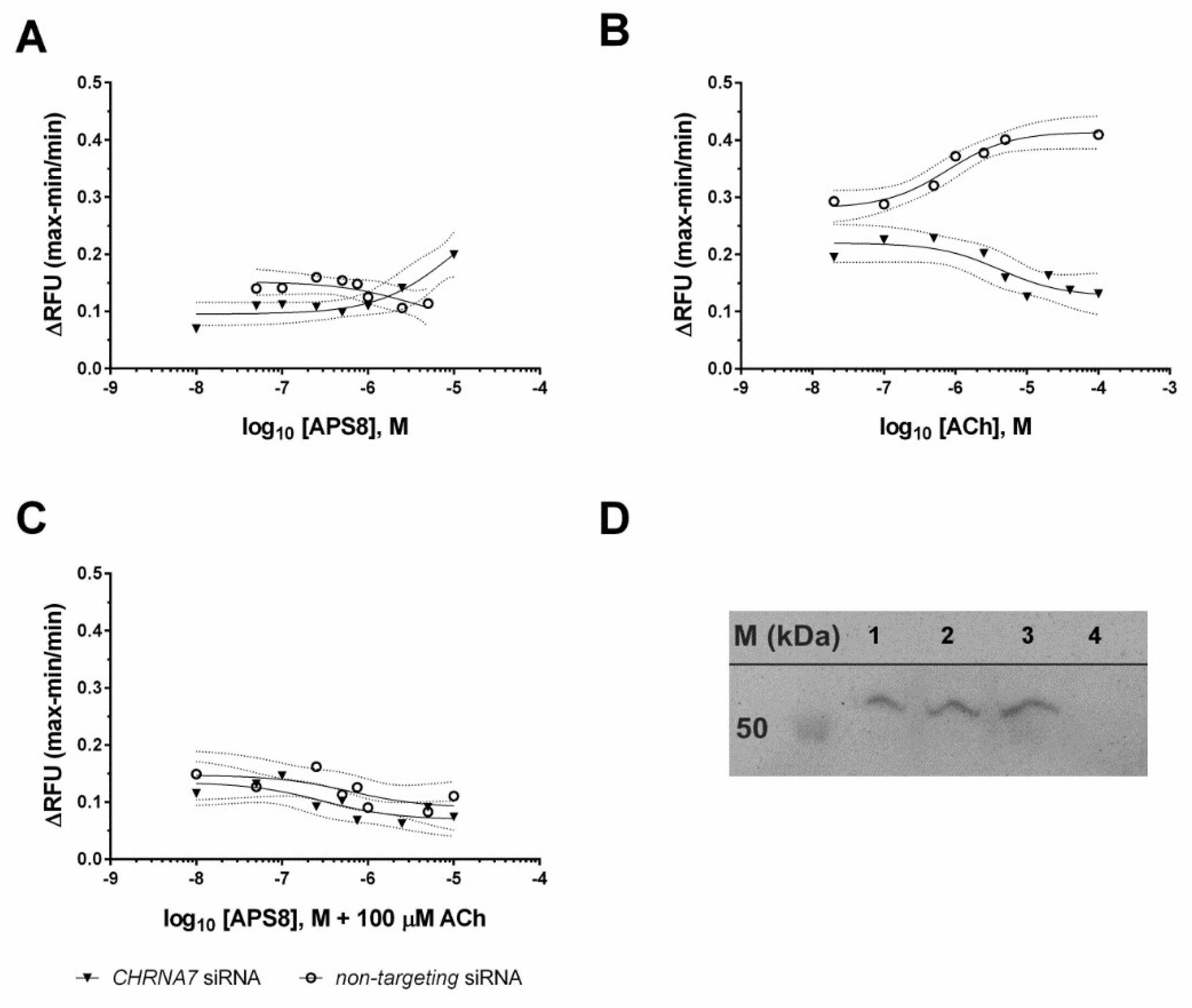
2.2. Ca2+ Imaging
2.3. Determination of LD50 in Mice
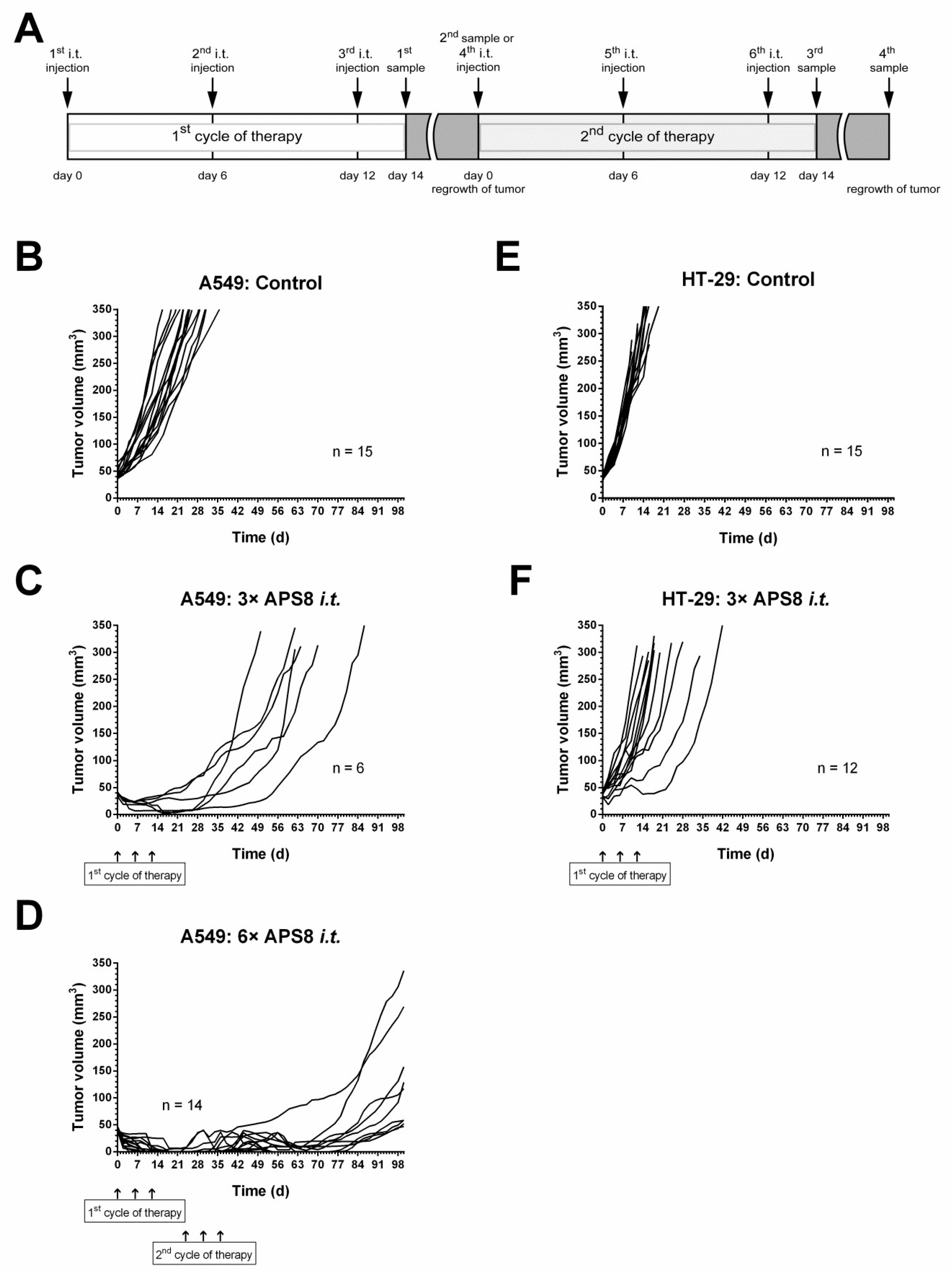
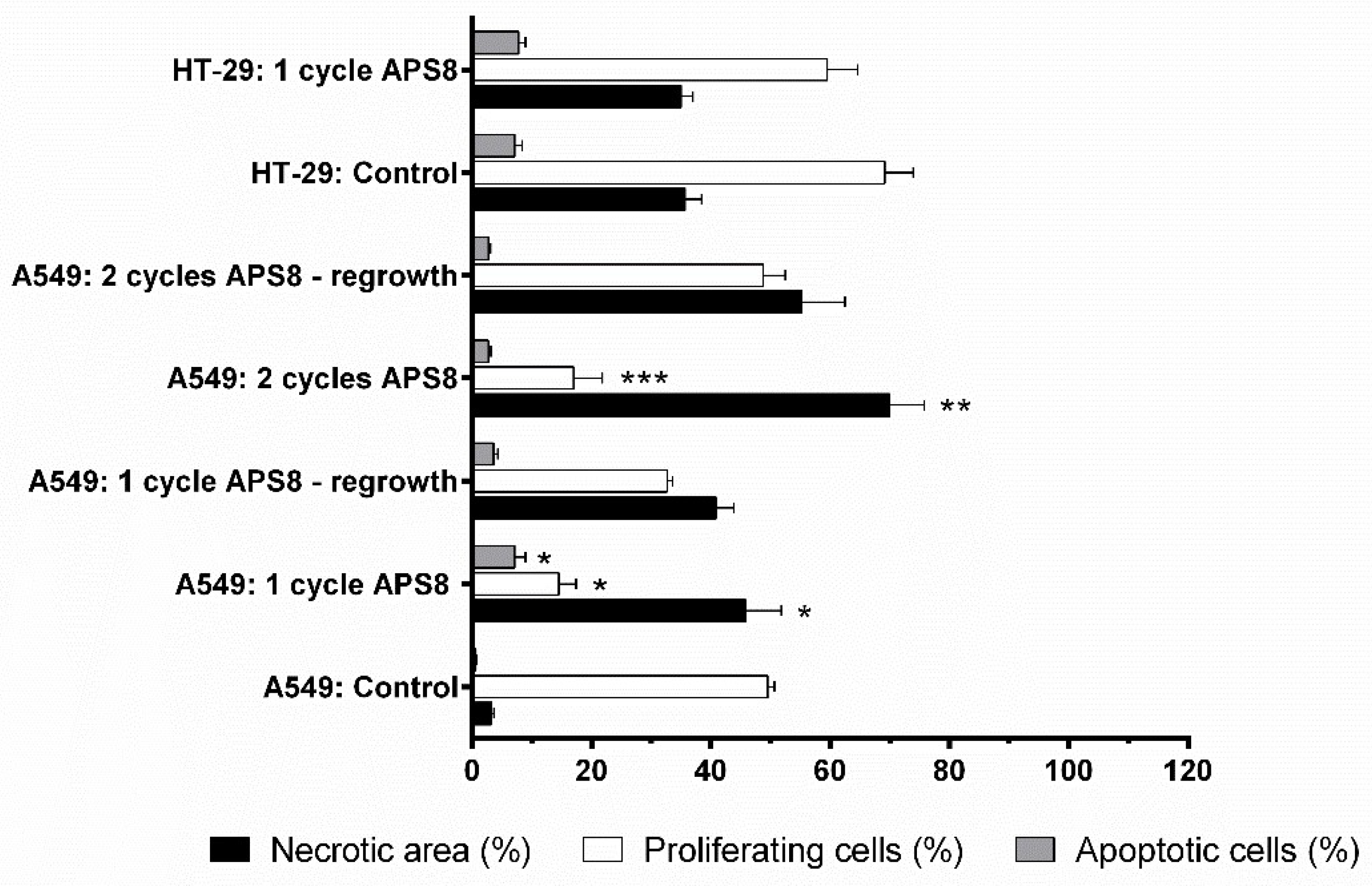
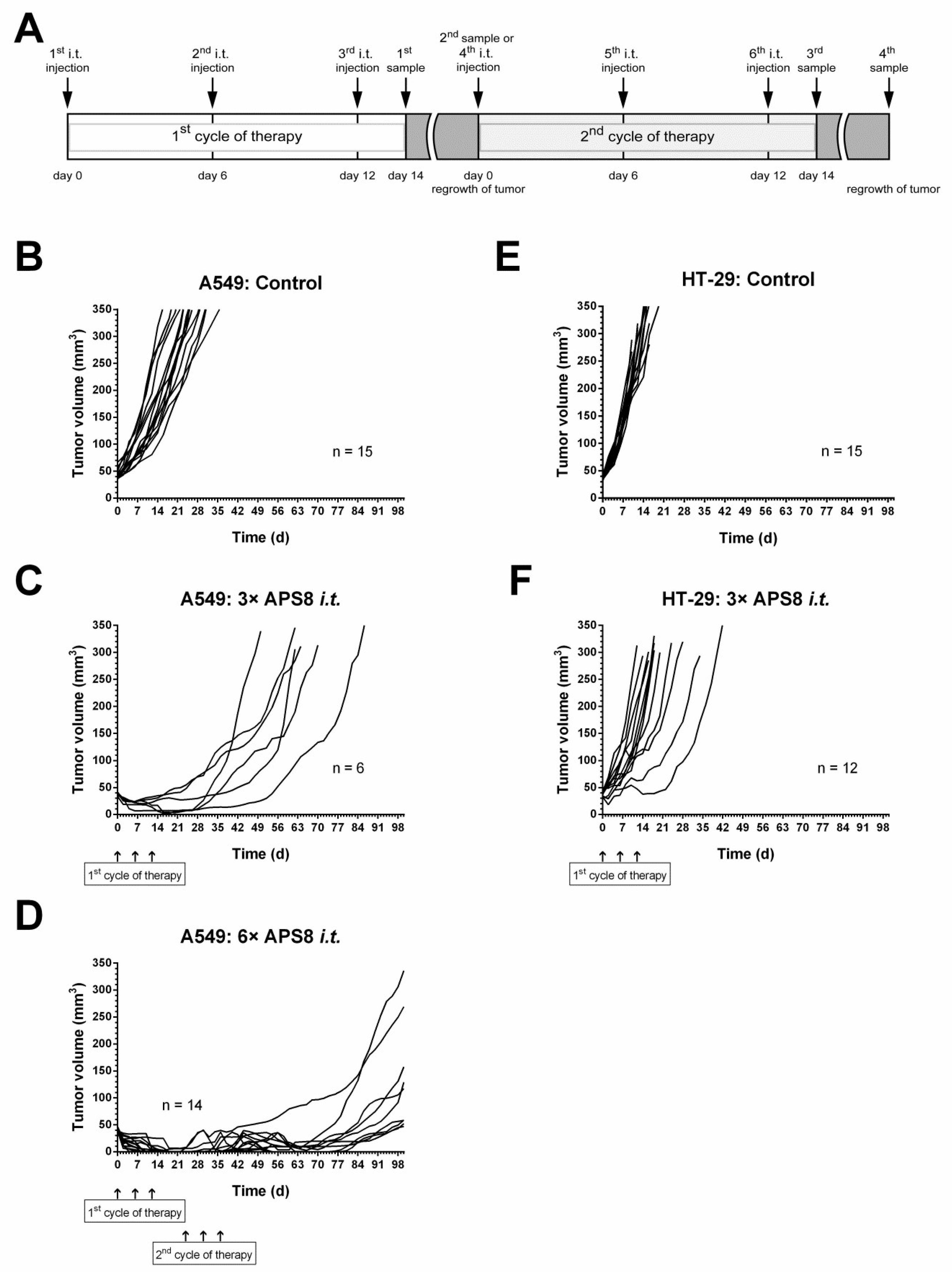
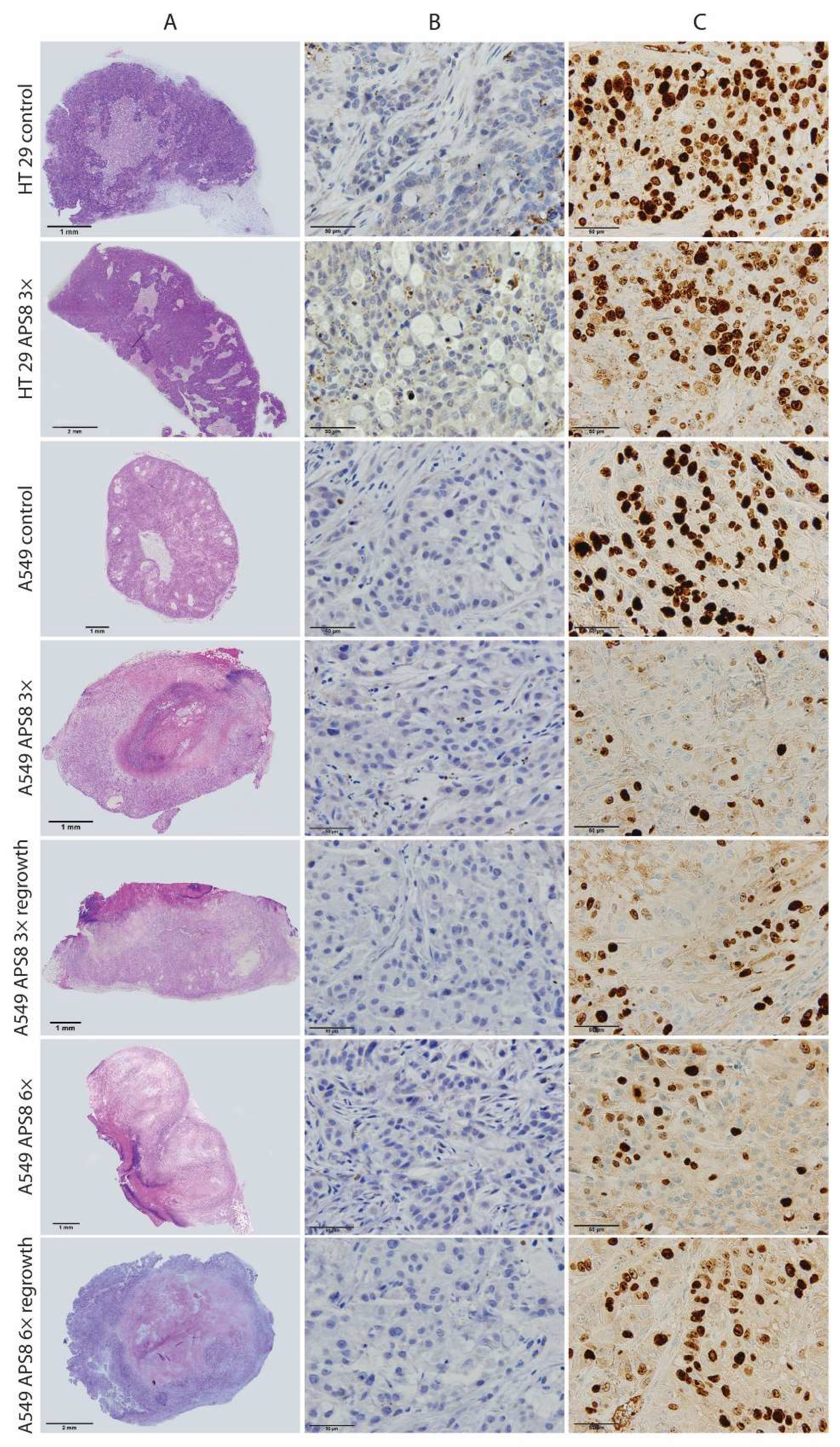
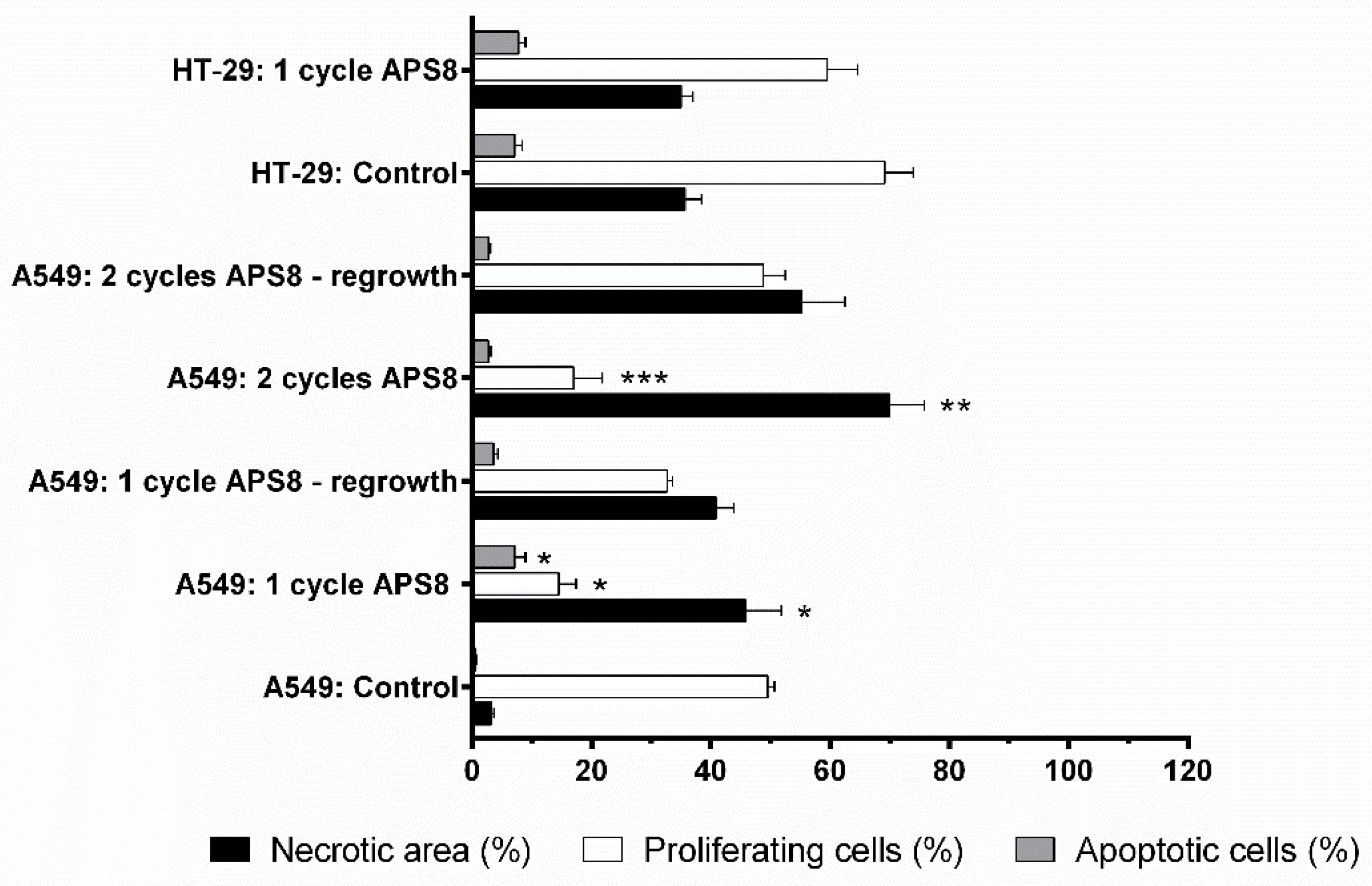
2.4. Antitumor Effectivness
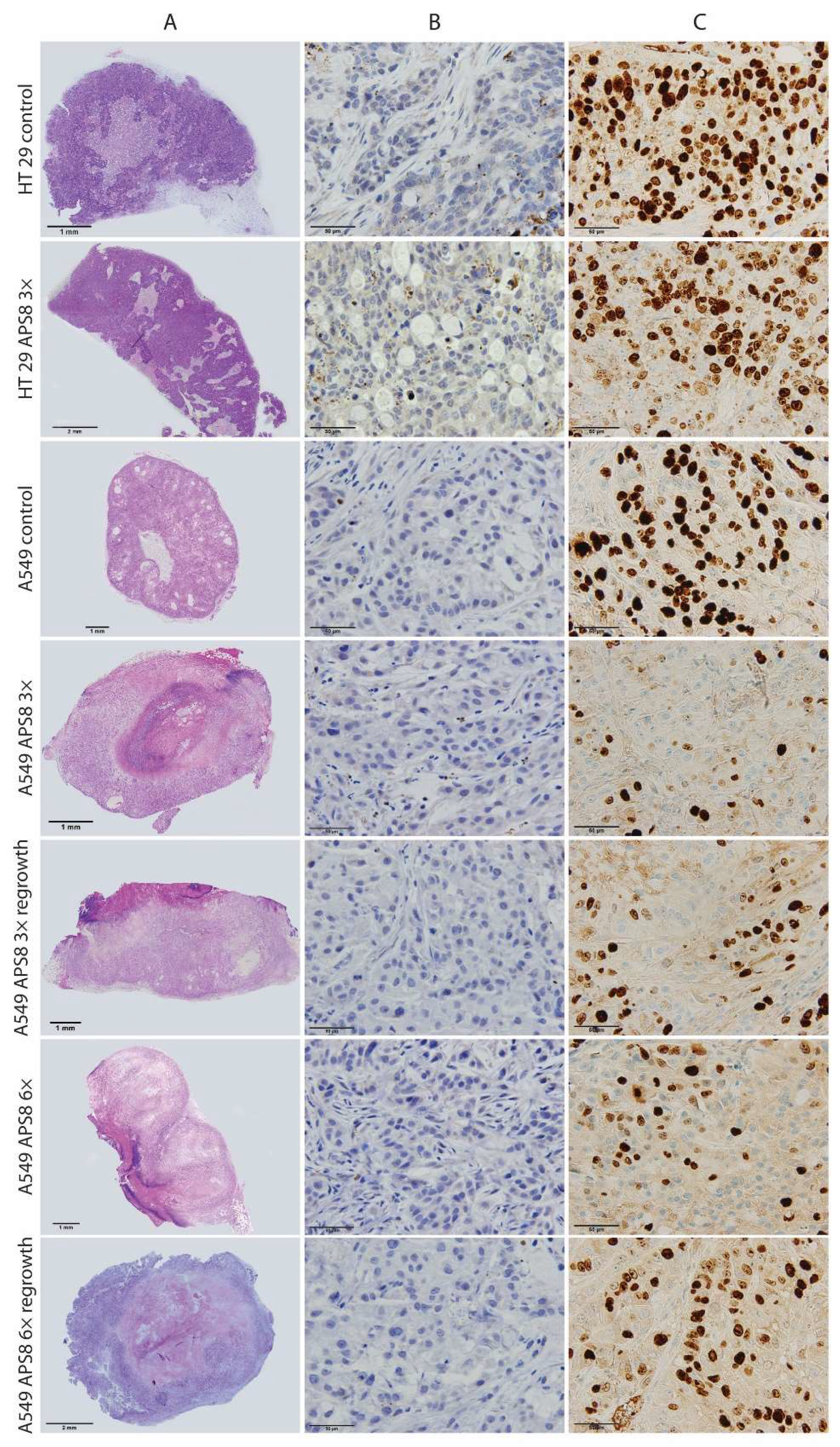
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. APS8 Stock Solution
4.3. Transient SiRNA Silencing and Quantitative Real-Time PCR Confirmation of Knock-Down
4.4. Protein Extraction and Western Blotting
4.5. Mechanism of APS8 Cell Toxicity
4.6. Microplate-Based Ca2+ Measurements
4.7. Ca2+ Imaging with Confocal Microscopy
4.8. Toxicity Study for Determination of LD50 in Mice
4.9. Antitumor Effectiveness
4.10. Histopathology
