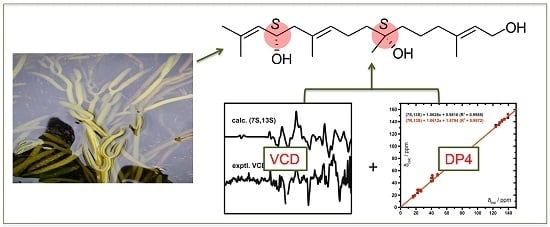
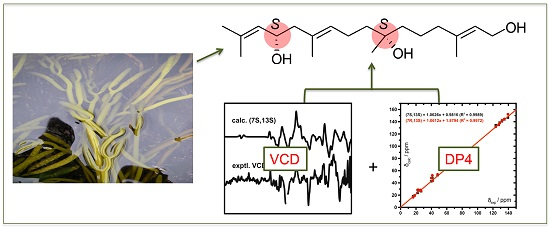
Bifurcatriol, a New Antiprotozoal Acyclic Diterpene from the Brown Alga Bifurcaria bifurcata


Abstract
:
1. Introduction
2. Results
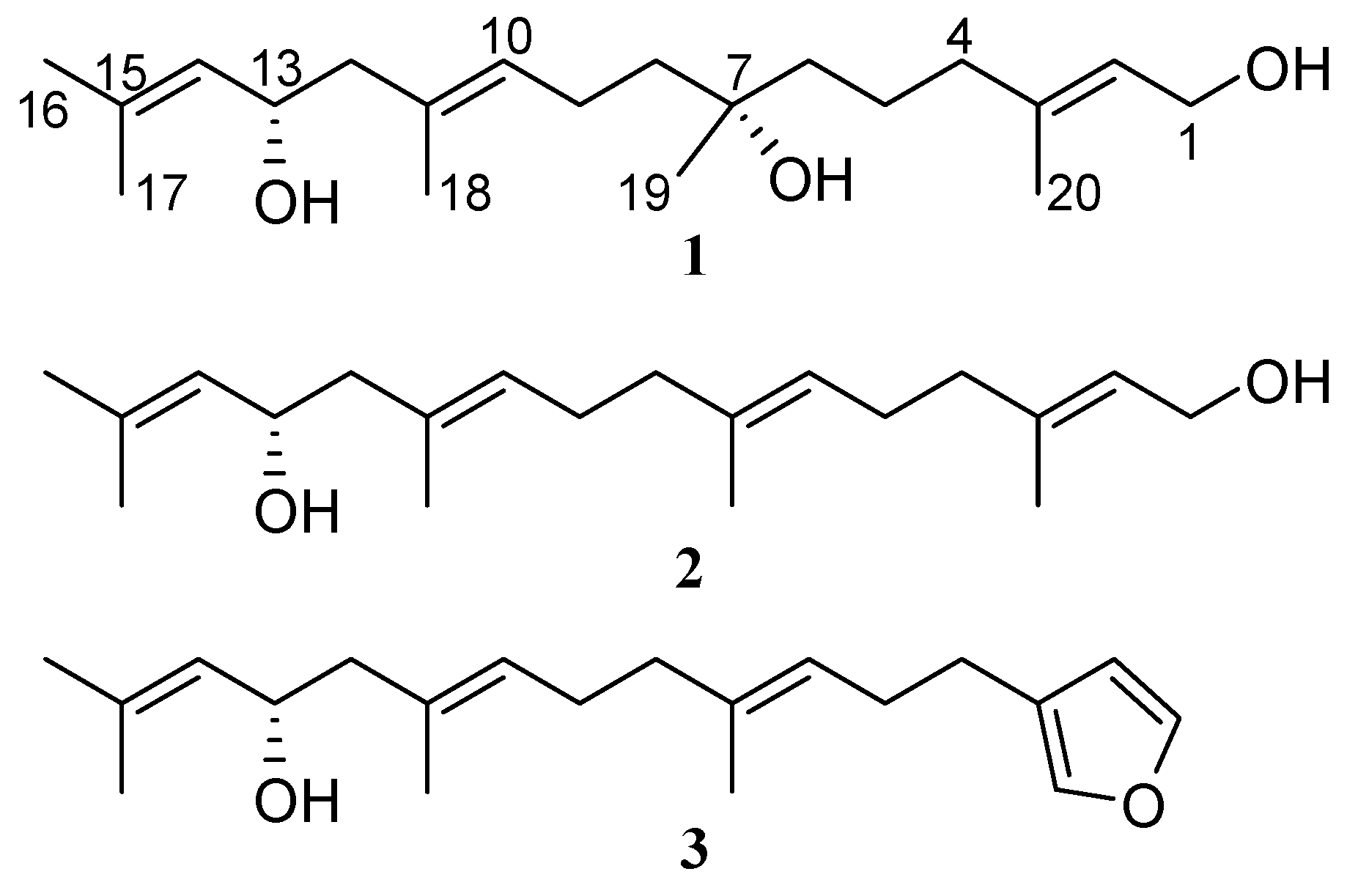
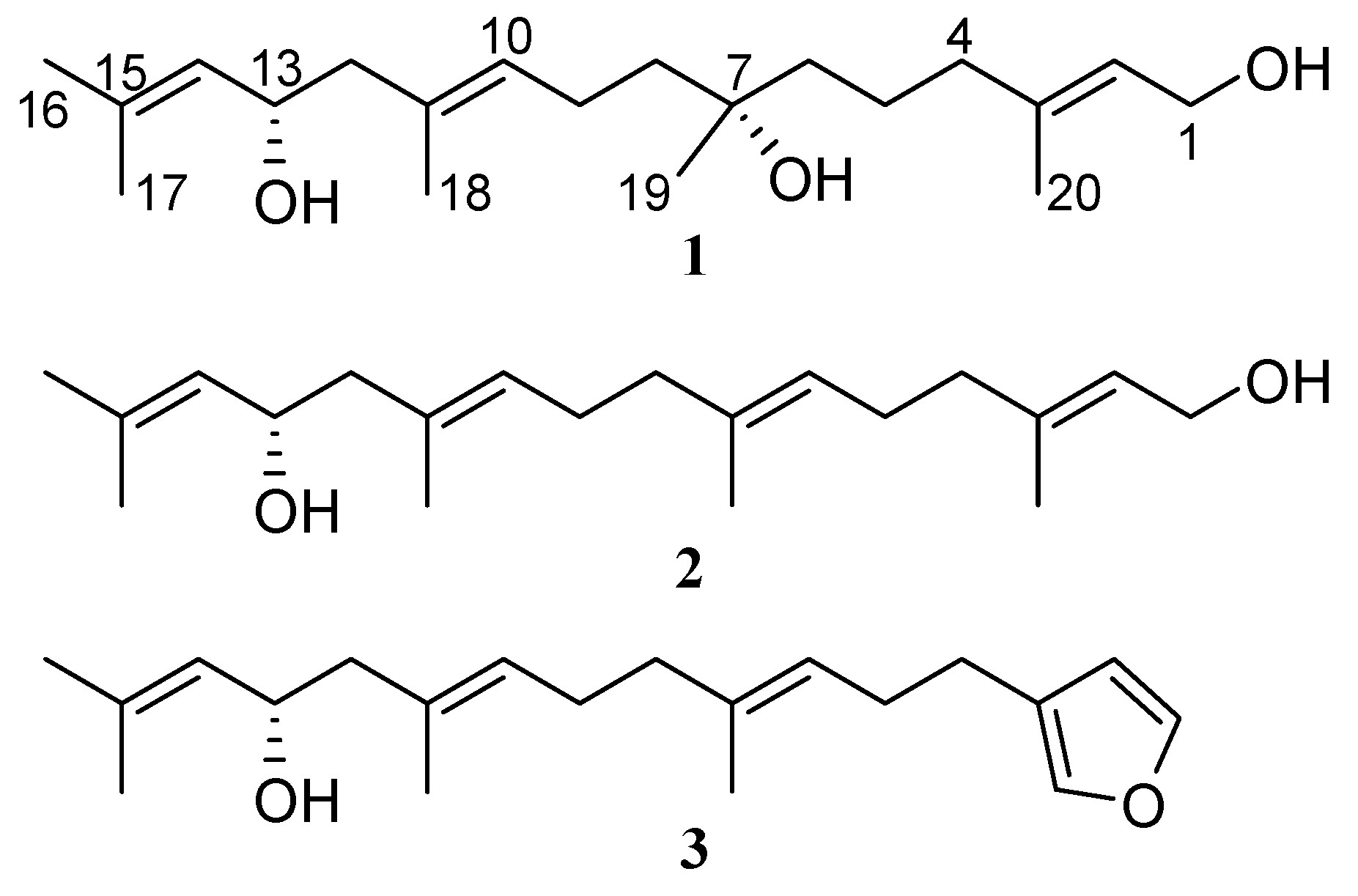
2.1. Isolation and Structure Elucidation
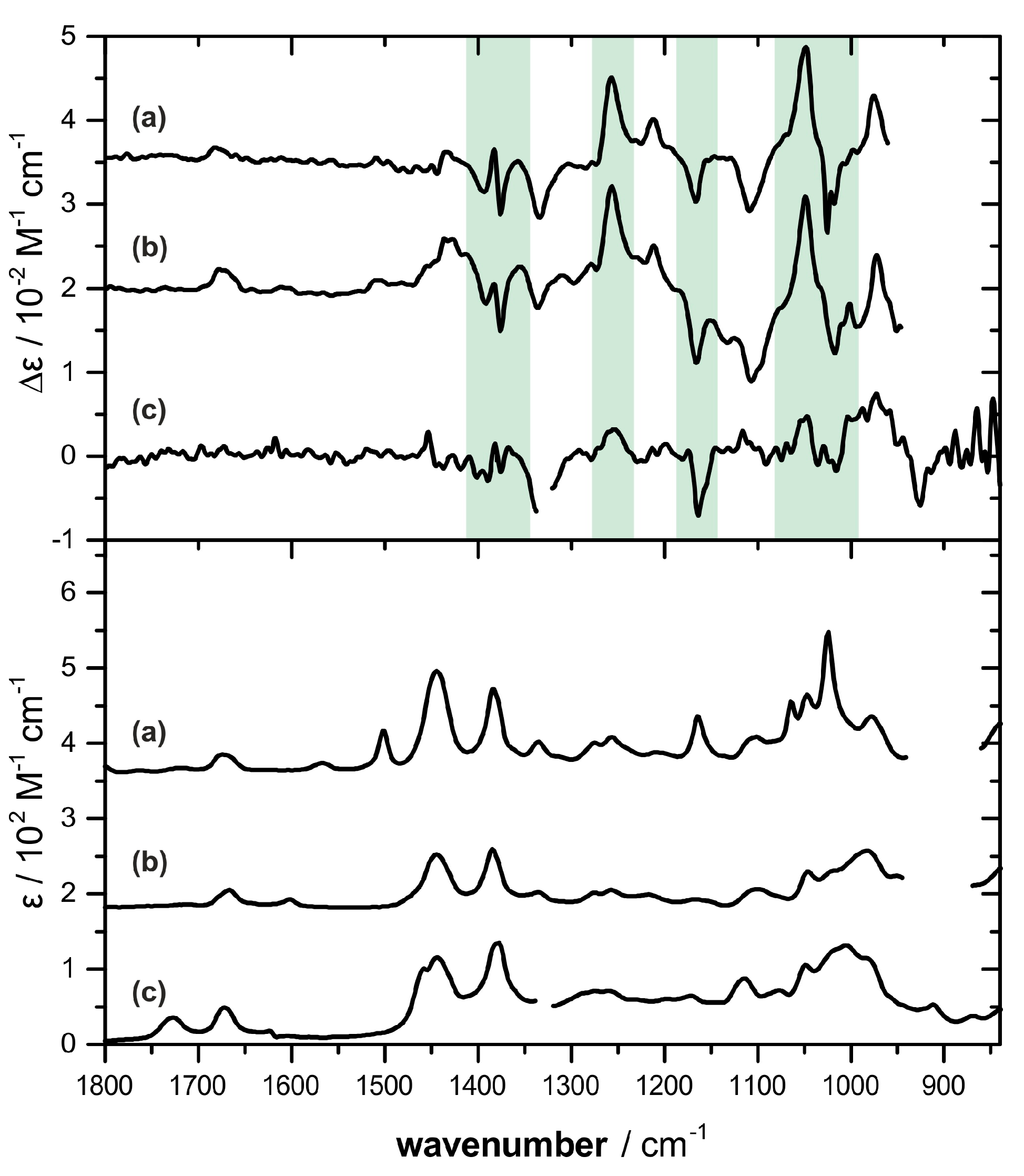
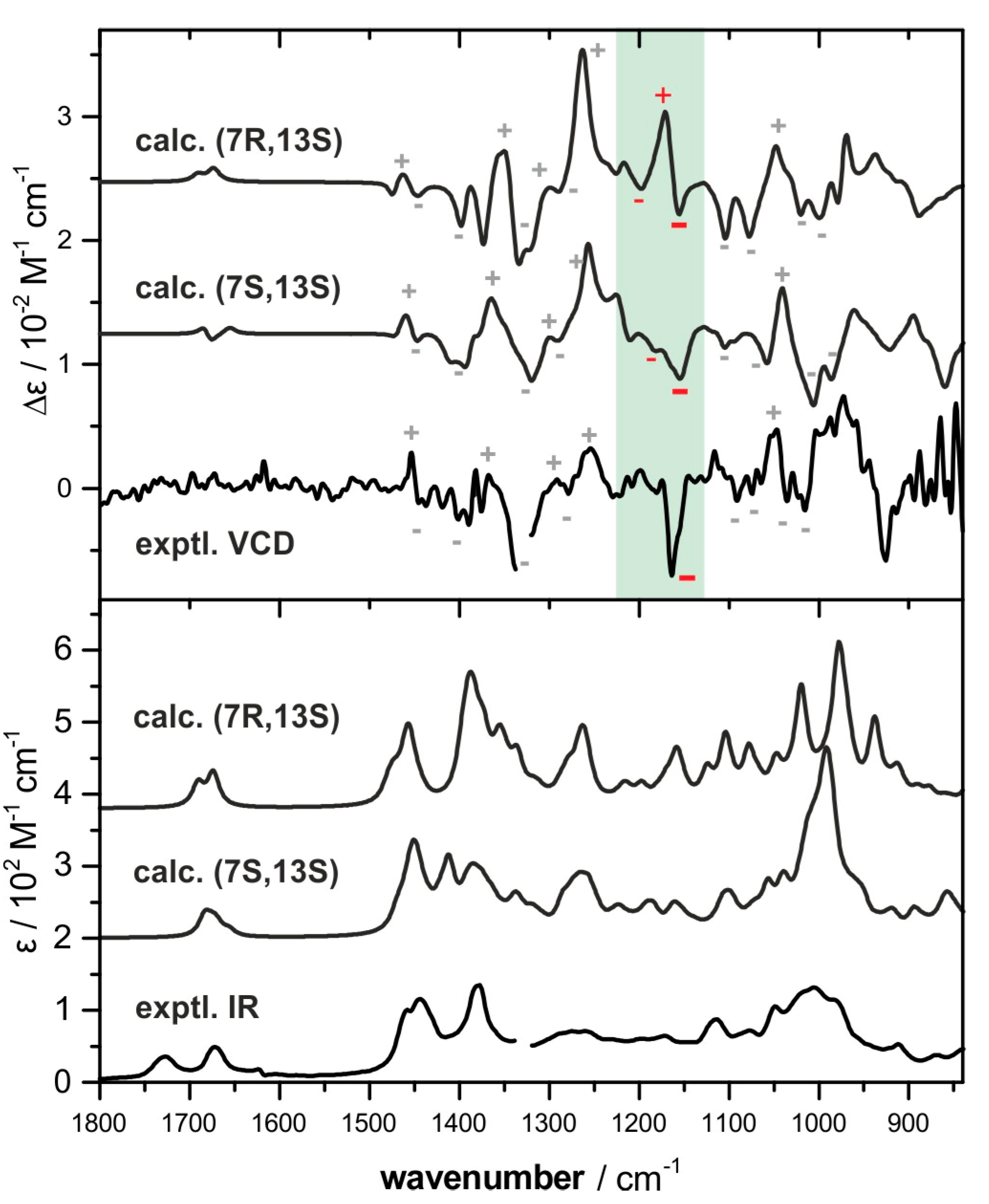
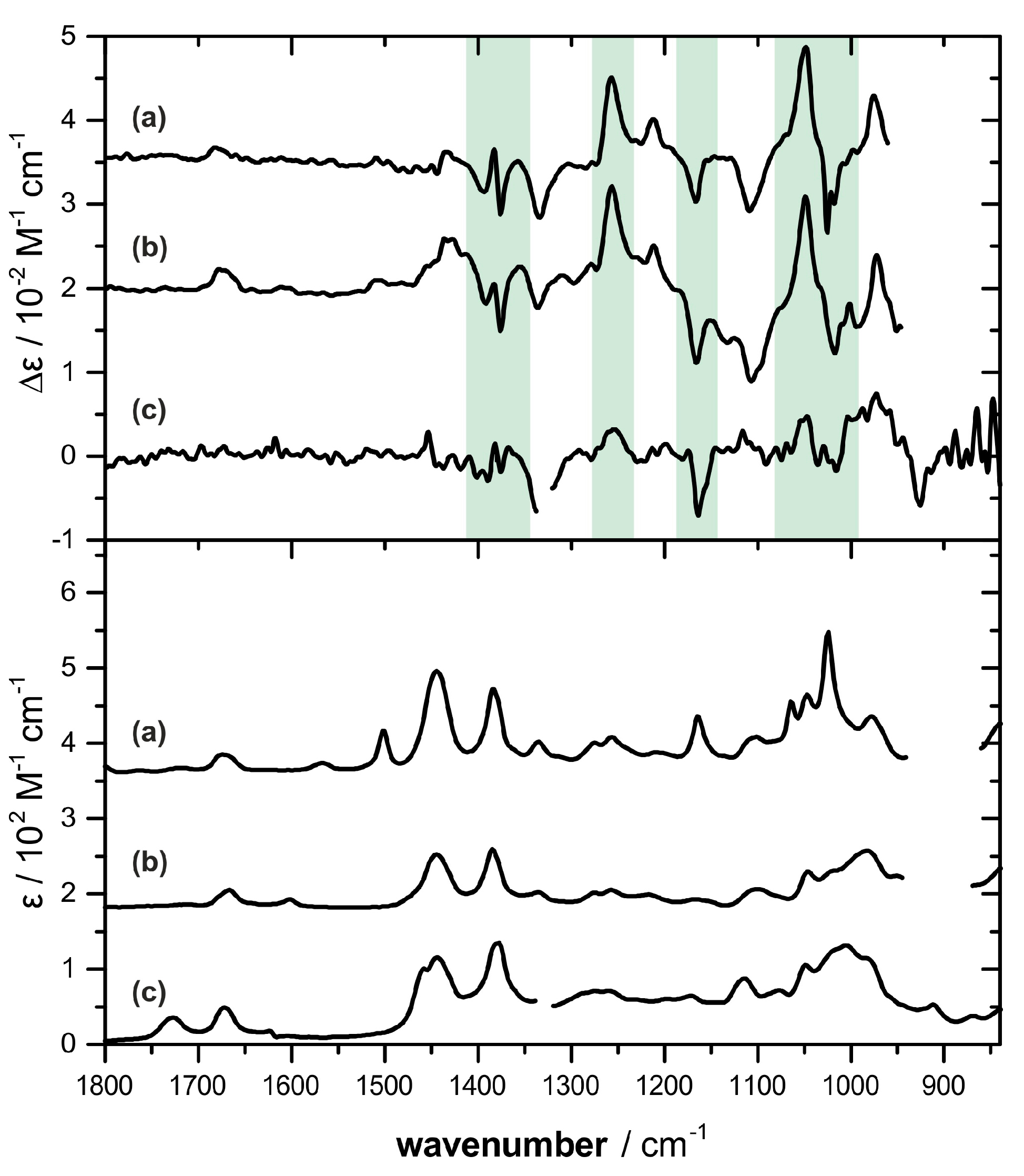
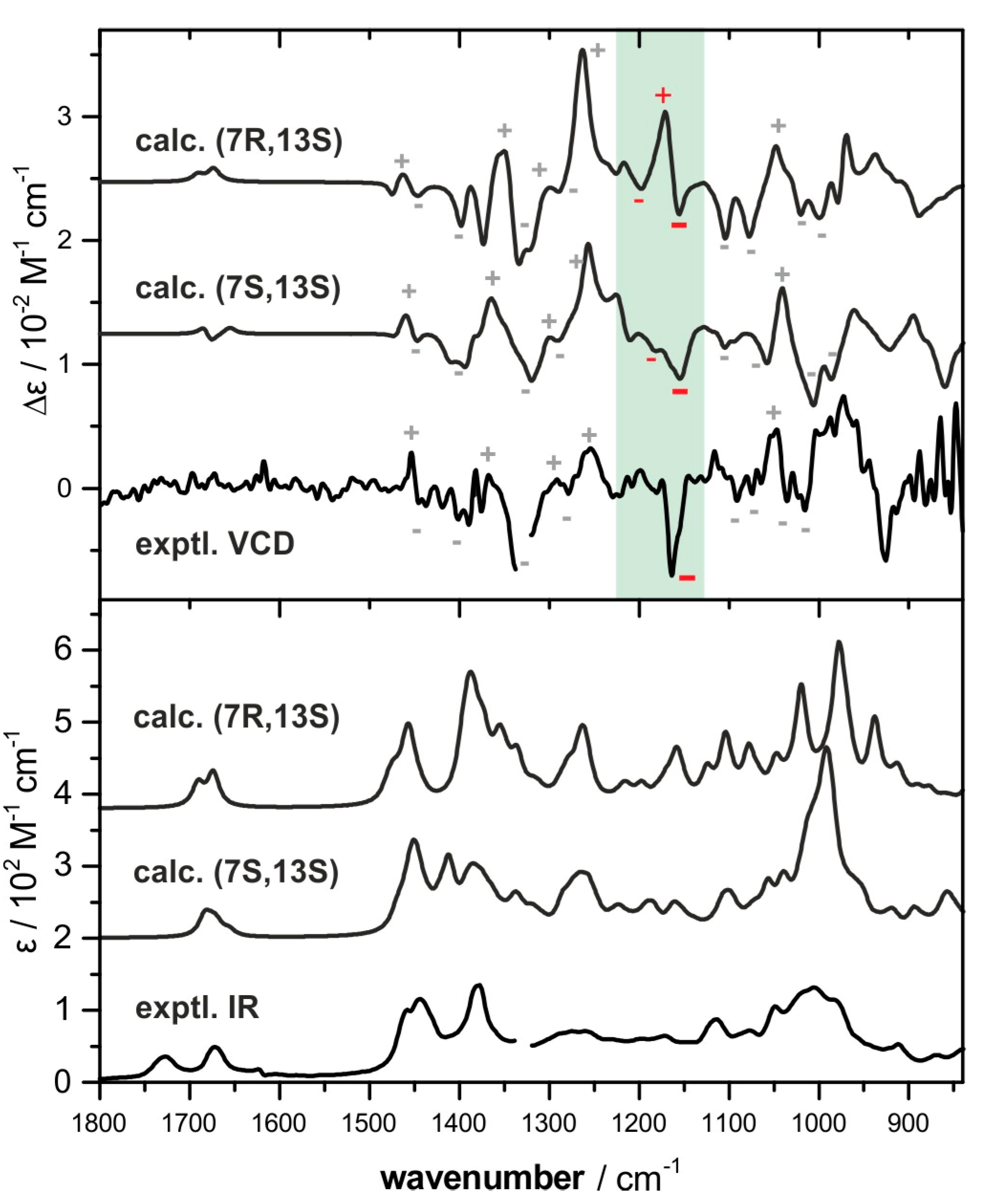
2.2. Absolute Configuration Assignment by VCD and 13C-NMR Spectroscopy
2.3. Bioactivity
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Algal Material
4.3. Extraction and Isolation
4.4. IR and VCD Spectroscopy
4.5. Computational Analysis
4.6. Antiprotozoal Activity
