
Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
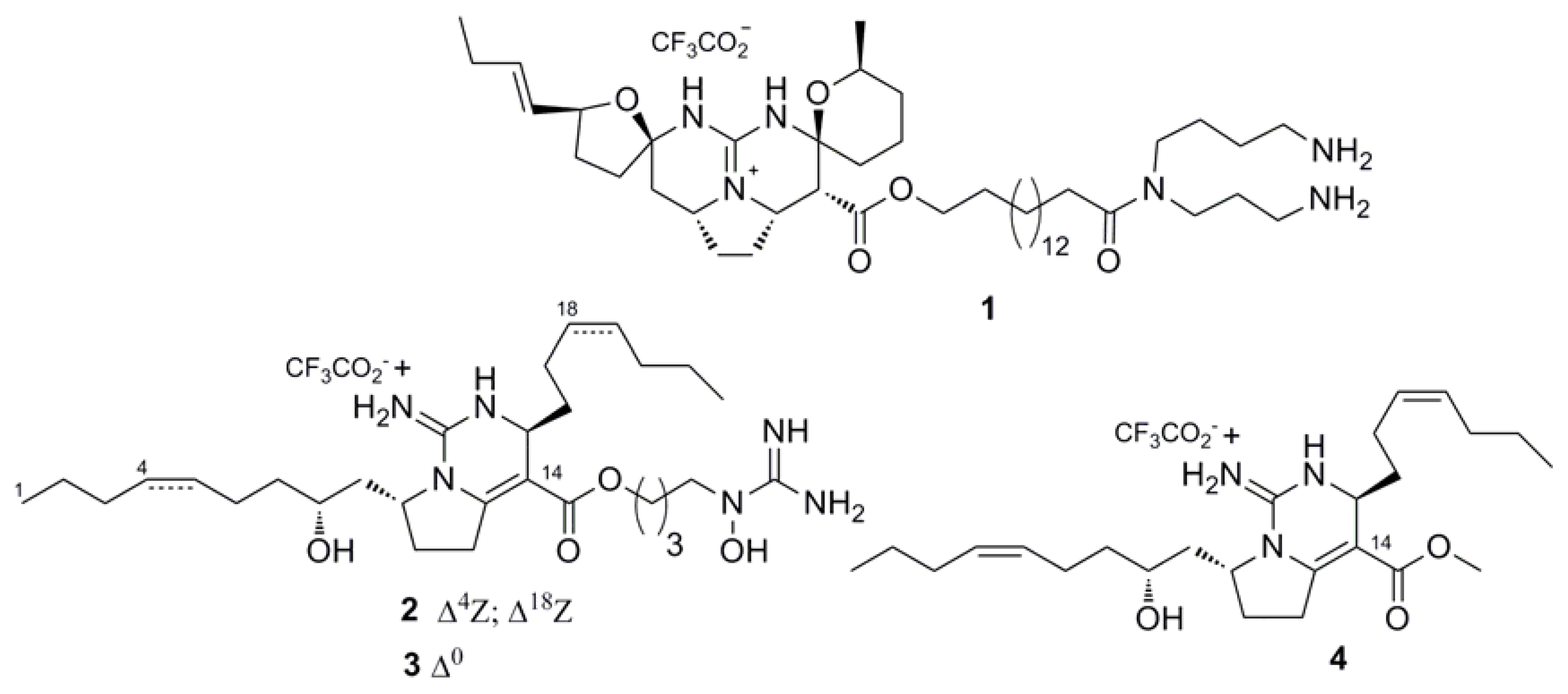
2.1. Isolation and Stucture of Individual Compounds
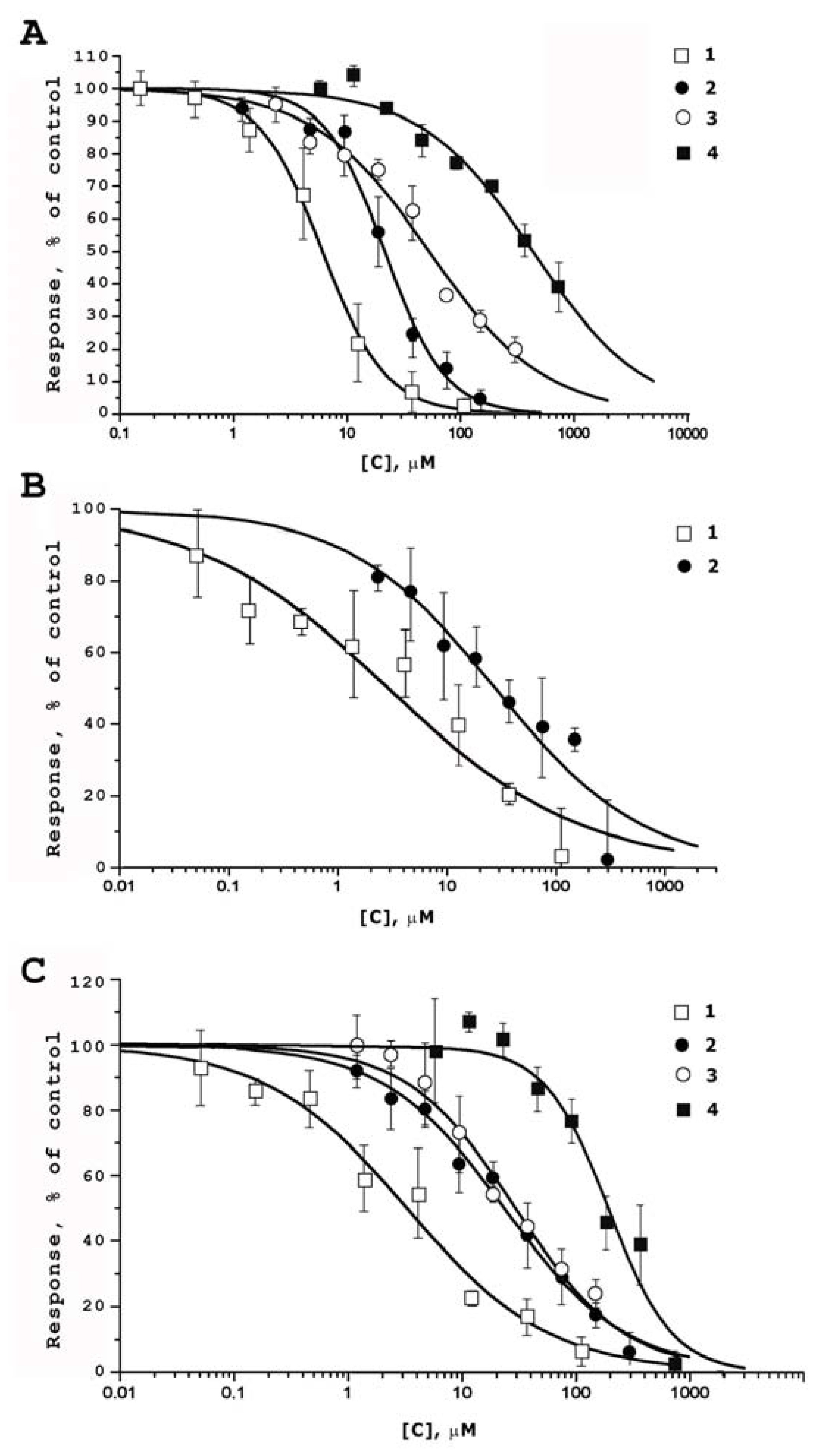
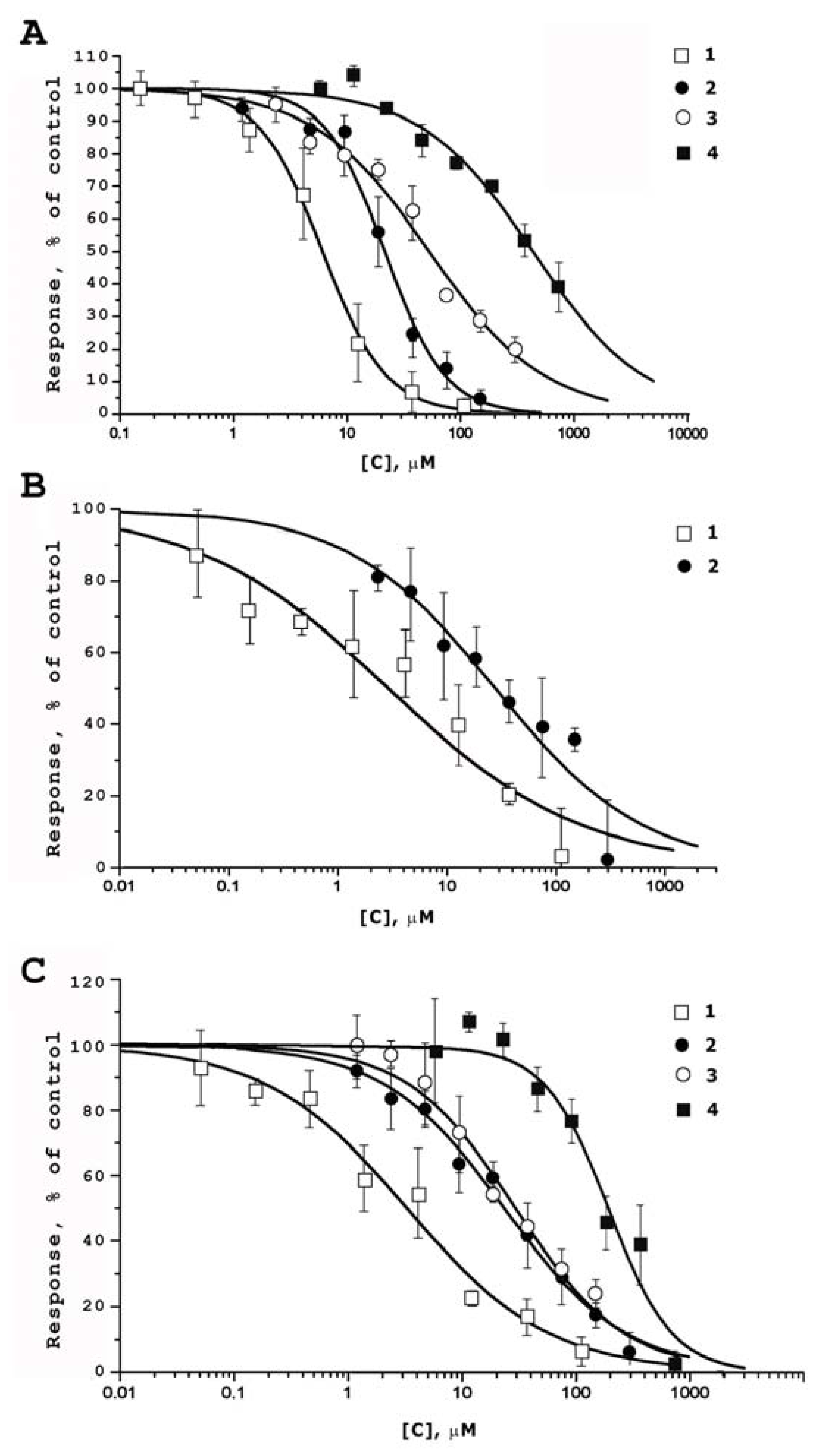
2.2. Functional Activity Study on TRP Receptors
3. Materials and Methods
3.1. General
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Inhibitory Activities on TRPV1, TRPV2, TRPV3 and TRPA1
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Romminger, S. The chemistry and biology of guanidine natural products. Nat. Prod. Rep. 2016, 33, 456–490. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.S.; Stonik, V.A. Monanchocidins B-E: Polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Lee, H.S.; Stonik, V.A. Monanchomycalins A and B, unusual guanidine alkaloids from the sponge Monanchora pulchra. Tetrahedron Lett. 2012, 53, 4228–4231. [Google Scholar] [CrossRef]
- Tabakmaher, K.M.; Denisenko, V.A.; Guzii, A.G.; Dmitrenok, P.S.; Lee, H.S.; Makarieva, T.N. Monanchomycalin C, a new pentacyclic guanidine alkaloid from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2013, 8, 1399–1402. [Google Scholar]
- Tabakmakher, K.M.; Makarieva, T.N.; Denisenko, V.A.; Guzii, A.G.; Dmitrenok, P.S.; Kuzmich, A.S.; Stonik, V.A. Normonanchocidins A, B and D, new pentacyclic guanidine alkaloids from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2015, 10, 913–916. [Google Scholar] [PubMed]
- Makarieva, T.N.; Ogurtsova, E.K.; Denisenko, V.A.; Dmitrenok, P.S.; Tabakmakher, K.M.; Guzii, A.G.; Pislyagin, E.A.; Es’kov, A.A.; Kozhemyako, V.B.; Aminin, D.L.; et al. Urupocidin A: A new, inducing iNOS expression bicyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2014, 16, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Denisenko, V.A.; Dmitrenok, P.S.; Ogurtsova, E.K.; Antonov, A.S.; et al. Pulchranin A, isolated from the Far-Eastern marine sponge, Monanchora pulchra: The first marine non-peptide inhibitor of TRPV-1 channels. Tetrahedron Lett. 2013, 54, 1247–1250. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Ogurtsova, E.K.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Lee, H.S.; et al. Pulchranins B and C, new acyclic guanidine alkaloids from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2013, 8, 1229–1232. [Google Scholar] [PubMed]
- Dyshlovoy, S.A.; Hauschild, J.; Amann, K.; Tabakmakher, K.M.; Venz, S.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; Fedorov, S.N.; et al. Marine alkaloid Monanchocidin A overcomes drug resistance by induction of autophagy and lysosomal membrane permeabilization. Oncotarget 2015, 6, 17328–17341. [Google Scholar] [CrossRef] [PubMed]
- Ogurtsova, E.K.; Makarieva, T.N.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Denisenko, V.A.; Dmitrenok, P.S.; Lee, Y.J.; Grishin, E.V.; Stonik, V.A. New derivatives of natural acyclic guanidine alkaloids with TRPV receptor-regulating properties. Nat. Prod. Commun. 2015, 10, 1171–1173. [Google Scholar] [PubMed]
- Kudryavtsev, D.; Makarieva, T.; Utkina, N.; Santalova, E.; Kryukova, E.; Methfessel, C.; Tsetlin, V.; Stonik, V.; Kasheverov, I. Marine natural products acting on the acetylcholine-binding protein and nicotinic receptors: From computer modeling to binding studies and electrophysiology. Mar. Drugs 2014, 12, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Owsianik, G.; Nilius, B.; Voets, T. TRP channels. Compr. Physiol. 2012, 2, 563–608. [Google Scholar] [PubMed]