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: Multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.-M.; Lu, S. Acetylcholine receptor pathway in lung cancer: New twists to an old story. World J. Clin. Oncol. 2014, 5, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Plummer, H.K.; Dhar, M.; Schuller, H.M. Expression of the alpha7 nicotinic acetylcholine receptor in human lung cells. Respir. Res. 2005, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.L.; Chang, Y.J.; Ho, Y.S.; Lee, C.H.; Yang, Y.Y.; An, J.; Lin, S.Y. Tobacco-specific carcinogen enhances colon cancer cell migration through alpha7-nicotinic acetylcholine receptor. Ann. Surg. 2009, 249, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Huang, C.S.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chang, Y.J.; Tam, K.W.; Wei, P.L.; Cheng, T.C.; Chu, J.S.; et al. Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, M.H.; Al-Wadei, H.A.N.; Schuller, H.M. Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors α3, α5, and α7. Mol. Cancer Res. 2012, 10, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Ho, Y.S.; Guo, H.R.; Wang, Y.J. Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol. Sci. 2008, 104, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Chellappan, S.P. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Improgo, M.R.; Soll, L.G.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 2013, 4, 251. [Google Scholar] [CrossRef] [PubMed]
- Medjber, K.; Freidja, M.L.; Grelet, S.; Lorenzato, M.; Maouche, K.; Nawrocki-Raby, B.; Birembaut, P.; Polette, M.; Tournier, J.M. Role of nicotinic acetylcholine receptors in