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jackson, M.B. Nature’s chemicals. The natural products that shaped our world. Ann. Bot. 2010, 106, vi–vii. [Google Scholar] [CrossRef]
- Salmon, M.; Laurendon, C.; Vardakou, M.; Cheema, J.; Defernez, M.; Green, S.; Faraldos, J.A.; O’Maille, P.E. Emergence of terpene cyclization in Artemisia annua. Nat. Commun. 2015, 6, 6143. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Morii, H. Recent advances in structural research on ether lipids from Archaea including comparative and physiological aspects. Biosci. Biotechnol. Biochem. 2005, 69, 2019–2034. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, D.J.; Croteau, R. Terpenoid metabolism. Plant Cell 1995, 7, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cal, K. Across skin barrier: Known methods, new performances. In Frontiers in Drug Design and Discovery; Rahman, A.U., Caldwell, G.W., Eds.; Bentham Science Publishers Ltd.: Sharjah, UAE, 2009; Volume 4, pp. 162–188. [Google Scholar]
- Maschek, J.A.; Baker, B.J. The chemistry of algal secondary metabolism. In Algal Chemical Ecology; Amsler, C.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1–24. [Google Scholar]
- Young, R.M.; Schoenrock, K.M.; von Salm, J.L.; Amsler, C.D.; Baker, B.J. Structure and function of macroalgal natural products. In Natural Products from Marine Algae: Methods and Protocols; Stengel, B.D., Connan, S., Eds.; Springer: New York, NY, USA, 2015; pp. 39–73. [Google Scholar]
- Amico, V. Marine brown algae of family Cystoseiraceae: Chemistry and chemotaxonomy. Phytochemistry 1995, 39, 1257–1279. [Google Scholar] [CrossRef]
