Angiotensin I-Converting Enzyme (ACE) Inhibitory Activity, Antioxidant Properties, Phenolic Content and Amino Acid Profiles of Fucus spiralis L. Protein Hydrolysate Fractions



Abstract
:1. Introduction
2. Results and Discussion
2.1. F. spiralis Protein Content and In Vitro Protein Digestibility Evaluation
2.2. Preparation of F. spiralis Protein Hydrolysate (FSPH) and Its Fractionation by Ultrafiltration
2.3. Protein and Peptide Contents of FSPH Fractions
2.4. Amino Acids Composition of FSPH Fractions
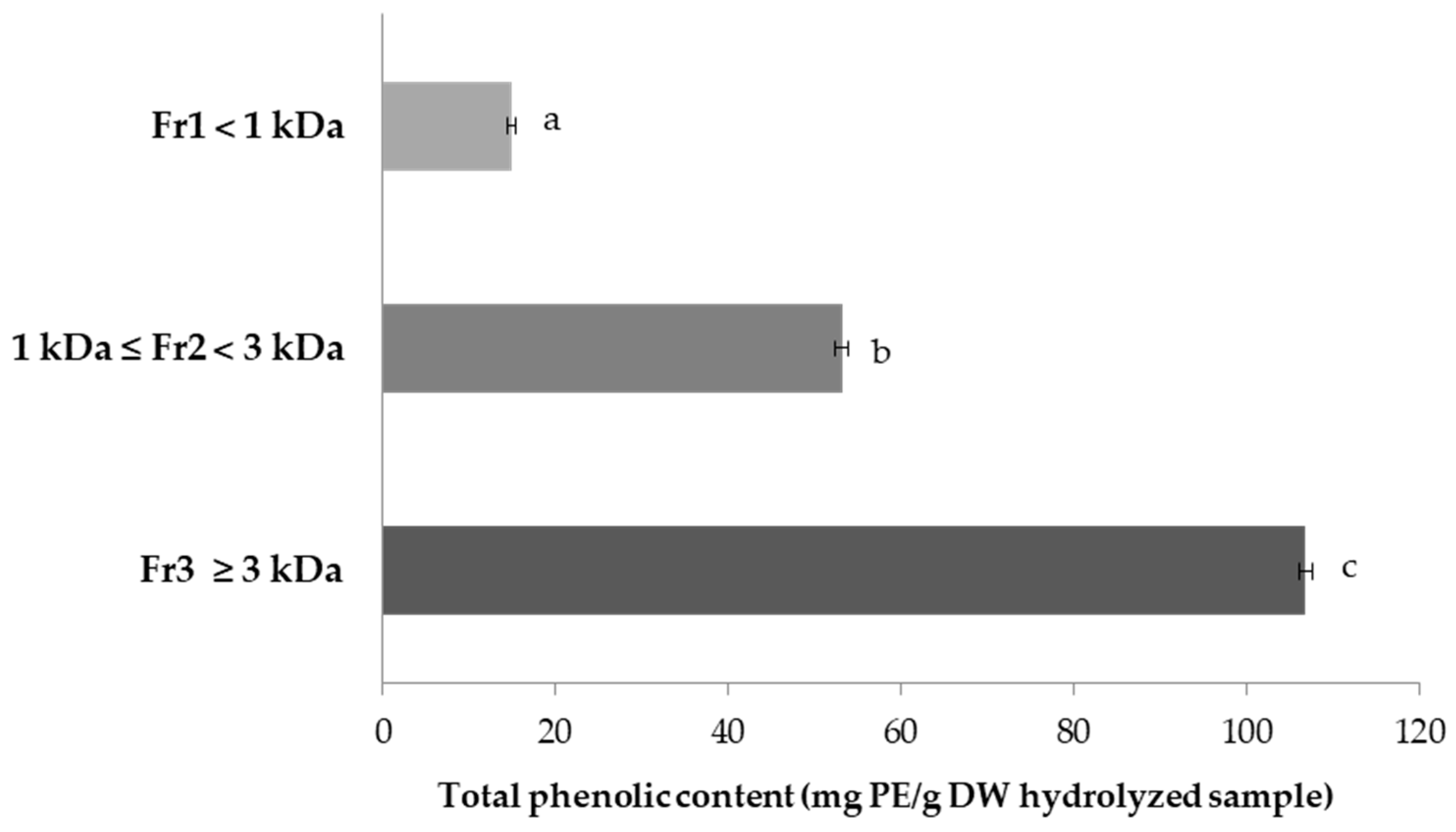
2.5. Total Phenolic Content (TPC) of FSPH Fractions
2.6. ACE-Inhibitory Activity of FSPH Fractions
2.7. Antioxidant Activities of FSPH Fractions
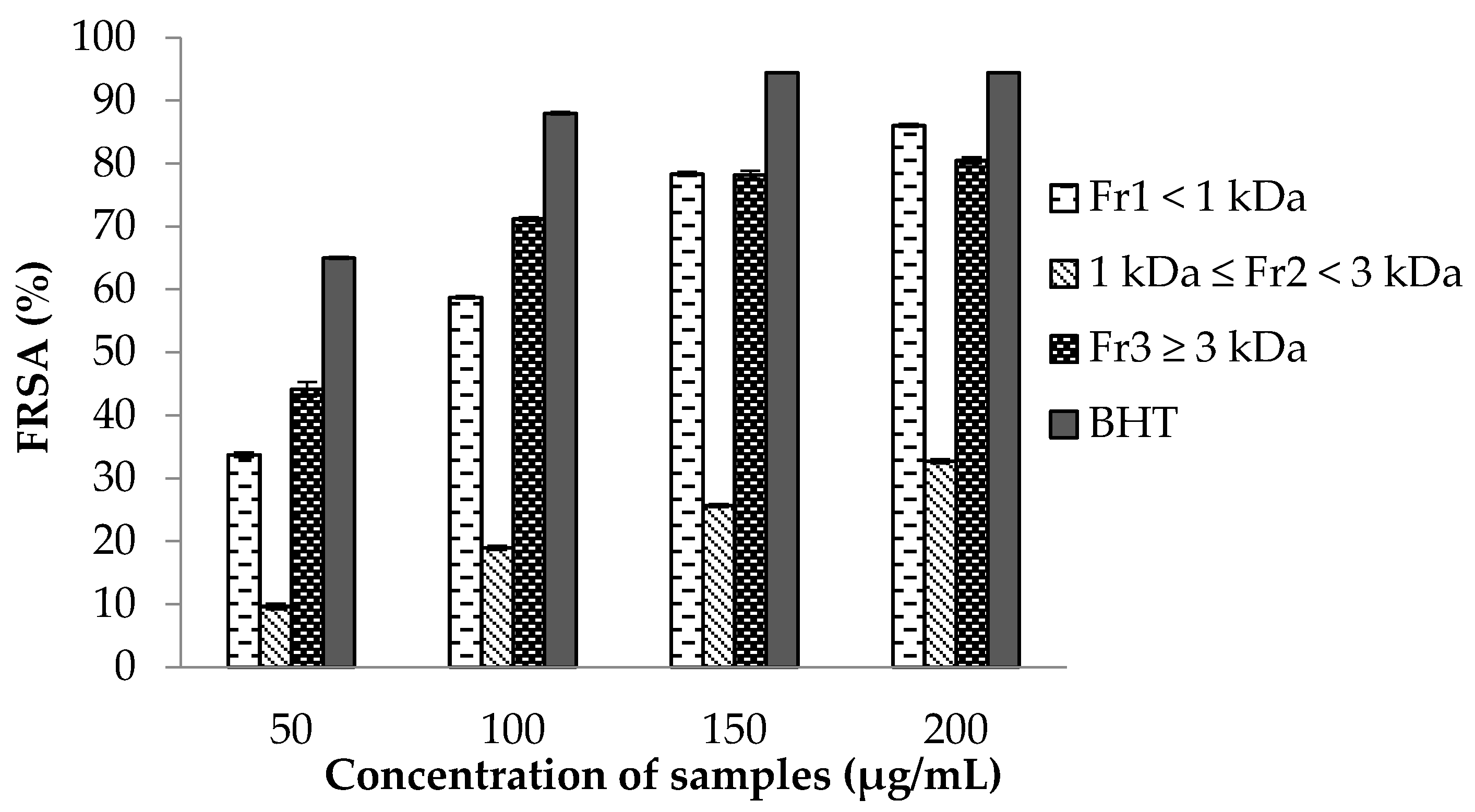
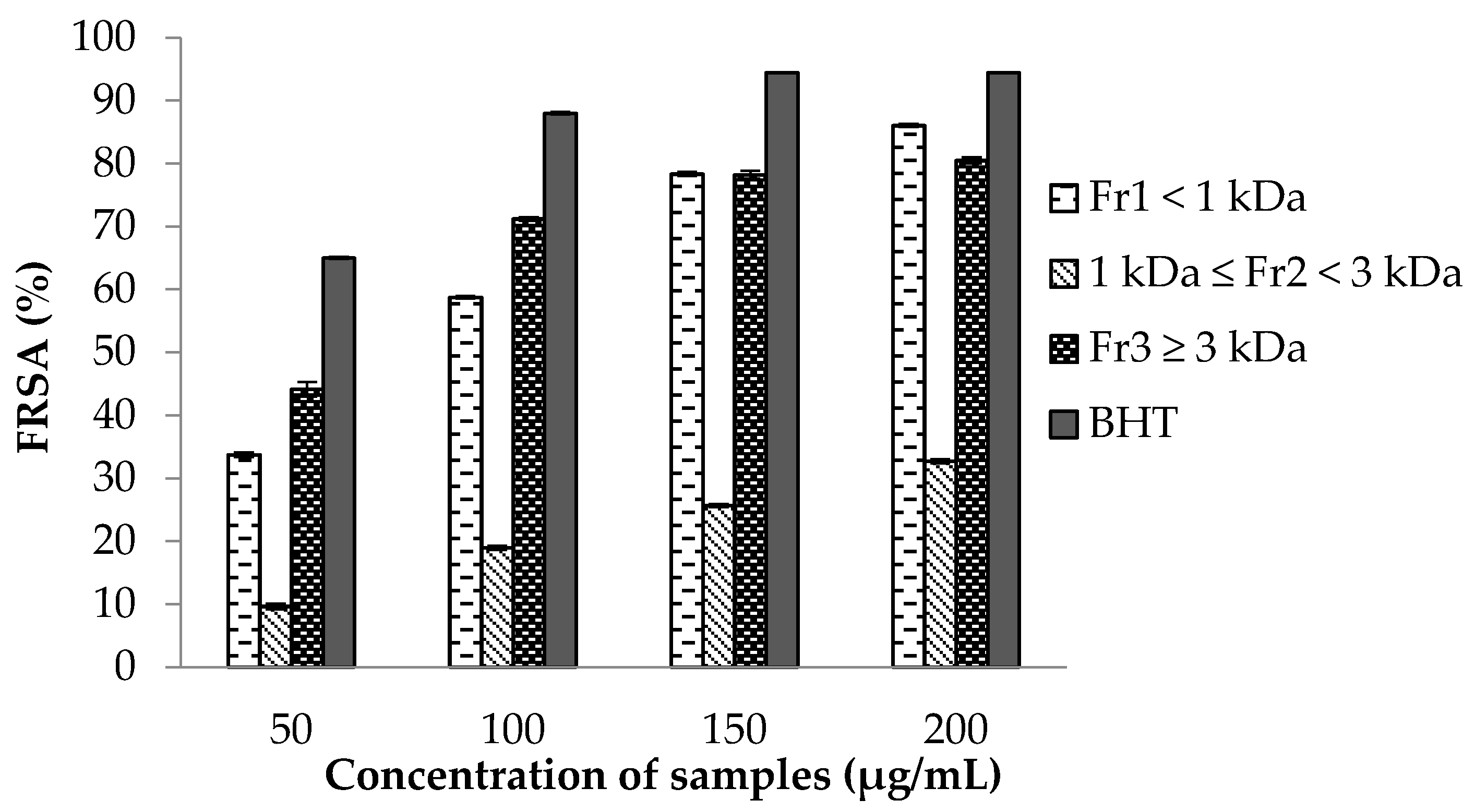
2.7.1. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) Free Radical Scavenging Activity (FRSA) Assay
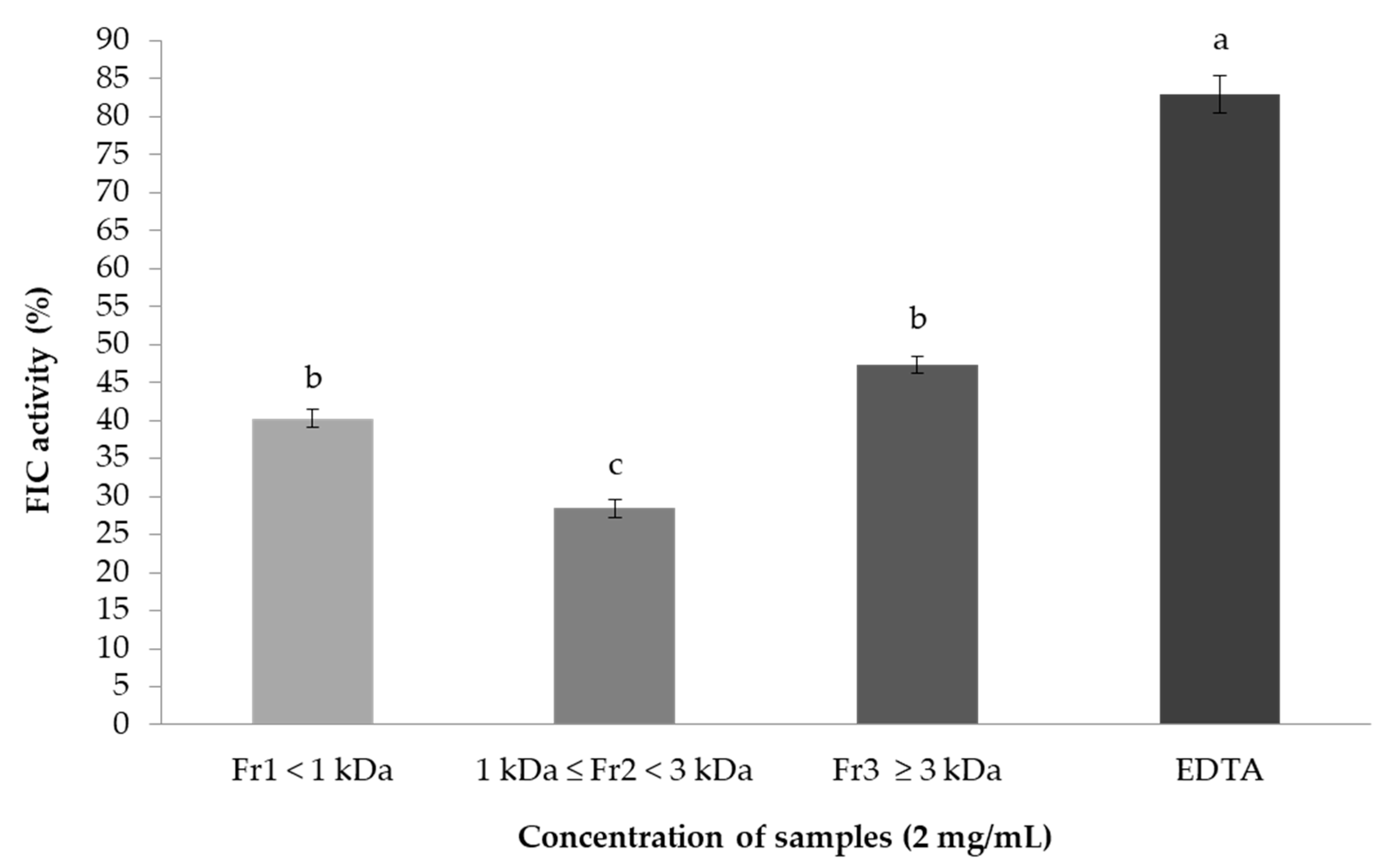
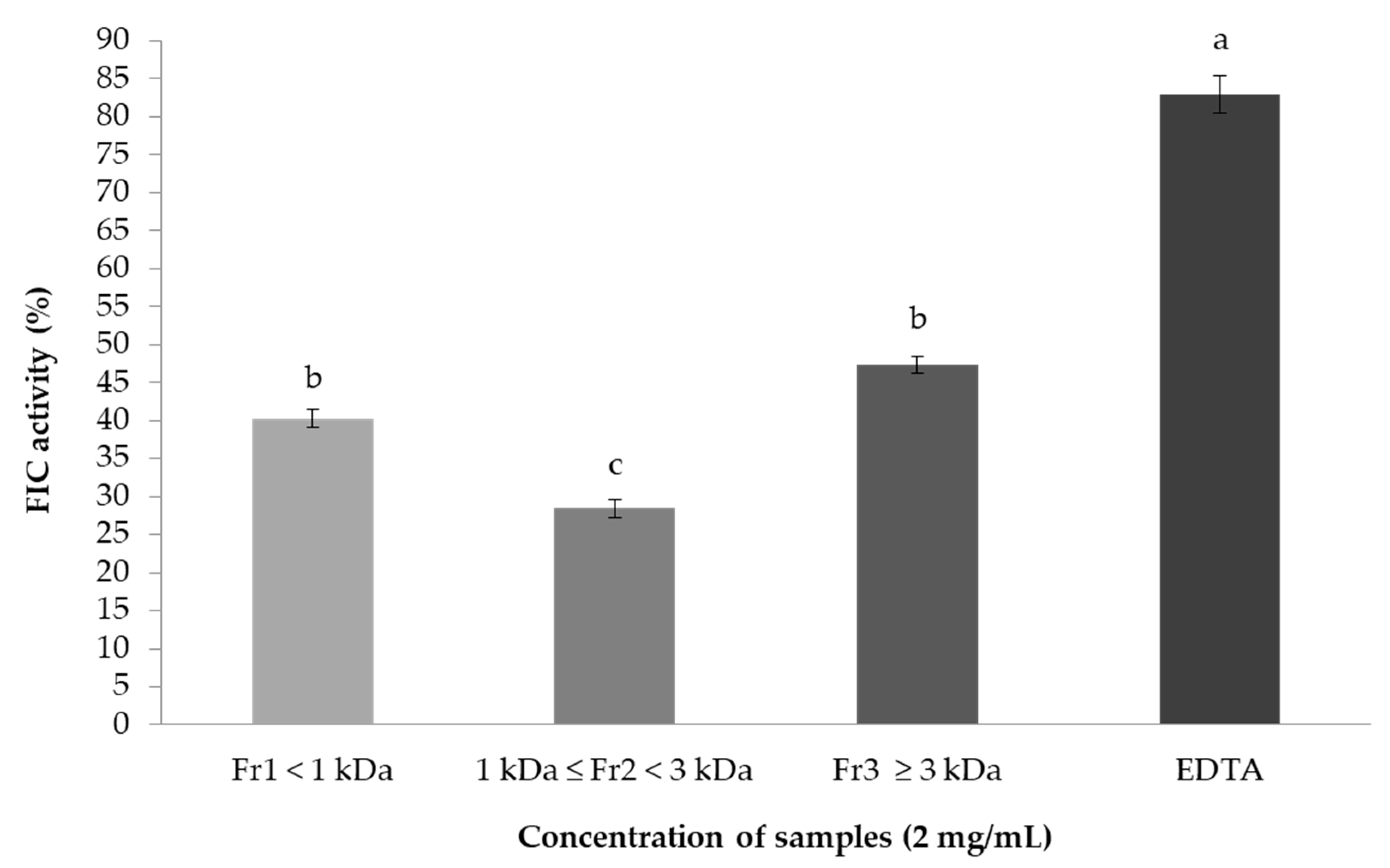
2.7.2. Ferrous Ion-Chelating (FIC) Activity Assay
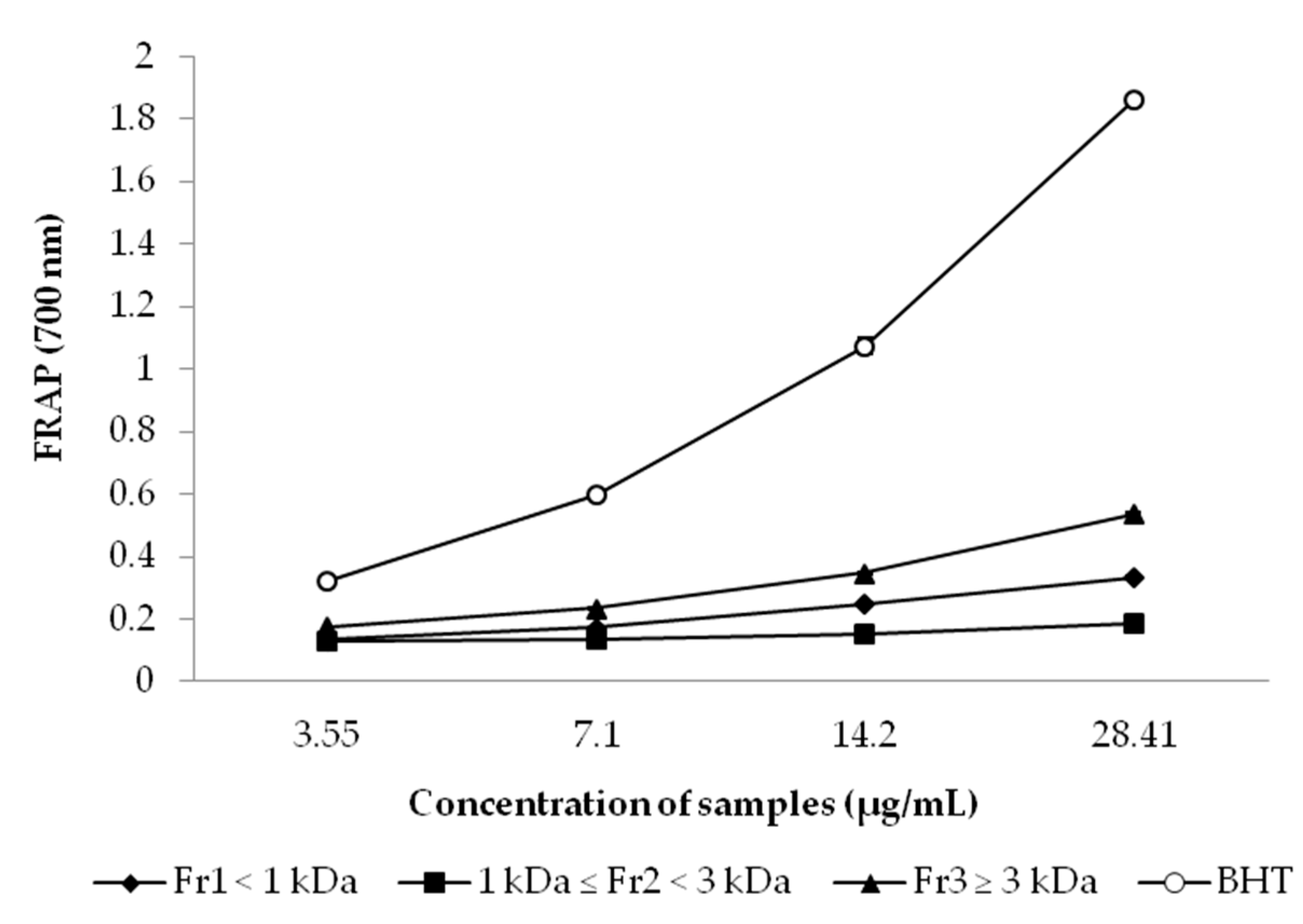
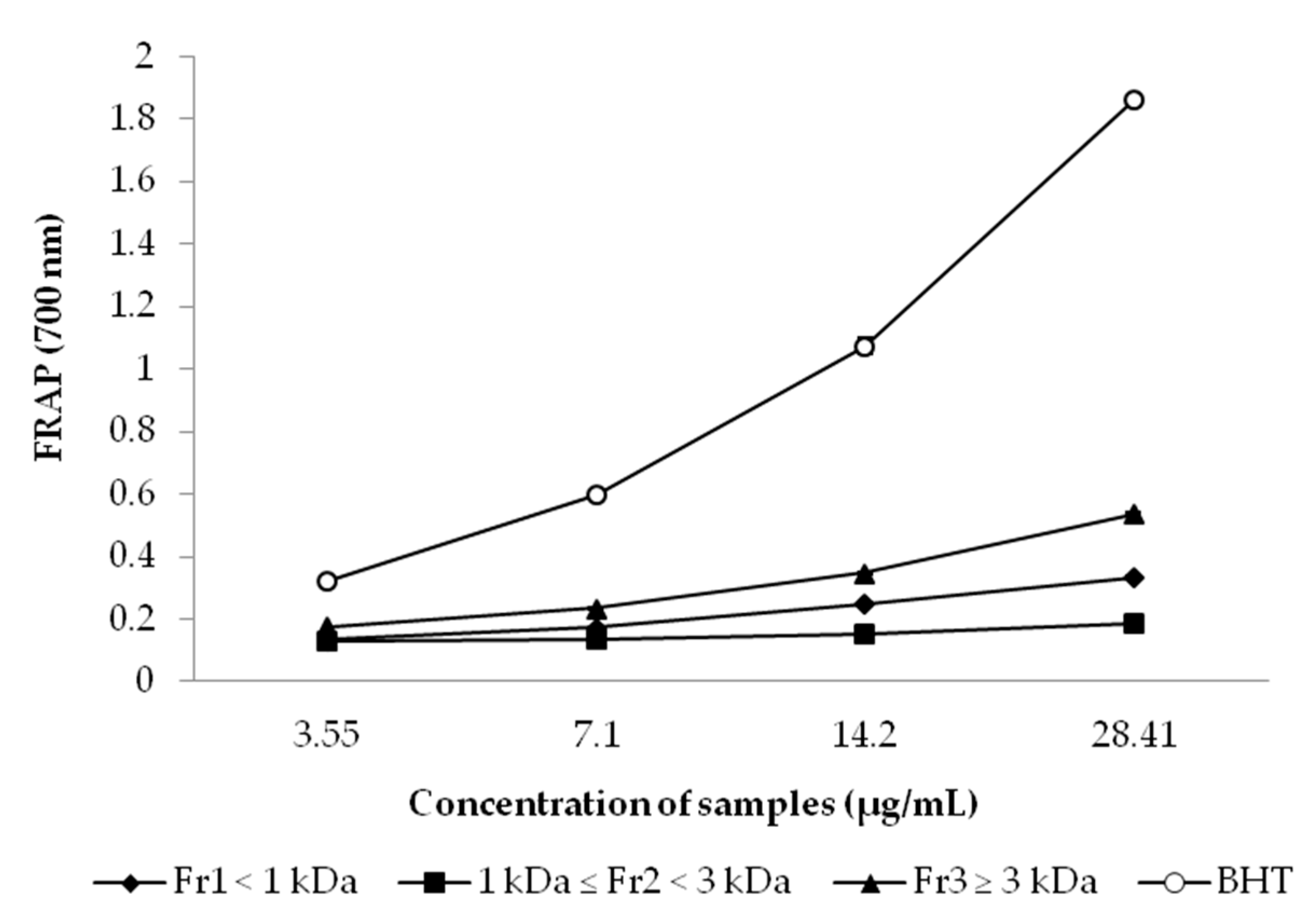
2.7.3. Ferric Reducing Antioxidant Power (FRAP) Assay
2.8. Pearson Correlation between Parameters
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Collection and Preparation of F. spiralis Sample
3.3. Extraction of Protein from F. spiralis
3.4. In Vitro F. spiralis Protein Digestibility Evaluation
3.5. Preparation of F. spiralis Protein Hydrolysate (FSPH)
3.6. Fractionation of FSPH by Ultrafiltration
3.7. Protein and Peptide Contents Analysis of FSPH Fractions
3.8. Amino Acids Composition of FSPH Fractions
3.9. Total Phenolic Content (TPC) Determination of FSPH Fractions
3.10. ACE-Inhibitory Activity Determination of FSPH Fractions
3.11. Antioxidant Activity Assays on FSPH Fractions
3.11.1. DPPH Free Radical Scavenging Activity (FRSA) Assay
3.11.2. Ferrous Ion-Chelating (FIC) Activity Assay
3.11.3. Ferric Reducing Antioxidant Power (FRAP) Assay
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lordan, S.; Ross, R.P.; Stanton, C. Marine bioactives as functional food ingredients: Potential to reduce the incidence of chronic diseases. Mar. Drugs 2011, 9, 1056–1100. [Google Scholar] [PubMed]
- Hata, Y.; Nakajima, K.; Uchida, J.I.; Hidaka, H.; Nakano, T. Clinical effects of brown seaweed, Undaria pinnatifida (wakame), on blood pressure in hypertensive subjects. J. Clin. Biochem. Nutr. 2001, 30, 43–53. [Google Scholar] [CrossRef]
- Paiva, L.; Lima, E.; Neto, A.I.; Baptista, J. Isolation and characterization of angiotensin I-converting enzyme (ACE) inhibitory peptides from Ulva rigida C. Agardh protein hydrolysate. J. Funct. Foods 2016, 26, 65–76. [Google Scholar] [CrossRef]