- Smani, T.; Shapovalov, G.; Skryma, R.; Prevarskaya, N.; Rosado, J.A. Functional and physiopathological implications of TRP channels. Biochim. Biophys. Acta 2015, 1853, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Szallasi, A. Transient receptor potential (TRP) channels: A clinical perspective. Br. J. Pharmacol. 2014, 171, 2474–2507. [Google Scholar] [CrossRef] [PubMed]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Sensory TRP channels: The key transducers of nociception and pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 73–118. [Google Scholar] [PubMed]
- Brederson, J.D.; Kym, P.R.; Szallasi, A. Targeting TRP channels for pain relief. Eur. J. Pharmacol. 2013, 716, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Colsoul, B.; Nilius, B.; Vennekens, R. Transient receptor potential (TRP) cation channels in diabetes. Curr. Top. Med. Chem. 2013, 13, 258–269. [Google Scholar] [CrossRef] [PubMed]
- De Logu, F.; Patacchini, R.; Fontana, G.; Geppetti, P. TRP functions in the broncho-pulmonary system. Semin. Immunopathol. 2016, 38, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Dhennin-Duthille, I.; Ay, A.S.; Rybarczyk, P.; Korichneva, I.; Ouadid-Ahidouch, H. New insights into pharmacological tools to TR(i)P cancer up. Br. J. Pharmacol. 2014, 171, 2582–2592. [Google Scholar] [CrossRef] [PubMed]
- Kojima, I.; Nagasawa, M. TRPV2. Handb. Exp. Pharmacol. 2014, 222, 247–272. [Google Scholar] [PubMed]
- Appendino, G.; Minassi, A.; Pagani, A.; Ech-Chadad, A. The role of natural products in the ligand deorphanization of TRP channels. Curr. Pharm. Des. 2008, 14, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- Heding, A.; Elling, C.E.; Schwartz, T.W. Novel method for the study of receptor Ca2+ signalling