1. Introduction
Cubitane-type diterpenoids are a rare group of compounds that are isolated from termites, soft corals, and plants [
1]. The first cubitanoid, (+)-cubitene, was discovered from the secretion gland of the termite
Cubitermes umbratus [
2]. Afterward, an array of cubitanoids, namely, calyculones A–I, were found from soft corals of genus
Eunicea [
3,
4,
5]. In some plants, cubitene was detected by gas chromatography–mass spectrometry (GC/MS) from their volatile oils [
6]. In the aforementioned three sources, cubitanoids were usually found in company with cembranoids [
1], which represent a large group of compounds in soft coral [
7,
8,
9,
10]. The origin of cubitanoids was speculated to be associated with the co-occurring cembranoids based on a photochemical interconversion [
4].
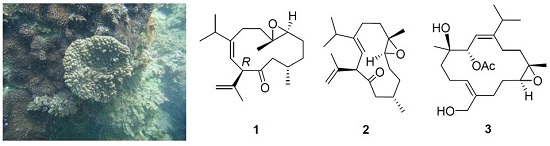
Our previous study on the soft coral
Sinularia nanolobata (Verseveldt), collected off the coast of the most southern tip of Taiwan, resulted in the characterization of caryophyllane- and xeniaphyllane-type terpenoids [
11]. Another collection of the same species off the coast of northern region of Taiwan led to the isolation of a novel C18 terpenoid [
12]. Herein, we report the investigation on the chemical constituents of
S. nanolobata, collected off the coast of the eastern region of Taiwan, which led to the isolation of two new cubitanoids, nanoculones A and B (
1 and
2), three new cembranoids, nanolobols A–C (
3–
5) (
Figure 1), as well as six known compounds (
6‒
11). The cytotoxicity of compounds
1–
11 against cancer cell lines P388 (murine leukemia), K562 (human erythromyeloblastoid leukemia), and HT-29 (human colon adenocarcinoma) was evaluated. In addition, the nitric oxide inhibitory activities of compounds
1–
4,
6–
8,
10, and
11 were further evaluated by assay of lipopolysaccharide (LPS)-stimulated NO production in activated RAW264.7 cells.
2. Results and Discussion
