
Asperentin B, a New Inhibitor of the Protein Tyrosine Phosphatase 1B



Abstract
:1. Introduction
2. Results
2.1. Origin and Classification of the Producer Strain LF660
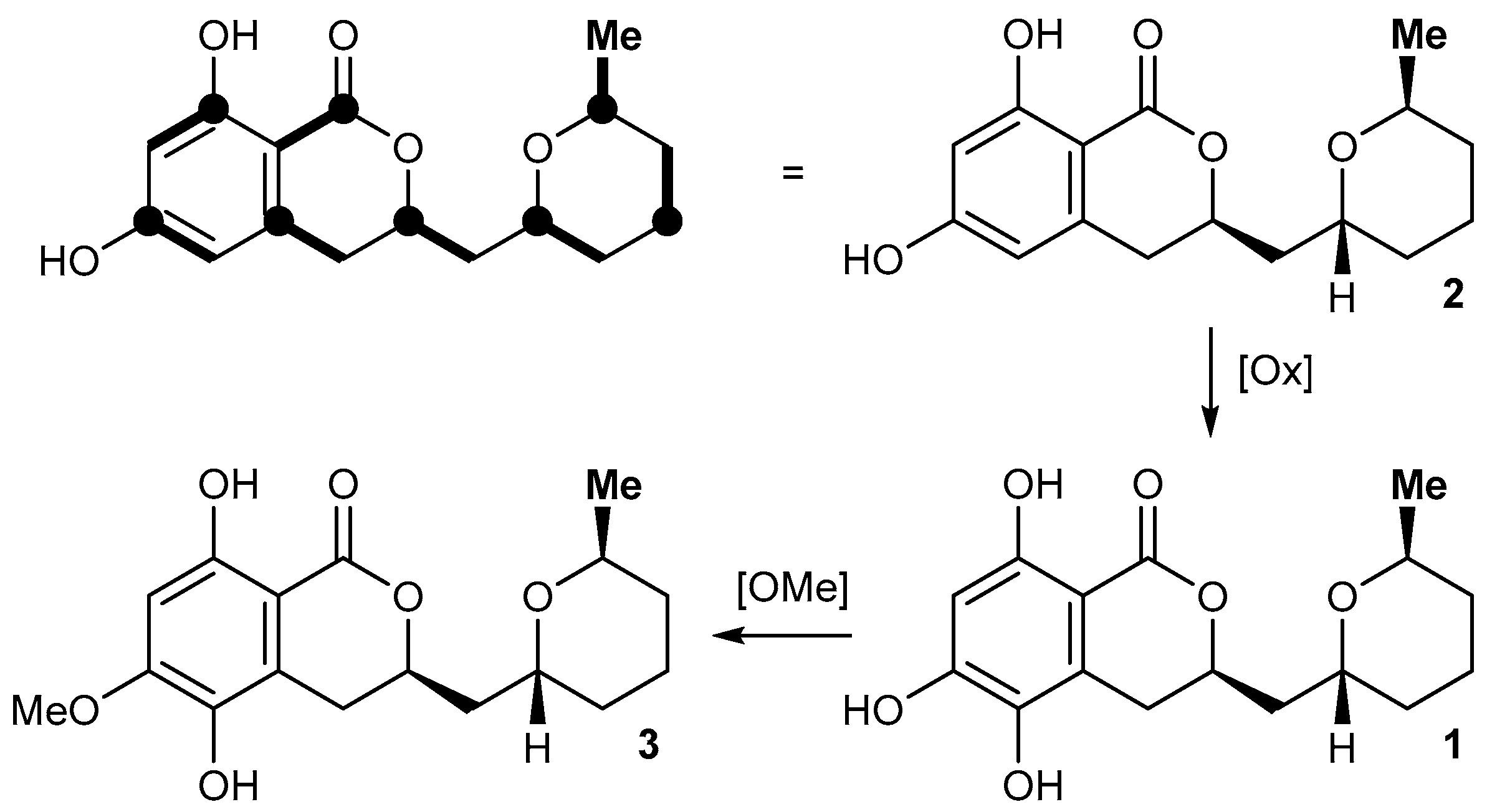
2.2. Isolation and Structure Elucidation of Asperentin B (1) by Aspergillus sydowii LF660
2.3. Biological Activities
3. Discussion
4. Materials and Methods
4.1. Isolation, Cultivation, Storage, and Classification of the Producer Strain LF660
4.2. Fermentation and Production of Extracts for the Purification of Compound 1
4.3. Isolation of Compound 1
4.4. Structure Elucidation of Compound 1
4.5. Biological Activities Assays
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Le Calvez, T.; Burgaud, G.; Mahé, S.; Barbier, G.; Vandenkoornhuyse, P. Fungal diversity in deep-sea hydrothermal ecosystems. Appl. Environ. Micobiol. 2009, 75, 6415–6421. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Pang, K.-L.; Luo, Z.-H. High fungal diversity and abundance recovered in the deep-sea sediments of the Pacific Ocean. Microb. Ecol. 2014, 68, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Natural products from marine fungi—Still an underrepresented resource. Mar. Drugs 2016, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Xue, Y.-R.; Liu, C.-H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-H.; Cai, S.-X.; Tian, L.; Lin, Z.-J.; Zhu, T.-J.; Fang, Y.-C.; Liu, P.-P.; Gu, Q.-Q.; Zhu, W.-M. Two new metabolites with cytotoxicities from deep-sea fungus, Aspergillus sydowi YH11–2. Arch. Pharmacal Res. 2007, 30, 1051–1054. [Google Scholar]
- Cai, S.; Zhu, T.; Du, L.; Zhao, B.; Li, D.; Gu, Q. Sterigmatocystins from the deep-sea-derived fungus Aspergillus versicolor. J. Antibiot. 2011, 64, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Cai, S.; Zhu, T.; Gu, Q.; Li, D.; Luan, Y. Secondary metabolites of a deep sea derived fungus Aspergillus versicolor CXCTD-06-6a and their bioactivity. J. Ocean Univ. China 2014, 13, 691–695. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed]
- Wiese, J.; Imhoff, J.F.; Gulder, T.A.M.; Labes, A.; Schmaljohann, R. Marine fungi as producers of benzocoumarins, a new class of inhibitors of glycogen-synthase-kinase 3β. Mar. Drugs 2016, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.M.; Van Walbeek, W.; MacLean, W.M. Cladosporin, a new antifungal metabolite from Cladosporium cladosporioides. J. Antibiot. 