- Soffer, R.L. Angiotensin-converting enzyme and the regulation of vasoactive peptides. Annu. Rev. Biochem. 1976, 45, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, E.T.; Devereux, R.B.; Yeh, J.; Best, L.G.; Fabsitz, R.R.; Howard, B.V. Prehypertension, diabetes, and cardiovascular disease risk in a population-based sample: The strong heart study. Hypertension 2006, 47, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.R.; Carson, J.M.; Caselli, A.; Spark, M.J.; Woods, R. Conformational analysis and active site modelling of angiotensin-converting enzyme inhibitors. J. Med. Chem. 1985, 28, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Suetsuna, K. Purification and identification of angiotensin I-converting enzyme inhibitors from the red alga Porphyra yezoensis. J. Mar. Biotechnol. 1998, 6, 163–167. [Google Scholar] [PubMed]
- Sato, M.; Hosokawa, T.; Yamaguchi, T.; Nakano, T.; Muramoto, K.; Kahara, T.; Funayama, K.; Kobayashi, A.; Nakano, T. Angiotensin I-converting enzyme inhibitory peptides derived from Wakame (Undaria pinnatifida) and their antihypertensive effect in spontaneously hypertensive rats. J. Agric. Food Chem. 2002, 50, 6245–6252. [Google Scholar] [CrossRef] [PubMed]
- Paiva, L.; Lima, E.; Neto, A.I.; Baptista, J. Angiotensin I-converting enzyme (ACE) inhibitory activity of Fucus spiralis macroalgae and influence of the extracts storage temperature: A short report. J. Pharm. Biomed. Anal. 2016, 131, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Hyun, S.K.; Kim, H.R.; Choi, J.S. Angiotensin-converting enzyme I inhibitory activity of phlorotannins from Ecklonia stolonifera. Fish. Sci. 2006, 72, 1292–1299. [Google Scholar] [CrossRef]
- Wijesekara, I.; Kim, S.-K. Angiotensin-I-converting enzyme (ACE) inhibitors from marine resources: Prospects in the pharmaceutical industry. Mar. Drugs 2010, 8, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, W.A.J.P.; Ko, S.-C.; Jeon, Y.-J. Effect of phlorotannins isolated from Ecklonia cava on angiotensin I-converting enzyme (ACE) inhibitory activity. Nutr. Res. Pract. 2011, 5, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Molina, A.; Fernández, K. Comparison of different extraction techniques for obtaining extracts from brown seaweeds and their potential effects as angiotensin I-converting enzyme (ACE) inhibitors. J. Appl. Phycol. 2016, 28, 1295–1302. [Google Scholar] [CrossRef]
- Catarino, M.D.; Silva, A.M.S.; Cardoso, S.M. Fucaceae: A source of bioactive phlorotannins. Int. J. Mol. Sci. 2017, 18, 1327. [Google Scholar] [CrossRef] [PubMed]
- Harnedy, P.A.; FitzGerald, R.J. Bioactive proteins, peptides and amino acids from macroalgae. J. Phycol. 2011, 47, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, K.; Jeon, Y.-J. Bio-functionalities of proteins derived from marine algae—A review. Food Res. Int. 2012, 48, 948–960. [Google Scholar] [CrossRef]
- Cian, R.E.; Alaiz, M.; Vioque, J.; Drago, S.R. Enzyme proteolysis enhanced extraction of ACE inhibitory and antioxidant compounds (peptides and polyphenols) from Porphyra columbina residual cake. J. Appl. Phycol. 2013, 25, 1197–1206. [Google Scholar] [CrossRef]