The EtOH extract of
S. nanolobata was concentrated and partitioned with EtOAc, successively. The resulting EtOAc layer was separated repeatedly by column chromatography and High-performance liquid chromatography (HPLC) to afford five new diterpenoids (
1–
5) (
Figure 1) and six known compounds (
6–
11). The known compounds were identified as calyculone I (
6) [
5], sinulariuol A (
7) [
13], sinulariols C, D, H, and J (
8–
11) [
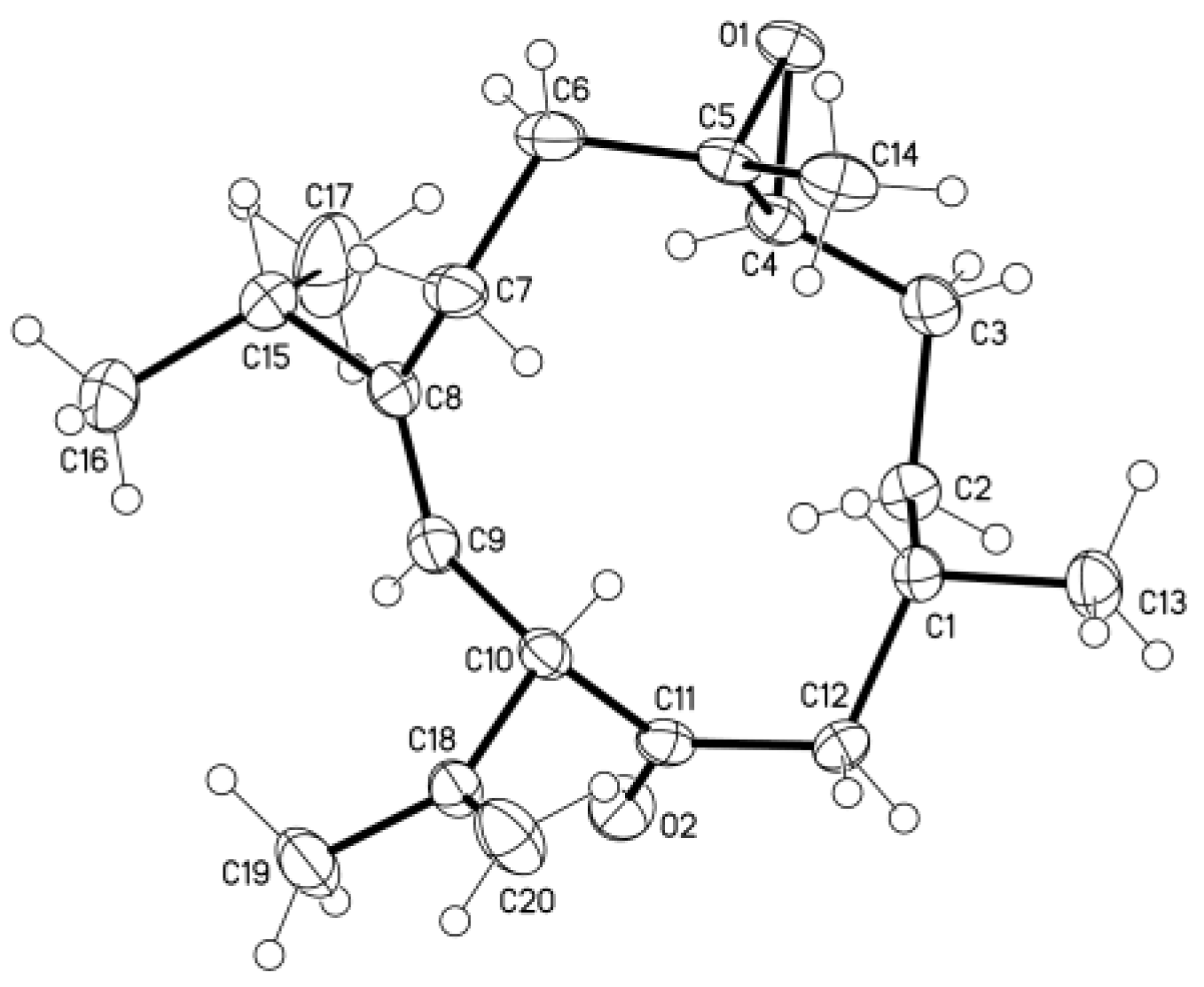
13], by comparison of their spectroscopic data with those reported in the literature. A single-crystal X-ray analysis was performed on
6, which led to the establishment of the absolute configuration of
6 (
Figure 2).
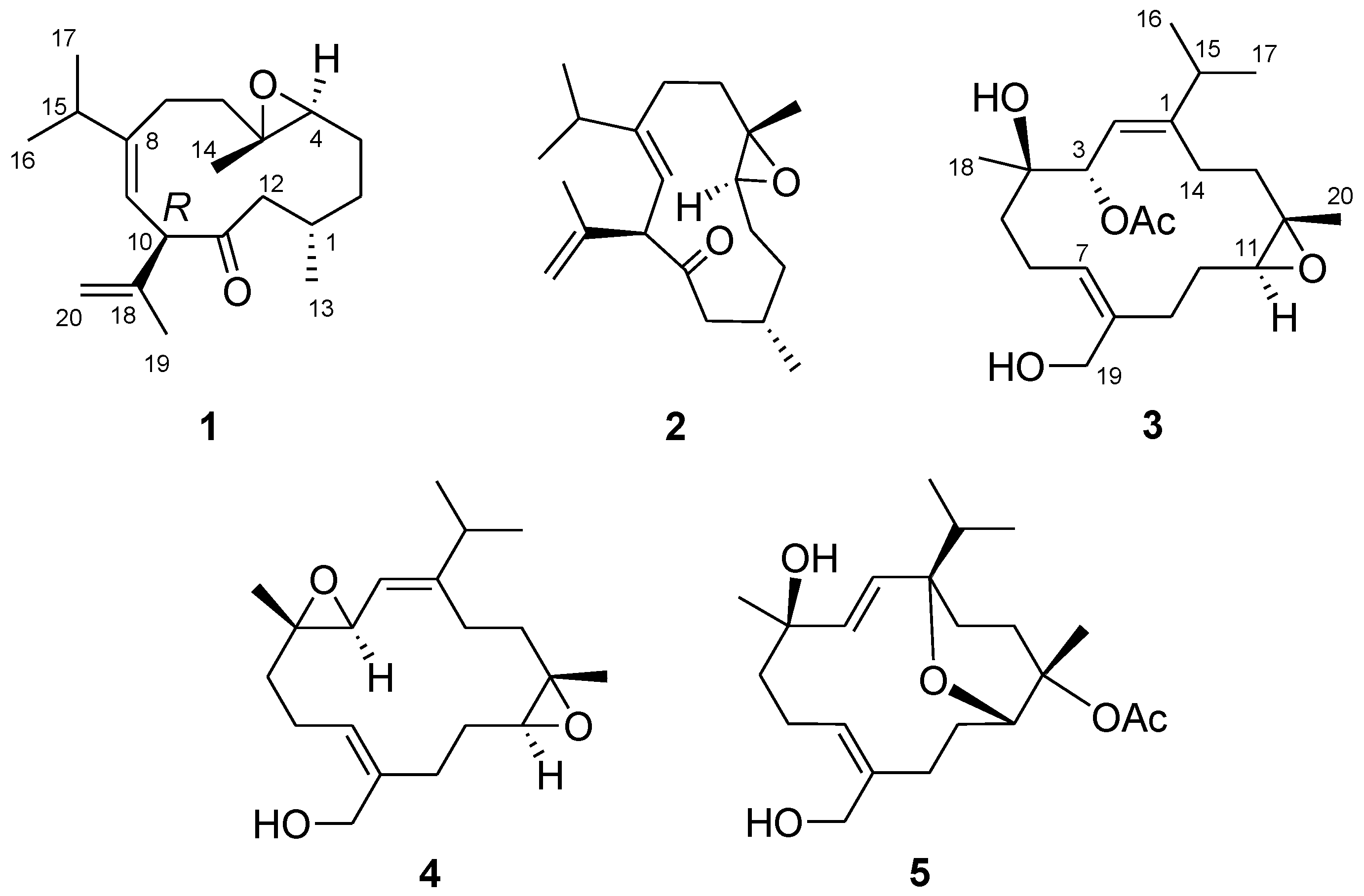
Inspection of the high resolution electrospray ionization mass spectroscopy (HRESIMS) (
Supplementary Materials, Figure S1) and
13C nuclear magnetic resonance (NMR) spectroscopic data allowed the establishment of a molecular formula of C
20H
32O