1971, 24, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Guo, K.; Li, X.Y.; Zheng, X.Y.; Kong, X.J.; Zheng, Z.H.; Xu, Q.Y.; Deng, X. Three new asperentin derivatives from the algicolous fungus Aspergillus sp. F00785. Mar. Drugs 2014, 12, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- McCain, D.F.; Wu, L.; Nickel, P.; Kassack, M.U.; Kreimeyer, A.; Gagliardi, A.; Collins, D.C.; Zhang, Z.Y. Suramin derivatives as inhibitors and activators of protein-tyrosine phosphatases. J. Biol. Chem. 2004, 279, 14713–14725. [Google Scholar] [CrossRef] [PubMed]
- Anke, H.; Zähner, H. Metabolic products of microorganisms. 170. On the antibiotic activity of cladosporin. Arch. Microbiol. 1978, 116, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Jacyno, J.M.; Harwood, J.S.; Cutler, H.G.; Lee, M.K. Isocladosporin, a biologically active isomer of cladosporin from Cladosporium cladosporioides. J. Nat. Prod. 1993, 56, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part 1: Diagnosis and Classification of Diabetes Mellitus; WHO: Geneva, Switzerland, 1999. [Google Scholar]
- World Health Organization. Global Report on Diabetes; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Qian, S.; Zhang, M.; He, Y.; Wang, W.; Liu, S. Recent advances in the development of protein tyrosine phosphatase 1B inhibitors for type 2 diabetes. Future Med. Chem. 2016, 8, 1239–1258. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Uetani, N.; Miranda-Saavedra, D.; Tremblay, M.L. PTP1B: A simple enzyme for a complex world. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Li, W.; Higai, K.; Koike, K. Canthinone alkaloids are novel protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1979–1981. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Jiang, B.; Wu, N.; Wang, S.Y.; Shi, D.Y. Small molecules as potent protein tyrosine phosphatase 1B (PTP1B) inhibitors documented in patents from 2009 to 2013. Mini Rev. Med. Chem. 2015, 15, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Ruberto, I.; Szoor, B.; Clark, R.; Matthews, K.R. Investigating mammalian tyrosine phosphatase inhibitors as potential ‘piggyback’ leads to target Trypanosoma brucei transmission. Chem. Biol. Drug Des. 2013, 81, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Wickerham, L.J. Taxonomy of Yeasts; U.S. Department of Agriculture: Washington, DC, USA, 1951; pp. 1–56.