- Oroian, M.; Escriche, I. Antioxidants: Characterization, natural sources, extraction and analysis. Food Res. Int. 2015, 74, 10–36. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, H.; Pihlanto, A. Food-derived bioactive peptides—Opportunities for designing future foods. Curr. Pharm. Des. 2003, 9, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, W.A.; Jeon, Y.-J. Enzyme-assistant extraction (EAE) of bioactive components: A useful approach for recovery of industrially important metabolites from seaweeds: A review. Fitoterapia 2012, 83, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.I.; Brotas, V.; Azevedo, J.M.N.; Patarra, R.F.; Álvaro, N.M.V.; Gameiro, C.; Prestes, A.C.L.; Nogueira, E.M. Qualidade de Águas Costeiras do Grupo Oriental do Arquipélago dos Açores e Proposta de Monitorização; Universidade dos Açores: Ponta Delgada, Portugal, 2009. [Google Scholar]
- Neto, A.I.; Tittley, I.; Raposeiro, P.M. Flora Marinha do Litoral dos Açores. Rocky Shore Marine Flora of the Azores; Secretaria Regional do Ambiente e do Mar: Horta, Portugal, 2005. [Google Scholar]
- Paiva, L.; Lima, E.; Patarra, R.F.; Neto, A.I.; Baptista, J. Edible Azorean macroalgae as source of rich nutrients with impact on human health. Food Chem. 2014, 164, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Cerantola, S.; Breton, F.; Ar Gall, E.; Deslandes, E. Co-occurrence and antioxidant activities of fucol and fucophlorethol classes of polymeric phenols in Fucus spiralis. Bot. Mar. 2006, 49, 347–351. [Google Scholar] [CrossRef]
- Tierney, M.S.; Smyth, T.J.; Rai, D.K.; Soler-Vila, A.; Croft, A.K.; Brunton, N. Enrichment of polyphenol contents and antioxidant activities of Irish brown macroalgae using food-friendly techniques based on polarity and molecular size. Food Chem. 2013, 139, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Pinteus, S.; Silva, J.; Alves, C.; Horta, A.; Fino, N.; Inês Rodrigues, A.; Mendes, S.; Pedrosa, R. Cytoprotective effect of seaweeds with high antioxidant activity from the Peniche coast (Portugal). Food Chem. 2017, 218, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Fleurence, J. Seaweed proteins: Biochemical nutritional aspects and potential uses. Trends Food Sci. Technol. 1999, 10, 25–28. [Google Scholar] [CrossRef]
- Quiroz-Castañeda, R.E.; Folch-Mallol, J.L. Hydrolysis of biomass mediated by cellulases for the production of sugars. In Sustainable Degradation of Lignocellulosic Biomass-Techniques, Applications and Commercialization; Chandel, A.K., da Silva, S.S., Eds.; InTech North America: New York, NY, USA, 2013. [Google Scholar]
- Paiva, L.; Lima, E.; Neto, A.I.; Baptista, J. Screening for angiotensin I-converting enzyme (ACE) inhibitory activity of enzymatic hydrolysates obtained from Azorean macroalgae. Arquipél. Life Mar. Sci. 2015, 32, 11–17. [Google Scholar]