- Muñoz, J.; Culioli, G.; Köck, M. Linear diterpenes from the marine brown alga Bifurcaria bifurcata: A chemical perspective. Phytochem. Rev. 2013, 12, 407–424. [Google Scholar] [CrossRef]
- Rindi, F.; Soler-Vila, A.; Guiry, M.D. Taxonomy of marine macroalgae used as sources of bioactive compounds. In Marine Bioactive Compounds: Sources, Characterization and Applications; Hayes, M., Ed.; Springer: Boston, MA, USA, 2012; pp. 1–53. [Google Scholar]
- Petrovic, A.G.; Berova, N.; Alonso-Gómez, J.L. Absolute configuration and conformational analysis of chiral compounds via experimental and theoretical prediction of chiroptical properties: ORD, ECD, and VCD. In Structure Elucidation in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 65–104. [Google Scholar]
- Blackmond, D.G. The origin of biological homochirality. Cold Spring Harb. Perspect. Biol. 2010, 2, a002147. [Google Scholar] [CrossRef] [PubMed]
- Batista, J.M., Jr.; da Silva Bolzani, V. Chapter 13: Determination of the absolute configuration of natural product molecules using Vibrational Circular Dichroism. In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 41, pp. 383–417. [Google Scholar]
- Batista, J.M., Jr.; Blanch, E.W.; da Silva Bolzani, V. Recent advances in the use of vibrational chiroptical spectroscopic methods for stereochemical characterization of natural products. Nat. Prod. Rep. 2015, 32, 1280–1302. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R. VCD Spectroscopy for Organic Chemists; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Sprenger, R.F.; Thomasi, S.S.; Ferreira, A.G.; Cass, Q.B.; Batista, J.M., Jr. Solution-state conformations of natural products from chiroptical spectroscopy: The case of isocorilagin. Org. Biomol. Chem. 2016, 14, 3369–3375. [Google Scholar] [CrossRef] [PubMed]
- Merten, C.; Li, F.; Bravo-Rodriguez, K.; Sanchez-Garcia, E.; Xu, Y.; Sander, W. Solvent-induced conformational changes in cyclic peptides: A vibrational circular dichroism study. Phys. Chem. Chem. Phys. 2014, 16, 5627–5633. [Google Scholar] [CrossRef] [PubMed]
- Kreienborg, N.M.; Pollok, C.H.; Merten, C. Towards an observation of active conformations in Asymmetric catalysis: Interaction-induced conformational preferences of a chiral thiourea model compound. Chem. Eur. J. 2016, 22, 12455–12463. [Google Scholar] [CrossRef] [PubMed]
- Osowski, T.; Golbek, J.; Merz, K.; Merten, C. Intermolecular interactions of a chiral amine borane adduct revealed by VCD spectroscopy. J. Phys. Chem. A 2016, 120, 4108–4115. [Google Scholar] [CrossRef] [PubMed]
- Merten, C.; Smyrniotopoulos, V.; Tasdemir, D. Assignment of absolute configurations of highly flexible linear diterpenes from the brown alga Bifurcaria bifurcata by VCD spectroscopy. Chem. Commun. 2015, 51, 16217–16220. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Spartan 14; Wavefunction Inc.: Irvine, CA, USA, 2014.
- The Goodman Group Highlights Page: Assignment of Stereochemistry and Structure Using NMR and DP4. Available online: http://www-jmg.ch.cam.ac.uk/tools/nmr/DP4/ (accessed on 15 May 2017).