cell proliferation and tumour invasion in broncho-pulmonary carcinomas. Lung Cancer 2015, 87, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Padmanabhan, J.; Chellappan, S. The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression. Cancers 2015, 7, 1447–1471. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.R.; Adhikari, A. Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation. Cell Commun. Signal. 2009, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Lee, C.H.; Ho, Y.S. Nicotinic acetylcholine receptor-based blockade: Applications of molecular targets for cancer therapy. Clin. Cancer Res. 2011, 17, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Improgo, M.R.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem. Pharmacol. 2011, 82, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011, 2011, 456743. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Investig. 2006, 116, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chang, Y.C.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chen, L.C.; Chang, Y.J.; Wei, P.L.; Chang, H.W.; Chang, C.H.; et al. Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces α9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 2011, 129, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Paleari, L.; Sessa, F.; Catassi, A.; Servent, D.; Mourier, G.; Doria-Miglietta, G.; Ognio, E.; Cilli, M.; Dominioni, L.; Paolucci, M.; et al. Inhibition of non-neuronal alpha7-nicotinic receptor reduces tumorigenicity in A549 NSCLC xenografts. Int. J. Cancer 2009, 125, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Alama, A.; Bruzzo, C.; Cavalieri, Z.; Forlani, A.; Utkin, Y.; Casciano, I.; Romani, M. Inhibition of the nicotinic acetylcholine receptors by cobra venom α-neurotoxins: Is there a perspective in lung cancer treatment? PLoS ONE 2011, 6, 20695. [Google Scholar] [CrossRef] [PubMed]
- Cesario, A.; Russo, P.; Nastrucci, C.; Granone, P. Is α7-nAChR a possible target for lung cancer and malignant pleural mesothelioma treatment? Curr. Drug Targets 2012, 13, 688–694. [Google Scholar] [PubMed]
- Van Soest, R.W.M.; Boury-Esnault, N.; Hooper, J.N.A.; Rützler, K.; de Voogd, N.J.; Alvarez de Glasby, B.; Hajdu, E.; Pisera, A.B.; Manconi, R.; Schoenberg, C.; et al. (2007) Haliclona (Rhizoniera) sarai (Pulitzer-Finali, 1969). Available online: http://marinespecies.org/porifera/porifera.php?p=taxdetails&id=244433 (accessed on 14 March 2016).