exemplified by the NK1 receptor. J. Recept. Signal Transduct. Res. 2002, 22, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Ogurtsova, E.; Makarieva, T.; Guzii, A.; Dmitrenok, P.; Denisenko, V.; Krasokhin, V.; Korolkova, Y.; Andreev, Y.; Mosharova, I.; Grishin, E. Inhibitory Activity on TRP Receptors of Pentacyclic Alkaloids from the Sponge Haliclona (Gellius) sp. Chem. Nat. Compd. 2015, 51, 194–196. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Gracheva, E.O.; Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl. Acad. Sci. USA 2011, 108, E1184–E1191. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.K.; Zhu, M.X. 2-Aminoethoxydiphenyl borate as a common activator of TRPV1, TRPV2, and TRPV3 channels. Handb. Exp. Pharmacol. 2007, 179, 173–187. [Google Scholar]
- Alpizar, Y.A.; Boonen, B.; Gees, M.; Sanchez, A.; Nilius, B.; Voets, T.; Talavera, K. Allyl isothiocyanate sensitizes TRPV1 to heat stimulation. Pflugers Arch. 2014, 466, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Iwasaki, Y.; Narukawa, M.; Iitsuka, Y.; Fukao, T.; Seki, T.; Ariga, T.; Watanabe, T. Diallyl sulfides in garlic activate both TRPA1 and TRPV1. Biochem. Biophys. Res. Commun. 2009, 382, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.; Lattrell, A.; Kronewald, S.; Niedermirtl, F.; Nau, C. Activation of TRPA1 by membrane permeable local anesthetics. Mol. Pain 2011, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Mihara, S.; Shibamoto, T. The role of flavor and fragrance chemicals in TRPA1 (transient receptor potential cation channel, member A1) activity associated with allergies. Allergy Asthma Clin. Immunol. 