- Elavarasan, K.; Kumar, V.N.; Shamasundar, B.A. Antioxidant and functional properties of fish protein hydrolysates from fresh water carp (Catla catla) as influenced by the nature of enzyme. J. Food Process. Preserv. 2014, 38, 1207–1214. [Google Scholar] [CrossRef]
- Gajanan, P.G.; Elavarasan, K.; Shamasundar, B.A. Bioactive and functional properties of protein hydrolysates from fish frame processing waste using plant proteases. Environ. Sci. Pollut. Res. 2016, 23, 24901–24911. [Google Scholar] [CrossRef] [PubMed]
- Auwal, S.M.; Zarei, M.; Abdul-Hamid, A.; Saari, N. Optimization of bromelain-aided production of angiotensin I-converting enzyme inhibitory hydrolysates from stone fish using response surface methodology. Mar. Drugs 2017, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Chalé, F.G.H.; Ruiz, J.C.R.; Fernández, J.J.A.; Ancona, D.A.B.; Campos, M.R.S. ACE inhibitory, hypotensive and antioxidant peptide fractions from Mucuna pruriens proteins. Process Biochem. 2014, 49, 1691–1698. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Aluko, R.E. Chemometric analysis of the amino acid requirements of antioxidant food protein hydrolysates. Int. J. Mol. Sci. 2011, 12, 3148–3161. [Google Scholar] [CrossRef] [PubMed]
- Fleurence, J. Seaweed proteins. In Proteins in Food Processing; Yada, R.Y., Ed.; Woodhead Publishing Limited: Cambridge, UK, 2004; pp. 197–213. [Google Scholar]
- Ge, Y.; Sun, A.; Ni, Y.; Cai, T. Study and development of a defatted wheat germ nutritive noodle. Eur. Food Res. Technol. 2001, 212, 344–348. [Google Scholar] [CrossRef]
- Athukorala, Y.; Jeon, Y.J. Screening for angiotensin I-converting enzyme inhibitory activity of Ecklonia cava. J. Food Sci. Nutr. 2005, 10, 134–139. [Google Scholar] [CrossRef]
- Tierney, M.S.; Soler-vila, A.; Croft, A.K.; Hayes, M. Antioxidant activity of the brown macroalgae Fucus spiralis Linnaeus harvested from the west coast of Ireland. Curr. Res. J. Biol. Sci. 2013, 5, 81–90. [Google Scholar]
- Wang, T.; Jónsdóttir, R.; Liu, H.; Gu, L.; Kristinsson, H.G.; Raghavan, S.; Ólafsdóttir, G. Antioxidant capacities of phlorotannins extracted from the brown algae Fucus vesiculosus. J. Agric. Food Chem. 2012, 60, 5874–5883. [Google Scholar] [CrossRef] [PubMed]
- Steevensz, A.J.; MacKinnon, S.L.; Hankinson, R.; Craft, C.; Connan, S.; Stengel, D.B.; Melanson, J.E. Profiling phlorotannins in brown macroalgae by liquid chromatography-high resolution mass spectrometry. Phytochem. Anal. 2012, 23, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Ferreres, F.; Lopes, G.; Gil-Izquierdo, A.; Andrade, P.B.; Sousa, C.; Mouga, T.; Valentão, P. Phlorotannin extracts from Fucales characterized by HPLC-DAD-ESI-MSn: Approaches to hyaluronidase inhibitory capacity and antioxidant properties. Mar. Drugs 2012, 10, 2766–2781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, B.; Dong, S.; Liu, Z.; Zhao, X.; Wang, J.; Zeng, M. A novel ACE inhibitory peptide isolated from Acaudina molpadioidea hydrolysate. Peptides 2009, 30, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.G.; Wei, L.G.; Liu, H.; Hui, S.Y. Mung-bean protein hydrolysates obtained with Alcalase exhibit angiotensin I-converting enzyme inhibitory activity. Food Sci. Technol. Int. 2005, 11, 281–287. [Google Scholar]