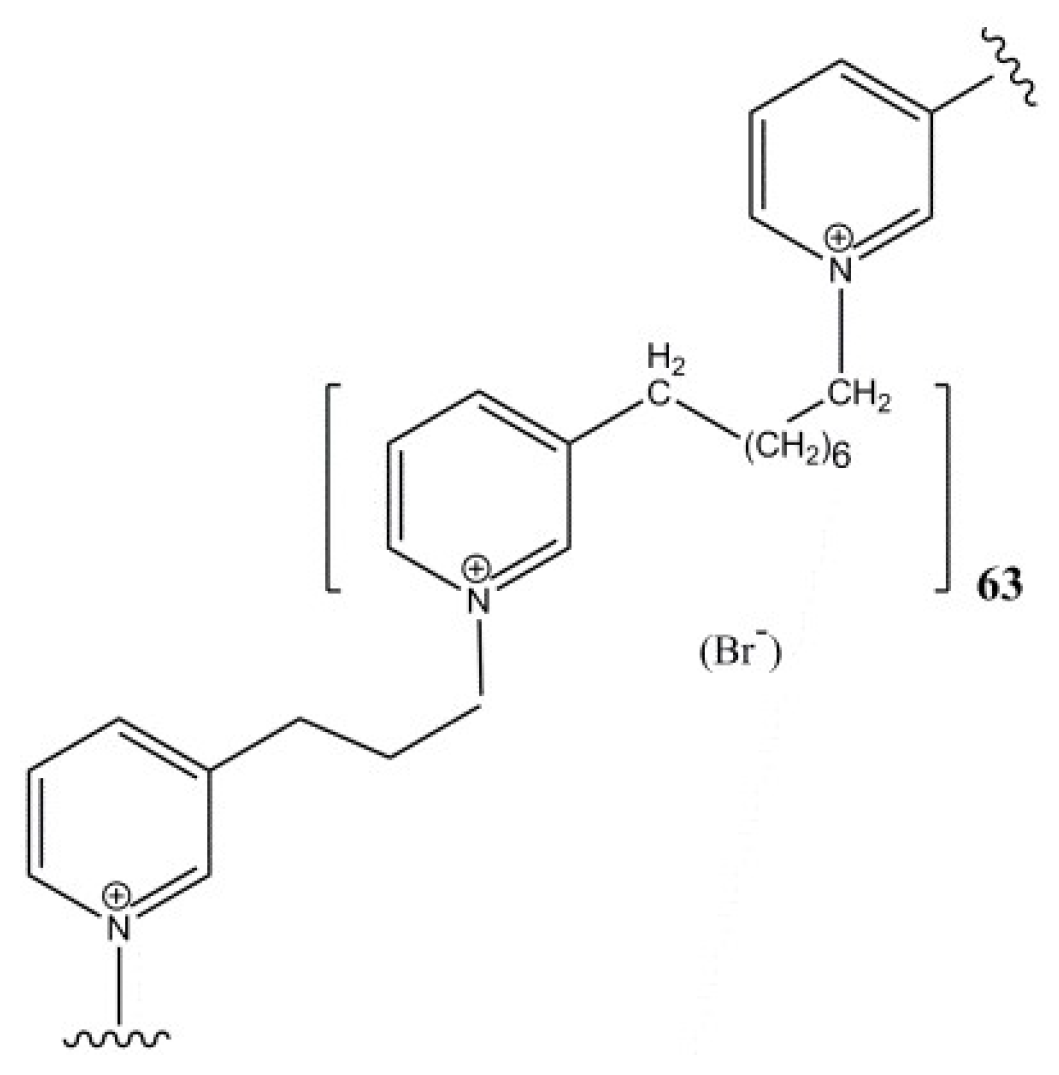
- Sepčić, K.; Guella, G.; Mancini, I.; Pietra, F.; Serra, M.D.; Menestrina, G.; Tubbs, K.; Maček, P.; Turk, T. Characterization of anticholinesterase-active 3-alkylpyridinium polymers from the marine sponge Reniera sarai in aqueous solutions. J. Nat. Prod. 1997, 60, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Sepčić, K.; Marcel, V.; Klaebe, A.; Turk, T.; Suput, D.; Fournier, D. Inhibition of acetylcholinesterase by an alkylpyridinium polymer from the marine sponge, Reniera sarai. Biochim. Biophys. Acta 1998, 1387, 217–225. [Google Scholar] [CrossRef]
- Mancini, I.; Sicurelli, A.; Guella, G.; Turk, T.; Maček, P.; Sepčić, K. Synthesis and bioactivity of linear oligomers related to polymeric alkylpyridinium metabolites from the Mediterranean sponge Reniera sarai. Org. Biomol. Chem. 2004, 2, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Lu, Z.; Edrada-Ebel, R.; Chatzi, C.; Tucker, S.J.; Sepčić, K.; Turk, T.; Zovko, A.; Shen, S.; Mancini, I.; et al. Chemical synthesis and biological activities of 3-alkyl pyridinium polymeric analogues of marine toxins. J. Chem. Biol. 2010, 3, 113–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zovko, A.; Viktorsson, K.; Lewensohn, R.; Kološa, K.; Filipič, M.; Xing, H.; Kem, W.R.; Paleari, L.; Turk, T. APS8, a polymeric alkylpyridinium salt blocks α7 nAChR and induces apoptosis in non-small cell lung carcinoma. Mar. Drugs 2013, 11, 2574–2594. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.J.; McClelland, D.; Jaspars, M.; Sepčić, K.; MacEwan, D.J.; Scott, R.H. The influence of alkyl pyridinium sponge toxins on membrane properties, cytotoxicity, transfection and protein expression in mammalian cells. Biochim. Biophys. Acta Biomembr. 2003, 1614, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Grandič, M.; Zovko, A.; Frangež, R.; Turk, T.; Sepčić, K. Binding and permeabilization of lipid bilayers by natural and synthetic 3-alkylpyridinium polymers. Bioorg. Med. Chem. 2012, 20, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S. Determining the Predictive Mechanism of Toxicity Using a Single-Well Multiplexed Assay. Available online: http://worldwide.promega.com/resources/pubhub/determining-the-predictive-mechanism-of-toxicity-with-apotox-glo/ (accessed on 20 August 2015).