2 for
1, requiring five degrees of unsaturation. The IR spectrum showed the presence of carbonyl group (1710 cm
−1). The NMR spectra of
1 (
Figures S2 and S3) displayed signals attributable to a ketone [δ
C 209.7 (qC)], a trisubstituted double bond [δ
C 147.1 (qC), δ
C 120.4 (CH); δ
H 5.45 (1H, d,
J = 10.0 Hz)], a disubstituted double bond [δ
C 143.0 (qC), δ
C 112.6 (CH
2); δ
H 4.85 and 4.80 (each 1H, s)], and a trisubstituted epoxide [δ
C 62.3 (qC), δ
C 62.5 (CH); δ
H 2.77 (1H, dd,
J = 9.2, 3.6 Hz)] (
Table 1), which accounted for four degrees of unsaturation and two oxygen atoms in the molecular formula. Thus, the structure of
1 should contain an additional ring. The above data, in conjunction with a highly deshielded proton at δ
H 4.21 (1H, d,
J = 10.0 Hz, H-10) coupled by the olefinic H-9, were reminiscent of a cubitane skeleton [
1,
2,
3,
4,
5].
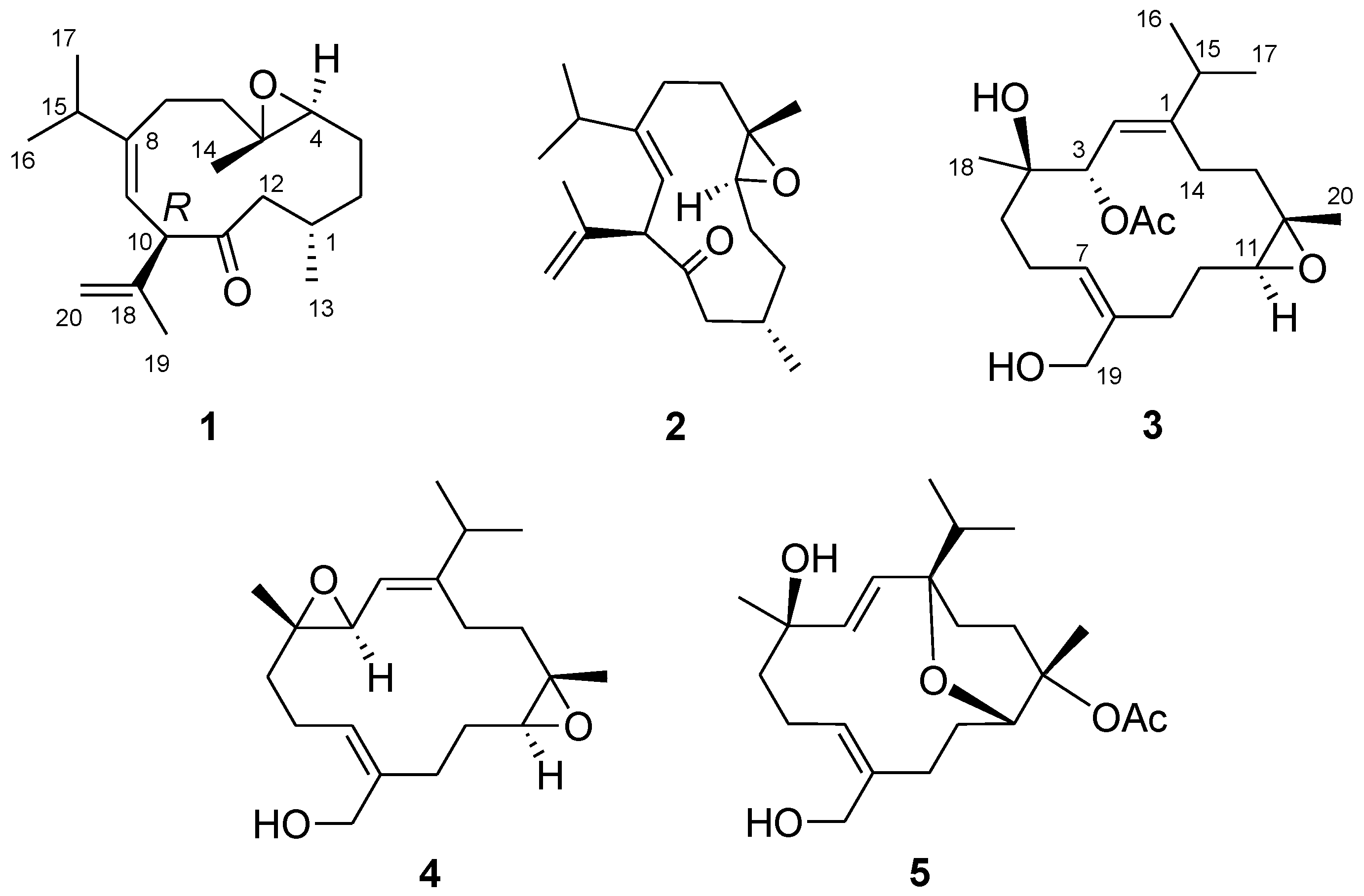
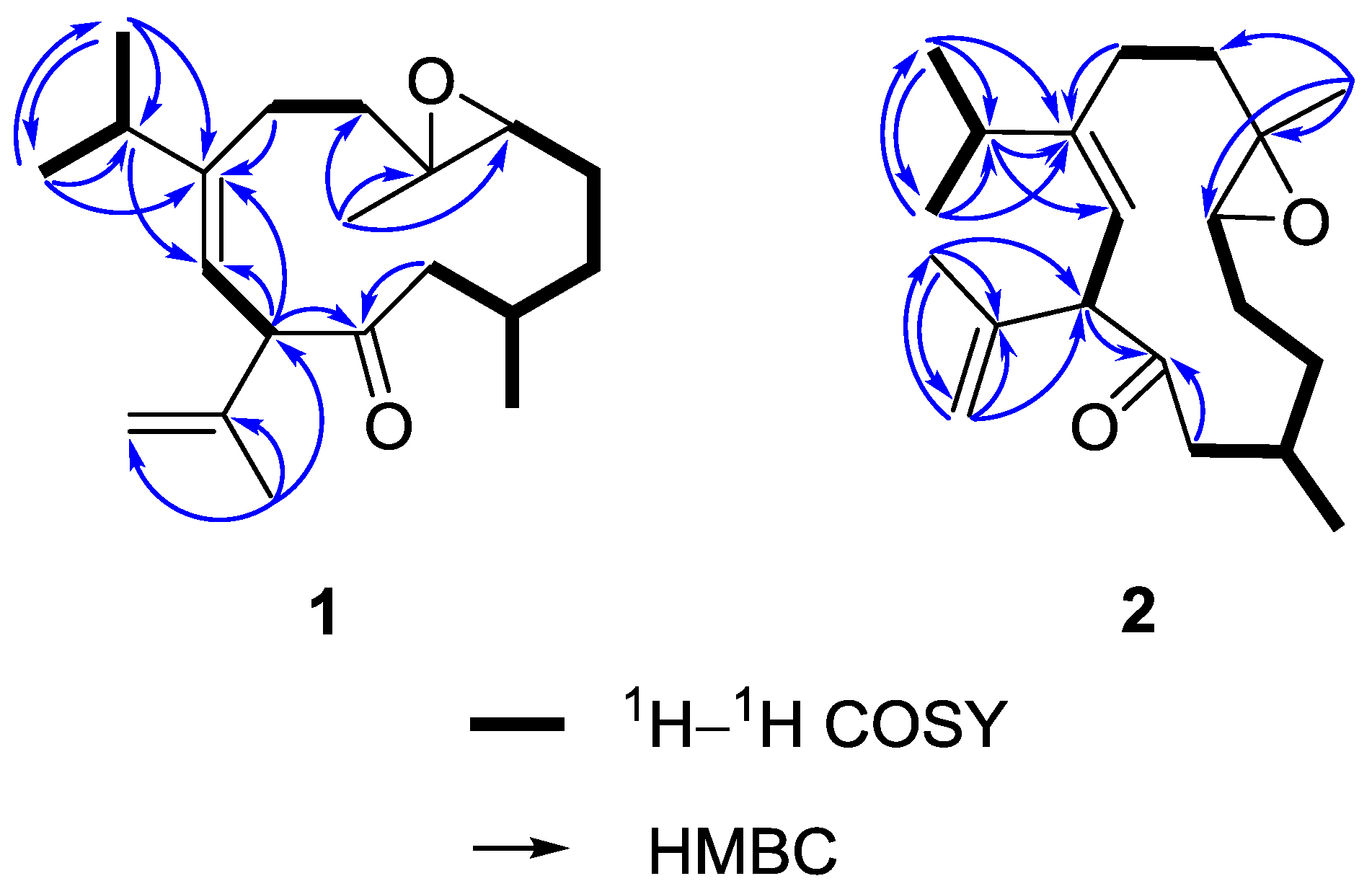
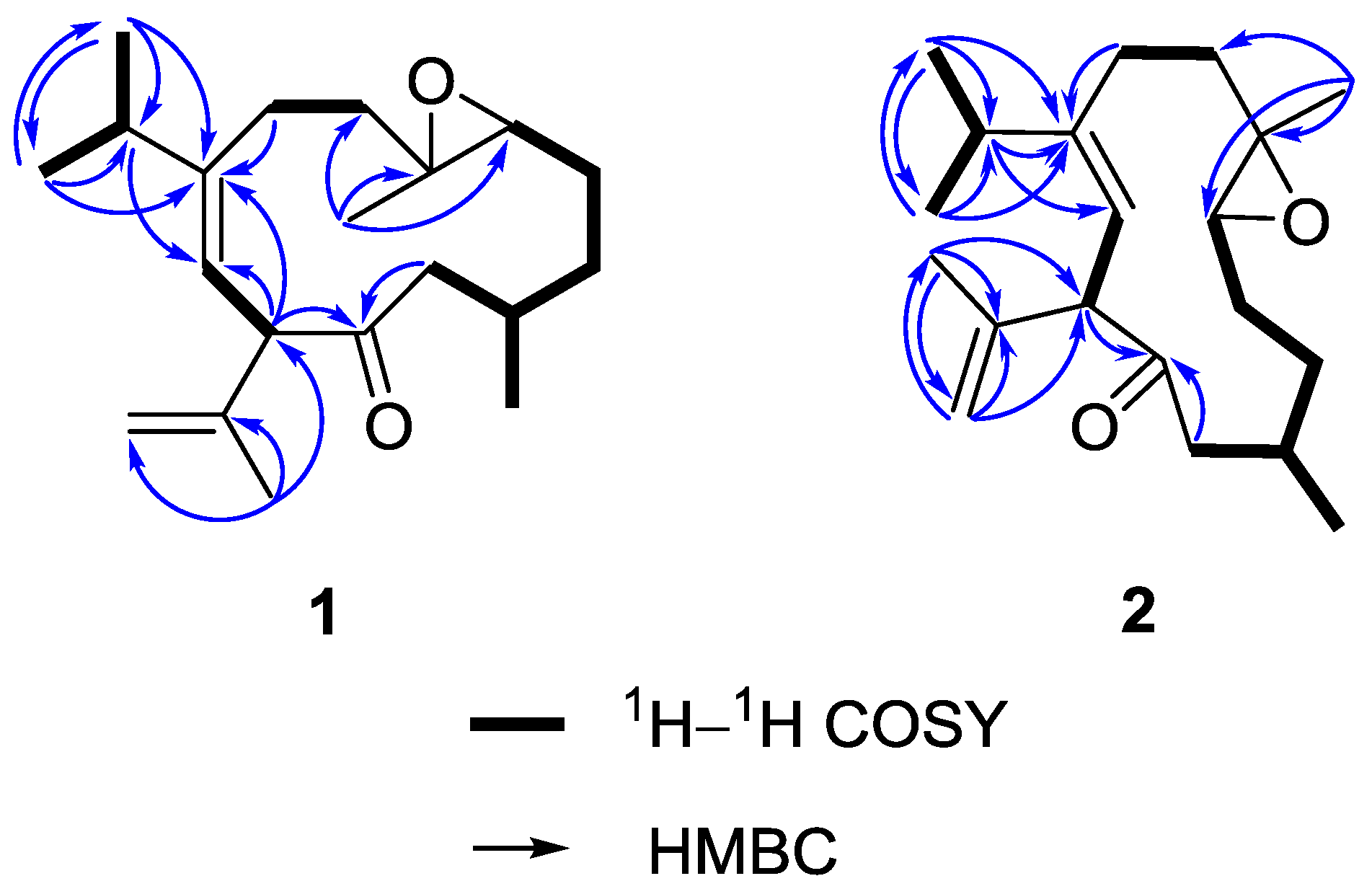