2015, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.F.; Kort, M.E.; Huth, J.R.; Sun, C.; Miesbauer, L.J.; Cassar, S.C.; Neelands, T.; Scott, V.E.; Moreland, R.B.; et al. Molecular determinants of species-specific activation or blockade of TRPA1 channels. J. Neurosci. 2008, 28, 5063–5071. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Compounds | EC50 (µM) | |||
|---|---|---|---|---|
| rTRPV1 | mTRPV2 | hTRPV3 | rTRPA1 | |
| 1 | 6.02 ± 0.36 | 2.84 ± 1.01 | 3.25 ± 0.60 | not active |
| 2 | 21.47 ± 2.51 | 28.06 ± 6.65 | 23.55 ± 2.03 | not active |
| 3 | 52.82 ± 5.31 | nd 1 | 29.19 ± 3.18 | not active |
| 4 | 435.07 ± 49.92 | nd 1 | 193.33 ± 25.47 | not active |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korolkova, Y.; Makarieva, T.; Tabakmakher, K.; Shubina, L.; Kudryashova, E.; Andreev, Y.; Mosharova, I.; Lee, H.-S.; Lee, Y.-J.; Kozlov, S. Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors. Mar. Drugs 2017, 15, 87. https://doi.org/10.3390/md15040087
Korolkova Y, Makarieva T, Tabakmakher K, Shubina L, Kudryashova E, Andreev Y, Mosharova I, Lee H-S, Lee Y-J, Kozlov S. Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors. Marine Drugs. 2017; 15(4):87. https://doi.org/10.3390/md15040087
Chicago/Turabian StyleKorolkova, Yuliya, Tatyana Makarieva, Kseniya Tabakmakher, Larisa Shubina, Ekaterina Kudryashova, Yaroslav Andreev, Irina Mosharova, Hyi-Seung Lee, Yeon-Ju Lee, and Sergey Kozlov. 2017. "Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors" Marine Drugs 15, no. 4: 87. https://doi.org/10.3390/md15040087
APA StyleKorolkova, Y., Makarieva, T., Tabakmakher, K., Shubina, L., Kudryashova, E., Andreev, Y., Mosharova, I., Lee, H.-S., Lee, Y.-J., & Kozlov, S. (2017). Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors. Marine Drugs, 15(4), 87. https://doi.org/10.3390/md15040087