- Grozio, A.; Paleari, L.; Catassi, A.; Servent, D.; Cilli, M.; Piccardi, F.; Paganuzzi, M.; Cesario, A.; Granone, P.; Mourier, G.; et al. Natural agents targeting the alpha7-nicotinic-receptor in NSCLC: A promising prospective in anti-cancer drug development. Int. J. Cancer 2008, 122, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Paleari, L.; Catassi, A.; Ciarlo, M.; Cavalieri, Z.; Bruzzo, C.; Servent, D.; Cesario, A.; Chessa, L.; Cilli, M.; Piccardi, F.; et al. Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif. 2008, 41, 936–959. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wu, C.H.; Ho, Y.S. From smoking to cancers: Novel targets to neuronal nicotinic acetylcholine receptors. J. Oncol. 2011, 2011, 693424. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, P.; Becchetti, A. Targeting neuronal nicotinic receptors in cancer: New ligands and potential side-effects. Recent Pat. Anticancer. Drug Discov. 2013, 8, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, B.J.; Williams, M.; Plummer, H.K.; Schuller, H.M. Activation of voltage-operated Ca2+-channels in human small cell lung carcinoma by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int. J. Oncol. 2000, 16, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Ee, P.L.R.; He, X.; Ross, D.D.; Beck, W.T. Modulation of breast cancer resistance protein (BCRP/ABCG2) gene expression using RNA interference. Mol. Cancer Ther. 2004, 3, 1577–1583. [Google Scholar] [PubMed]
- Niles, A.L.; Riss, T.L. Multiplexed viability, cytotoxicity, and caspase activity assays. Methods Mol. Biol. 2015, 1219, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.; Napier, J.; Balestra, M.; DeCory, T.; Hale, G.; Macor, J.; Mack, R.; Loch, J.; Wu, E.; Kover, A.; et al. (−)-Spiro[1-azabicyclo[2.2.2]octane-3,5’-oxazolidin-2’-one], a conformationally restricted analogue of acetylcholine, is a highly selective full agonist at the alpha 7 nicotinic acetylcholine receptor. J. Med. Chem. 2000, 43, 4045–4050. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing. the R Foundation for Statistical Computing: Vienna, Austria, 2011; ISBN 3-900051-07-0. [Google Scholar]
- Wickham, H. ggplot2; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 120–132. [Google Scholar] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Serša, G.; Čemažar, M.; Miklavčič, D. Antitumor effectiveness of electrochemotherapy with cis-diamminedichloroplatinum(II) in mice. Cancer Res. 1995, 55, 3450–3455. [Google Scholar] [PubMed]
- Grandič, M.; Sepčić, K.; Turk, T.; Juntes, P.; Frangež, R. In vivo toxic and lethal cardiovascular effects of a synthetic polymeric 1,3-dodecylpyridinium salt in rodents. Toxicol. Appl. Pharmacol. 2011, 255, 86–93. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berne, S.; Čemažar, M.; Frangež, R.; Juntes, P.; Kranjc, S.; Grandič, M.; Savarin, M.; Turk, T. APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors. Mar. Drugs 2018, 16, 367. https://doi.org/10.3390/md16100367
Berne S, Čemažar M, Frangež R, Juntes P, Kranjc S, Grandič M, Savarin M, Turk T. APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors. Marine Drugs. 2018; 16(10):367. https://doi.org/10.3390/md16100367
Chicago/Turabian StyleBerne, Sabina, Maja Čemažar, Robert Frangež, Polona Juntes, Simona Kranjc, Marjana Grandič, Monika Savarin, and Tom Turk. 2018. "APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors" Marine Drugs 16, no. 10: 367. https://doi.org/10.3390/md16100367
APA StyleBerne, S., Čemažar, M., Frangež, R., Juntes, P., Kranjc, S., Grandič, M., Savarin, M., & Turk, T. (2018). APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors. Marine Drugs, 16(10), 367. https://doi.org/10.3390/md16100367