- Wilson, J.; Hayes, M.; Carney, B. Angiotensin-I-converting enzyme and prolyl endopeptidase inhibitory peptides from natural sources with a focus on marine processing by-products. Food Chem. 2011, 129, 235–244. [Google Scholar] [CrossRef]
- Segura-Campos, M.R.; Peralta-González, F.; Castellanos-Ruelas, A.; Chel-Guerrero, L.A.; Betancur-Ancona, D.A. Effect of Jatropha curcas peptide fractions on the angiotensin I-converting enzyme inhibitory activity. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, R.; Zarei, M.; Ebrahimpour, A.; Abdul-Hamid, A.; Ismail, A.; Saari, N. Angiotensin-I converting enzyme (ACE) inhibitory and anti-oxidant activities of sea cucumber (Actinopyga lecanora) hydrolysates. Int. J. Mol. Sci. 2015, 16, 28870–28885. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-C.; Hsu, F.-L.; Tsai, J.-C.; Chan, P.; Liu, J.Y.; Thomas, G.N.; Tomlinson, B.; Lo, M.-Y.; Lin, J.-Y. Antihypertensive effects of tannins isolated from traditional Chinese herbs as non-specific inhibitors of angiotensin converting enzyme. Life Sci. 2003, 73, 1543–1555. [Google Scholar] [CrossRef]
- Wang, T.; Jónsdóttir, R.; Kristinsson, H.G.; Hreggvidsson, G.O.; Jónsson, J.O.; Thorkelsson, G.; Ólafsdóttir, G. Enzyme-enhanced extraction of antioxidant ingredients from red algae Palmaria palmata. LWT—Food Sci. Technol. 2010, 43, 1387–1393. [Google Scholar] [CrossRef]
- Fan, J.; He, J.; Zhuang, Y.; Sun, L. Purification and identification of antioxidant peptides from enzymatic hydrolysates of Tilapia (Oreochromis niloticus) frame protein. Molecules 2012, 17, 12836–12850. [Google Scholar] [CrossRef] [PubMed]
- Udenigwe, C.C.; Lu, Y.-L.; Han, C.-H.; Hou, W.-C.; Aluko, R.E. Flaxseed protein-derived peptide fractions: Antioxidant properties and inhibition of lipopolysaccharide-induced nitric oxide production in murine macrophages. Food Chem. 2009, 116, 277–284. [Google Scholar] [CrossRef]
- Chen, H.-M.; Muramoto, K.; Yamauchi, F.; Fujimoto, K.; Nokihara, K. Antioxidative properties of histidine-containing peptides designed from peptide fragments found in the digests of a soybean protein. J. Agric. Food. Chem. 1998, 46, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kouzuma, Y.; Yonekura, M. Structures and properties of antioxidative peptides derived from royal jelly protein. Food Chem. 2009, 113, 238–245. [Google Scholar] [CrossRef]
- Rajapakse, N.; Mendis, E.; Jung, W.-K.; Je, J.-Y.; Kim, S.-K. Purification of a radical scavenging peptide from fermented mussel sauce and its antioxidant properties. Food. Res. Int. 2005, 38, 175–182. [Google Scholar] [CrossRef]