- Wiese, J.; Ohlendorf, B.; Blümel, M.; Schmaljohann, R.; Imhoff, J.F. Phylogenetic identification of fungi isolated from the marine sponge Tethya aurantium and identification of their secondary metabolites. Mar. Drugs 2011, 9, 561–585. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tatsuova, T.; Madden, T.L. Blast 2 sequences—A new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999, 174, 247–250. [Google Scholar] [CrossRef]
- Schneemann, I.; Kajahn, I.; Ohlendorf, B.; Zinecker, H.; Erhard, A.; Nagel, K.; Wiese, J.; Imhoff, J.F. Mayamycin, a cytotoxic polyketide from a marine Streptomyces strain isolated from the marine sponge Halichondria panicea. J. Nat. Prod. 2010, 73, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Ohlendorf, B.; Schulz, D.; Erhard, A.; Nagel, K.; Imhoff, J.F. Geranylphenazinediol, an acetylcholinesterase inhibitor produced by a Streptomyces species. J. Nat. Prod. 2012, 75, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Nagel, K.; Schneemann, I.; Kajahn, I.; Labes, A.; Wiese, J.; Imhoff, J.F. Proposed beneficial effects of 2,4-diacetylphloroglucinol-producing pseudomonads on the marine alga Saccharina latissima. Aquat. Microb. Ecol. 2012, 67, 239–249. [Google Scholar] [CrossRef]
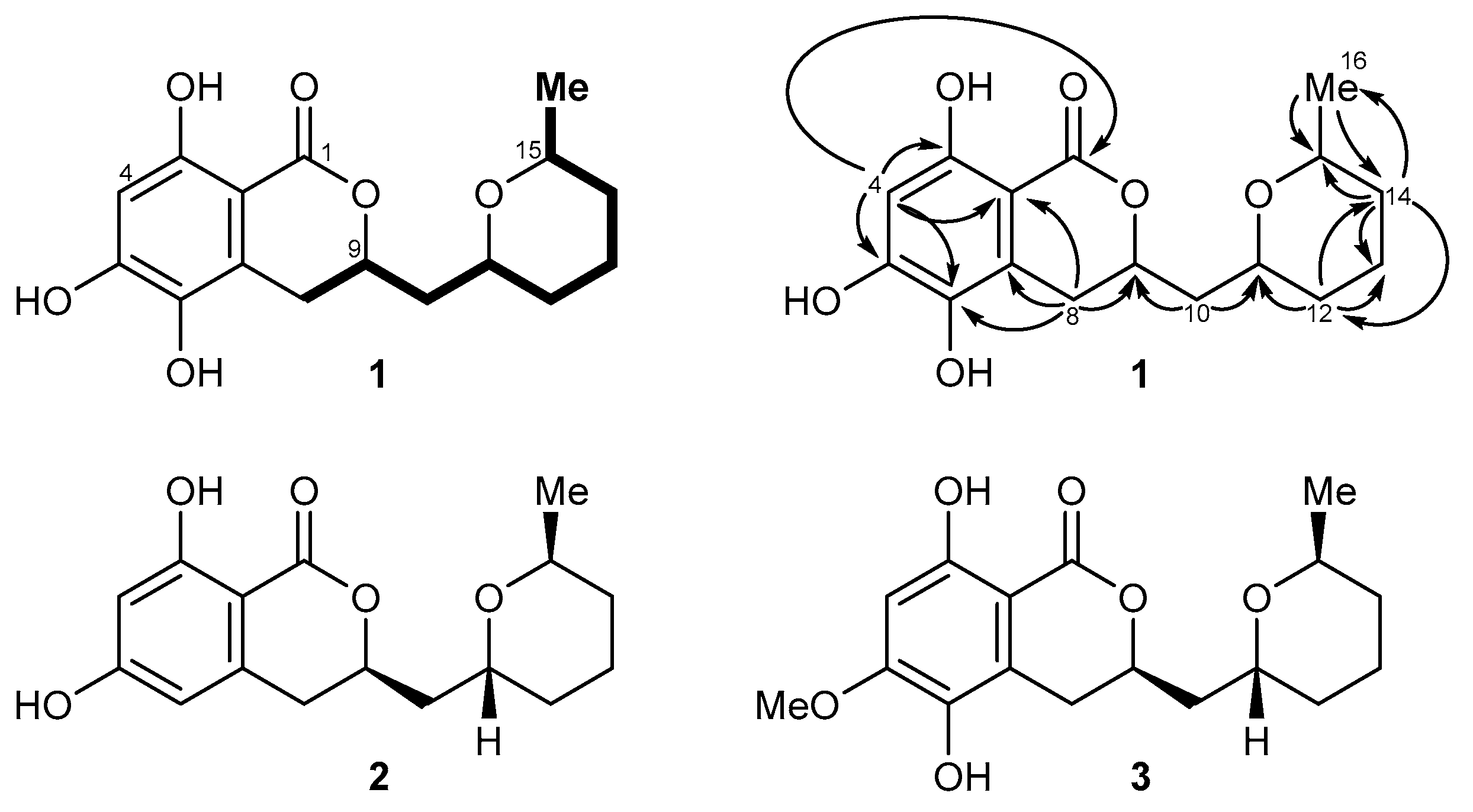

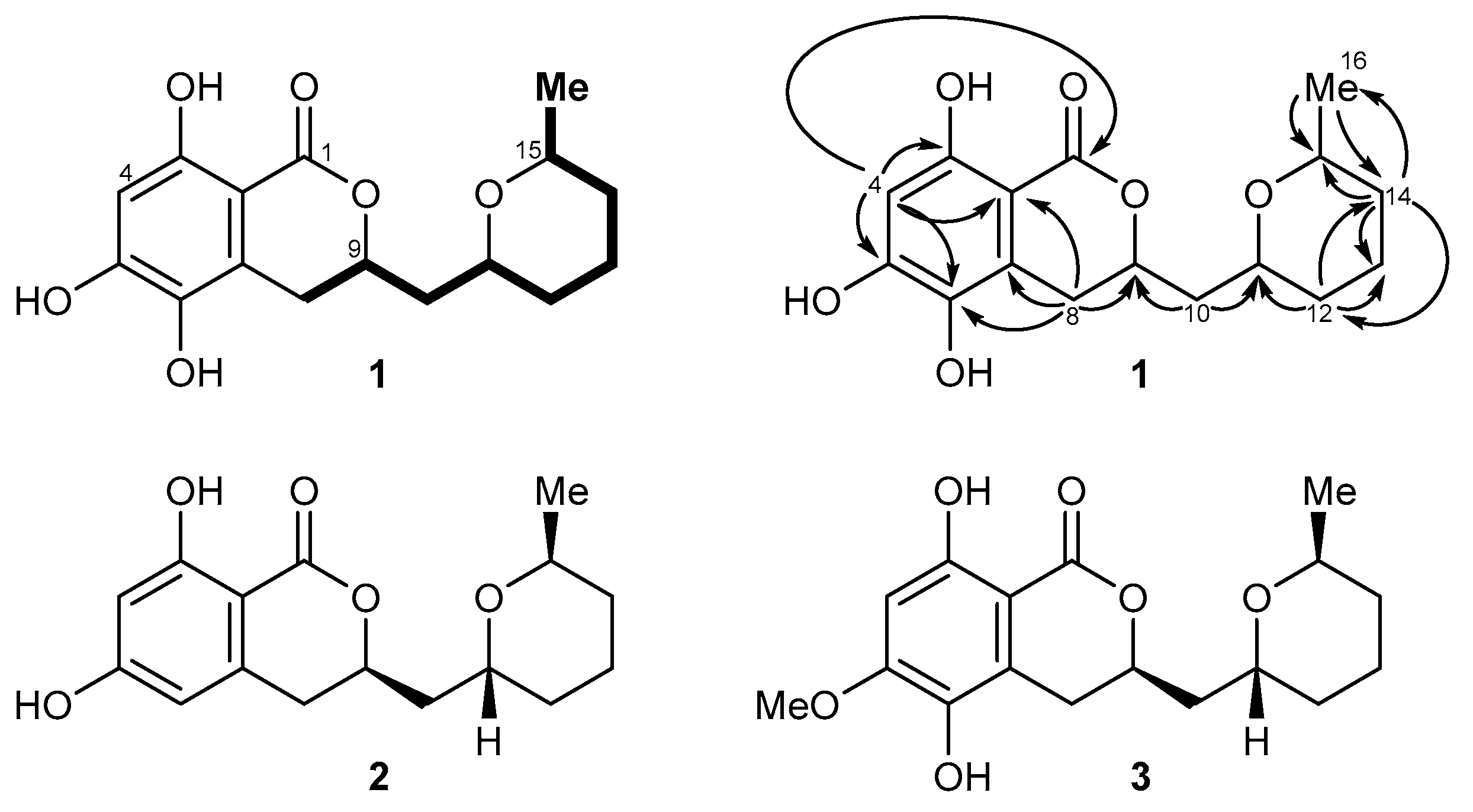{kind=link}
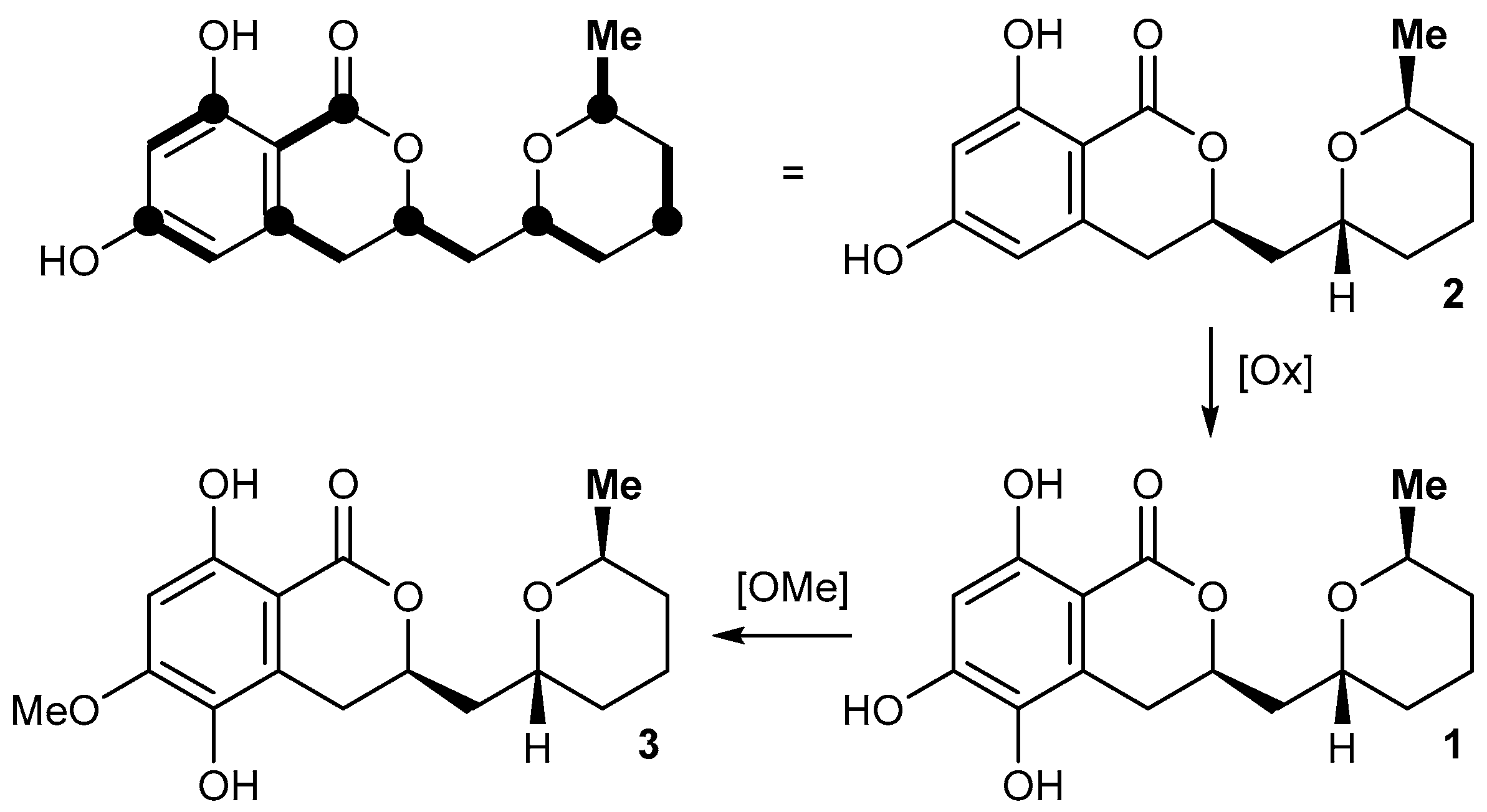{kind=link}
| Signal | 1H | I, Mult, J | 13C | COSY | HMBC |
|---|---|---|---|---|---|
| 1 | - | 171.9 | |||
| 2 | - | 100.1 | |||
| 3 | - | 158.1 | |||
| 4 | 6.26 | 1H, s | 101.9 | - | 1, 2, 3, 5, 6 |
| 5 | - | 155.9 | |||
| 6 | - | 135.9 | |||
| 7 | - | 126.0 | |||