On the basis of H–H correlation spectroscopy (COSY) correlations of H
2-12/H-1/H
2-2/H
2-3/H-4, H-1/H
3-13, H
2-6/H
2-7, H
3-16/H-15/H
3-17, and H-9/H-10, the spin systems were established as shown in
Figure 3. An isopropyl was deduced to be attached at C-8 according to the heteronuclear multiple bond correlation (HMBC) correlations from H
3-16 (or H
3-17) to C-15, C-17 (or C-16), and C-8. A propen-2-yl fragment was assigned at C-10 based on the HMBC correlations from H
3-19 to C-10, C-18, and C-20. The methyl-substituted epoxide was assigned according to the HMBC correlations from H
3-14 to C-4, C-5, and C-6. Accordingly, the planar structure of
1 was established and found to be the same as that of calyculone I (
6) [
5].
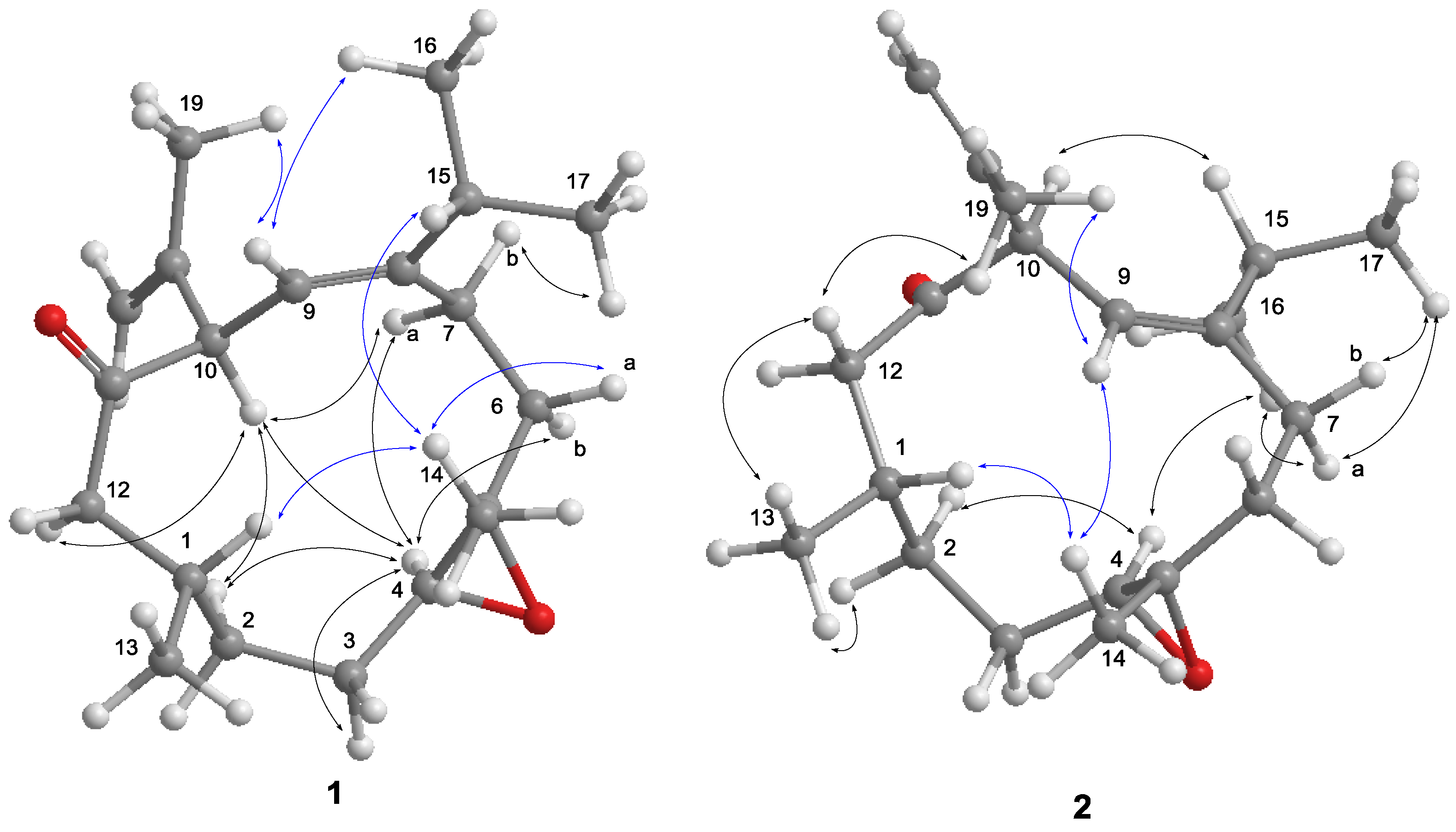
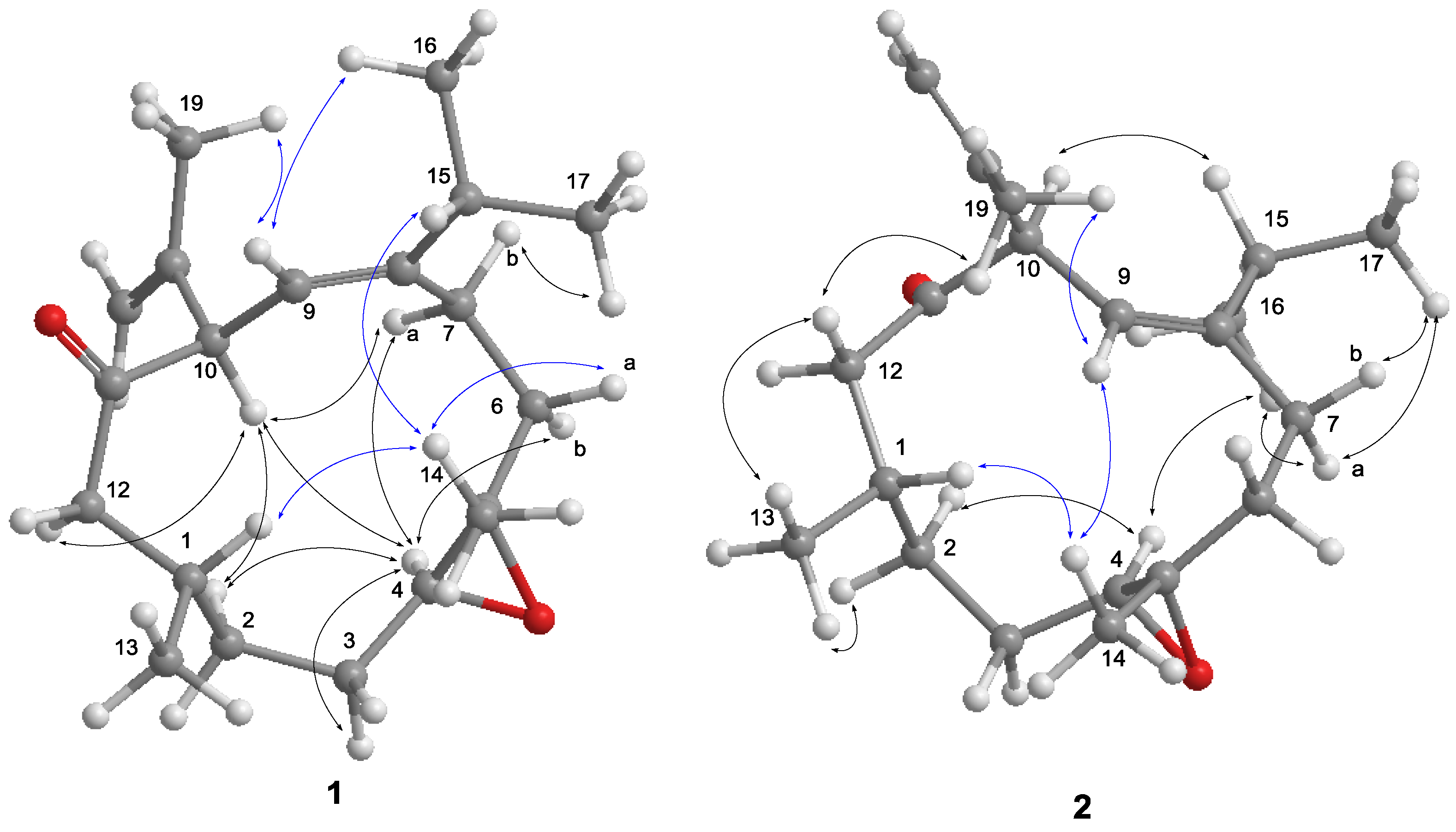
The nuclear Overhauser effect spectroscopy (NOESY) experiment was performed and its correlations were interpreted for the elucidation of the relative configuration of