- Nimalaratne, C.; Bandara, N.; Wu, J. Purification and characterization of antioxidant peptides from enzymatically hydrolyzed chicken egg white. Food Chem. 2015, 188, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Alemán, A.; Pérez-Santín, E.; Bordenave-Juchereau, S.; Arnaudin, I.; Gómez-Guillén, M.C.; Montero, P. Squid gelatin hydrolysates with antihypertensive, anticancer and antioxidant activity. Food Res. Int. 2011, 44, 1044–1051. [Google Scholar] [CrossRef]
- Heffernan, N.; Smyth, T.J.; Soler-Villa, A.; Fitzgerald, R.J.; Brunton, N.P. Phenolic content and antioxidant activity of fractions obtained from selected Irish macroalgae species (Laminaria digitata, Fucus serratus, Gracilaria gracilis and Codium fragile). J. Appl. Phycol. 2015, 27, 519–530. [Google Scholar] [CrossRef]
- Kim, K.N.; Heo, S.-J.; Song, C.B.; Lee, J.; Heo, M.S.; Yeo, I.K.; Kang, K.; Hyun, J.W.; Jeon, Y.-J. Protective effect of Ecklonia cava enzymatic extracts on hydrogen peroxide-induced cell damage. Process Biochem. 2006, 41, 2393–2401. [Google Scholar] [CrossRef]
- Wang, T.; Jónsdóttir, R.; Ólafsdóttir, G. Total phenolic compounds, radical scavenging and metal chelation of extracts from Icelandic seaweeds. Food Chem. 2009, 116, 240–248. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Alaiz, M.; Vioque, J. Affinity purification and characterisation of chelating peptides from chickpea protein hydrolysates. Food Chem. 2011, 129, 485–490. [Google Scholar] [CrossRef]
- Girgih, A.T.; Udenigwe, C.C.; Aluko, R.E. In vitro antioxidant properties of hemp seed (Cannabis sativa L.) protein hydrolysate fractions. J. Am. Oil Chem. Soc. 2011, 88, 381–389. [Google Scholar] [CrossRef]
- Senevirathne, M.; Kim, S.-H.; Siriwardhana, N.; Ha, J.-H.; Lee, K.-W.; Jeon, Y.-J. Antioxidant potential of Ecklonia cava on reactive oxygen species scavenging, metal chelating, reducing power and lipid peroxidation inhibition. Revista de Agaroquimica y Tecnologia de Alimentos 2006, 12, 27–38. [Google Scholar]
- Santoso, J.; Yoshie-Stark, Y.; Suzuki, T. Anti-oxidant activity of methanol extracts from Indonesian seaweeds in an oil emulsion model. Fish. Sci. 2004, 70, 183–188. [Google Scholar] [CrossRef]
- Duh, P.-D. Antioxidant activity of burdock (Arctium lappa Linné): It’s scavenging effect on free radical and active oxygen. J. Am. Oil Chem. Soc. 1998, 75, 455–461. [Google Scholar] [CrossRef]
- Chandini, S.-K.; Ganesan, P.; Bhaskar, N. In vitro antioxidant activities of three selected brown seaweeds of India. Food Chem. 2008, 107, 707–713. [Google Scholar] [CrossRef]
- Ganesan, P.; Kumar, C.S.; Bhaskar, N. Antioxidant properties of methanol extract and its solvent fractions obtained from selected Indian red seaweeds. Bioresour. Technol. 2008, 99, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Zubia, M.; Fabre, M.S.; Kerjean, V.; Lann, K.L.; Stiger-Pouvreau, V.; Fauchon, M.; Deslandes, E. Antioxidant and antitumoural activities of some Phaeophyta from Brittany coasts. Food Chem. 2009, 116, 693–701. [Google Scholar] [CrossRef]