| 8a | 3.18 | 1H, dd, 16.8, 3.4 | 28.6 | 8b, 9 | 2, 6, 7 |
| 8b | 2.66 | 1H, dd, 16.8, 11.3 | 28.6 | 8a, 9 | 2, 6, 7, 9 |
| 9 | 4.62 | 1H, dddd, 11.3, 9.1, 3.4, 3.4 | 77.7 | 8a/b, 10a/b | - |
| 10a | 2.15 | 1H, ddd, 14.7, 10.4, 3.4 | 39.4 | 9, 10b, 11 | 11 |
| 10b | 1.80 | 1H, ddd, 14.7, 9.1, 3.4 | 39.4 | 9, 10a, 11 | 9 |
| 11 | 4.15 | 1H, m | 68.4 | 10a/b, 12a/b | - |
| 12a | 1.72 | 1H, m | 31.5 | 11, 12b, 13 | 11, 13 |
| 12b | 1.41 | 1H, m | 31.5 | 11, 12a, 13 | 11, 13, 14 |
| 13 | 1.70 | 2H, m | 19.3 | 12, 14 | * |
| 14a | 1.70 | 1H, m | 32.8 | 13, 15 | * |
| 14b | 1.33 | 1H, m | 32.8 | 13, 15 | 12, 13, 15, 16 |
| 15 | 3.92 | 1H, m | 68.4 | 16 | 13 |
| 16 | 1.19 | 3H, d, 6.5 | 20.0 | 15 | 14, 15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiese, J.; Aldemir, H.; Schmaljohann, R.; Gulder, T.A.M.; Imhoff, J.F. Asperentin B, a New Inhibitor of the Protein Tyrosine Phosphatase 1B. Mar. Drugs 2017, 15, 191. https://doi.org/10.3390/md15060191
Wiese J, Aldemir H, Schmaljohann R, Gulder TAM, Imhoff JF. Asperentin B, a New Inhibitor of the Protein Tyrosine Phosphatase 1B. Marine Drugs. 2017; 15(6):191. https://doi.org/10.3390/md15060191
Chicago/Turabian StyleWiese, Jutta, Hülya Aldemir, Rolf Schmaljohann, Tobias A. M. Gulder, and Johannes F. Imhoff. 2017. "Asperentin B, a New Inhibitor of the Protein Tyrosine Phosphatase 1B" Marine Drugs 15, no. 6: 191. https://doi.org/10.3390/md15060191
APA StyleWiese, J., Aldemir, H., Schmaljohann, R., Gulder, T. A. M., & Imhoff, J. F. (2017). Asperentin B, a New Inhibitor of the Protein Tyrosine Phosphatase 1B. Marine Drugs, 15(6), 191. https://doi.org/10.3390/md15060191