- Merten, C.; Dirkmann, M.; Schulz, F. Stereochemical assignment of fusiccocadiene from NMR shielding constants and vibrational circular dichroism spectroscopy. Chirality 2017. [Google Scholar] [CrossRef] [PubMed]
- De Gussem, E.; Herrebout, W.; Specklin, S.; Meyer, C.; Cossy, J.; Bultinck, P. Strength by joining methods: Combining synthesis with NMR, IR, and Vibrational Circular Dichroism spectroscopy for the determination of the relative configuration in hemicalide. Chem. Eur. J. 2014, 20, 17385–17394. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Atay, I.; Kirmizibekmez, H.; Kaiser, M.; Akaydin, G.; Yesilada, E.; Tasdemir, D. Evaluation of in vitro antiprotozoal activity of Ajuga laxmannii and its secondary metabolites. Pharm. Biol. 2016, 54, 1808–1814. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, Mult (J in Hz) | COSY | NOESY | HMBC (1H → 13C) |
|---|---|---|---|---|---|
| 1 | 59.3, CH2 | 4.13, d (6.8) | 2, 20 | 20 | 2, 3 |
| 2 | 123.6, CH | 5.39, t (6.8) | 1, 20 | 4 | 1, 4, 20 |
| 3 | 139.5, C | ||||
| 4 | 39.9, CH2 | 2.00, t (7.0) | 5 | 2 | 2, 3, 5, 6, 20 |
| 5 | 22.0, CH2 | 1.45, m | 4 | 19 | 3, 4, 6, 7 |
| 6 | 41.7, CH2 | 1.40, m | 19 | 4, 5, 7, 8, 19 | |
| 7 | 72.7, C | ||||
| 8 | 41.3, CH2 | 1.49, t (7.8) | 9 | 10, 19 | 6, 7, 9, 10, 19 |
| 9 | 22.8, CH2 | 2.07, dt (7.0, 7.8) | 8, 10 | 19 | 7, 8, 10, 11 |
| 10 | 128.6, CH | 5.25, t (7.0) | 9, 18 | 8, 12 | 8, 9, 12, 13, 18 |
| 11 | 131.6, C | ||||
| 12 | 48.1, CH2 | 2.11, d (6.9) | 13 | 10, 14 | 10, 11, 13, 14, 18 |
| 13 | 65.9, CH | 4.41, dt (8.2, 6.9) | 12, 14 | 17, 18 | 11, 12, 14, 15 |
| 14 | 127.5, CH | 5.13, d (8.2) | 13, 16, 17 | 12, 16 | 12, 13, 16, 17 |
| 15 | 134.8, C | ||||
| 16 | 25.7, CH3 | 1.69, br.s | 14 | 14 | 14, 15, 17, |
| 17 | 18.2, CH3 | 1.66, br.s | 14 | 13 | 14, 15, 16, |
| 18 | 16.1, CH3 | 1.65, br.s | 10 | 13 | 10, 11, 12 |
| 19 | 26.7, CH3 | 1.14, s | 5, 6, 8, 9 | 6, 7, 8 | |
| 20 | 16.2, CH3 | 1.65, br.s | 2 | 1 | 2, 3, 4 |
| Compound | P. falciparum | T. b. rhodesiense | T. cruzi | L. donovani | Cytotoxicity |
|---|---|---|---|---|---|
| 1 | 0.65 | 11.8 | 47.8 | 18.8 | 56.6 |
| Reference | 0.05 a | 0.01 b | 0.59 c | 0.12 d | 0.004 e |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyrniotopoulos, V.; Merten, C.; Kaiser, M.; Tasdemir, D. Bifurcatriol, a New Antiprotozoal Acyclic Diterpene from the Brown Alga Bifurcaria bifurcata. Mar. Drugs 2017, 15, 245. https://doi.org/10.3390/md15080245
Smyrniotopoulos V, Merten C, Kaiser M, Tasdemir D. Bifurcatriol, a New Antiprotozoal Acyclic Diterpene from the Brown Alga Bifurcaria bifurcata. Marine Drugs. 2017; 15(8):245. https://doi.org/10.3390/md15080245
Chicago/Turabian StyleSmyrniotopoulos, Vangelis, Christian Merten, Marcel Kaiser, and Deniz Tasdemir. 2017. "Bifurcatriol, a New Antiprotozoal Acyclic Diterpene from the Brown Alga Bifurcaria bifurcata" Marine Drugs 15, no. 8: 245. https://doi.org/10.3390/md15080245
APA StyleSmyrniotopoulos, V., Merten, C., Kaiser, M., & Tasdemir, D. (2017). Bifurcatriol, a New Antiprotozoal Acyclic Diterpene from the Brown Alga Bifurcaria bifurcata. Marine Drugs, 15(8), 245. https://doi.org/10.3390/md15080245