- Vinayak, R.C.; Sabu, A.S.; Chatterji, A. Bio-prospecting of a few brown seaweeds for their cytotoxic and antioxidant activities. Evid. Based Complement. Altern. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Babbar, N.; Oberoi, H.S.; Uppal, D.S.; Patil, R.T. Total phenolic content and antioxidant capacity of extracts obtained from six important fruit residues. Food Res. Int. 2011, 44, 391–396. [Google Scholar] [CrossRef]
- Pal, R.; Rai, J.P. Phytochelatins: Peptides involved in heavy metal detoxification. Appl. Biochem. Biotechnol. 2010, 160, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.H.; Cheung, P.C.K.; Ang, P.O., Jr. Nutritional evaluation of protein concentrates isolated from two red seaweeds: Hypnea charoides and Hypnea japonica in growing rats. Hydrobiologia 2004, 512, 271–278. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Paiva, L.; Lima, E.; Neto, A.I.; Massimo, M.; Baptista, J. Nutritional and functional bioactivity value of selected Azorean macroalgae: Ulva compressa, Ulva rigida, Gelidium microdon, and Pterocladiella capillacea. J. Food Sci. 2017, 82, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Church, F.C.; Swaisgood, H.E.; Porter, D.H.; Catignani, G.L. Spectrophotometric assay using O-phthaldialdehyde for determination of proteolysis in milk and isolated milk proteins. J. Dairy Sci. 1983, 66, 1219–1227. [Google Scholar] [CrossRef]
- Waterhouse, A.L. Determination of total phenolics. In Current Protocols in Food Analytical Chemistry; Wrolstad, R.E., Ed.; John Wiley & Sons: New York, NY, USA, 2002. [Google Scholar]
- Cushman, D.W.; Cheung, H.S. Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochem. Pharmacol. 1971, 20, 1637–1648. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Corrêa, A.P.; Daroit, D.J.; Coelho, J.; Meira, S.M.; Lopes, F.C.; Risso, P.H.; Brandelli, A. Antioxidant, antihypertensive and antimicrobial properties of ovine milk caseinate hydrolyzed with a microbial protease. J. Sci. Food Agric. 2011, 91, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, M. Studies on products of browning reactions: Antioxidative activities of products of browning reaction prepared from glucosamine. Jpn. J. Nutr. 1986, 44, 307–315. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Yield (mg/g of DW FSPH) | ACE-Inhibition | Protein Content (mg/g of DW FSPH) | Peptide Content (mg/g of DW FSPH) | |
|---|---|---|---|---|---|
| Percentage | IC50 Value (mg/mL) | ||||
| Fr1 < 1 kDa | 206.28 | 45.08 ± 0.66 a | 1.850 ± 0.06 b | 123.15 ± 2.78 a | 43.81 ± 2.27 a |
| 1 kDa ≤ Fr2 < 3 kDa | 53.72 | 41.93 ± 1.62 a | 2.000 ± 0.06 b | 336.28 ± 4.96 b | 35.91 ± 1.58 a |
| Fr3 ≥ 3 kDa | 703.95 | 86.85 ± 1.89 b | 0.500 ± 0.03 a | 474.03 ± 4.44 b | 243.82 ± 2.9 b |
| Amino Acid (AA) | F. spiralis Protein Hydrolysate (FSPH) Fraction | ||
|---|---|---|---|
| Fr1 < 1 kDa | 1 kDa ≤ Fr2 < 3 kDa | Fr3 ≥ 3 kDa | |
| Alanine | tc | tc | 25.34 ± 0.13 |
| Glycine | tc | tc | 22.38 ± 0.13 |
| Valine | 155.53 ± 2.25 | 209.37 ± 3.35 | 236.83 ± 4.65 |
| Threonine | 1.22 ± 0.08 | 1.71 ± 0.03 | 17.42 ± 0.13 |
| Serine | 2.75 ± 0.06 | 3.18 ± 0.03 | 19.36 ± 0.16 |
| Leucine | 2.24 ± 0.12 | 2.85 ± 0.05 | 25.35 ± 0.17 |
| Isoleucine | 1.89 ± 0.03 | 2.84 ± 0.06 | 32.55 ± 0.43 |
| Proline | 1.34 ± 0.05 | 1.93 ± 0.03 | 12.47 ± 0.11 |
| Methionine | tc | tc | 20.07 ± 0.31 |
| Aspartic acid | 3.63 ± 0.07 | 4.53 ± 0.10 | 37.53 ± 0.35 |
| Phenylalanine | 1.80 ± 0.02 | 1.49 ± 0.02 | 15.13 ± 0.15 |
| Glutamic acid | 10.40 ± 0.10 | 7.01 ± 0.22 | 46.33 ± 0.42 |
| Lysine | 15.48 ± 0.20 | 7.57 ± 0.11 | 13.82 ± 0.13 |
| Tyrosine | tc | tc | 38.17 ± 0.21 |
| Arginine | tc | tc | tc |
| Histidine | tc | tc | tc |
| Tryptophan | ND | ND | ND |
| Total AA | 196.28 a | 242.48 a | 562.75 b |
| AA distribution (%) | |||
| Hydrophobic | 82.94 a | 90.10 a | 65.35 b |
| Hydrophilic | 15. 04 a | 7.88 b | 17.36 a |
| Neutral | 2.02 b | 2.02 b | 17.29 a |
| Aromatic | 0.92 b | 0.61 b | 9.47 a |
| Branched-side chains | 81.34 a | 88.69 a | 52.37 b |
| Negatively charged | 7.15 b | 4.76 c | 14.90 a |
| Positively charged | 7.89 a | 3.12 b | 2.46 b |
| FRSA | FIC Activity | FRAP | Total Phenolic | ACE-Inhibition | Peptide Content | |
|---|---|---|---|---|---|---|
| FRSA | 1 | - | - | - | - | - |
| FIC activity | 0.890 | 1 | - | - | - | |
| FRAP | 0.760 | 0.973 | 1 | - | - | - |
| Total phenolic | 0.003 | 0.458 | 0.652 | 1 | - | - |
| ACE-inhibition | 0.660 | 0.822 | 0.932 | 0.883 | 1 | - |
| Peptide content | 0.446 | 0.805 | 0.921 | 0.896 | 1 | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paiva, L.; Lima, E.; Neto, A.I.; Baptista, J. Angiotensin I-Converting Enzyme (ACE) Inhibitory Activity, Antioxidant Properties, Phenolic Content and Amino Acid Profiles of Fucus spiralis L. Protein Hydrolysate Fractions. Mar. Drugs 2017, 15, 311. https://doi.org/10.3390/md15100311
Paiva L, Lima E, Neto AI, Baptista J. Angiotensin I-Converting Enzyme (ACE) Inhibitory Activity, Antioxidant Properties, Phenolic Content and Amino Acid Profiles of Fucus spiralis L. Protein Hydrolysate Fractions. Marine Drugs. 2017; 15(10):311. https://doi.org/10.3390/md15100311
Chicago/Turabian StylePaiva, Lisete, Elisabete Lima, Ana Isabel Neto, and José Baptista. 2017. "Angiotensin I-Converting Enzyme (ACE) Inhibitory Activity, Antioxidant Properties, Phenolic Content and Amino Acid Profiles of Fucus spiralis L. Protein Hydrolysate Fractions" Marine Drugs 15, no. 10: 311. https://doi.org/10.3390/md15100311
APA StylePaiva, L., Lima, E., Neto, A. I., & Baptista, J. (2017). Angiotensin I-Converting Enzyme (ACE) Inhibitory Activity, Antioxidant Properties, Phenolic Content and Amino Acid Profiles of Fucus spiralis L. Protein Hydrolysate Fractions. Marine Drugs, 15(10), 311. https://doi.org/10.3390/md15100311