1 (
Figure 4). An NOE correlation between H-9 and H
3-16 suggested an
E geometry for C-8/C-9 double bond. The absence of an NOE cross peak between H
3-14 and H-4 along with the presence of a correlation between H-4 and H-6b conducted to assign a
trans geometry for 4,5-epoxide. The observed NOE correlations of H-10/H-4, H-4/H-7a and H-10/H-7a suggested the same orientation of H-10 and H-4 in the molecule, whereas a correlation of H
3-14/H-1 disclosed that H-1 is oriented opposite H-10. Thus,
1 was deduced as a C-10 epimer of calyculone I (
6).
Analysis of the
13C NMR and HRESIMS spectral data of
2 reveal the same molecular formula as that of
1 (
Figures S4 and S6). The interpretation of NMR data and COSY and HMBC correlations (
Figure 3) again showed that it is possibly a diastereomer or geometrical isomer of
1. The NOE experiment allowed the relative configuration of
2 to be defined. As shown in
Figure 4, the NOE correlation of H-10/H-15 as well as correlations of H-4/H
3-16 and H
3-16/H-7a conducted to assign an 8
Z olefin and a
trans epoxide. Moreover, correlations of H-1/H
3-14 and H
3-14/H-9 suggested that H-1 and H
3-14 were oriented on opposite face to H-10. Consequently,
2 was determined as an 8
Z epimer of
1.
The HRESIMS of
3 displayed a sodiated molecular ion peak at
m/z 403.2456 [M + Na]
+ (
Figure S7), indicative of a molecular formula of C
22H
36O
5, suggesting five degrees of unsaturation. Its IR spectrum showed characteristic absorptions due to the presence of hydroxy (3437 cm
−1) and ester carbonyl (1732 cm
−1) groups. The latter was determined as an acetoxy functionality based on the NMR resonances at δ
C 170.9 (C), δ
C 21.4 (CH
3), and δ
H 2.07 (
Table 2). The
13C NMR spectrum (
Figure S9) also showed characteristic signals composed of two olefins [δ
C 151.8 (C), 137.7 (C), 131.7 (CH), and 118.2 (CH)], an epoxide [δ
C 62.2 (CH) and 61.6 (C)], and three oxygen-bearing carbons [δ
C 74.8 (CH), 74.0 (C), and 59.5 (CH
2)]. The above functionalities accounted for four of the five degrees of unsaturation, implying
3 to be monocyclic. Except for the acetyl group, its NMR data resemble those of sinulariol L [
13], while 2D NMR interpretation revealed that
3 was a 3-
O-acetyl analogue of sinulariol L. Analysis of NOE correlations of
3 ascertained that they shared the same relative configurations at C-3, C-4, C-11, and C-12, as well as the same geometries of C-1 and C-7 double bonds.
The molecular formula of
4 was determined as C
20H
32O
3 by the analysis of its HRESIMS and
13C NMR spectrum (
Figures S10 and S12). Its NMR spectroscopic data (
Table 2) also were found to be mostly similar to those of sinulariol L [
13]. Inspection of
1H and
13C NMR data of
4 suggested the presence of two epoxides [δ
C 61.9 (CH), 61.3 (C × 2), and 59.5 (CH); δ
H 3.33 (1H, d,
J = 6.8 Hz) and 2.73 (1H, dd,
J = 10.0, 4.0 Hz)]. These data suggested
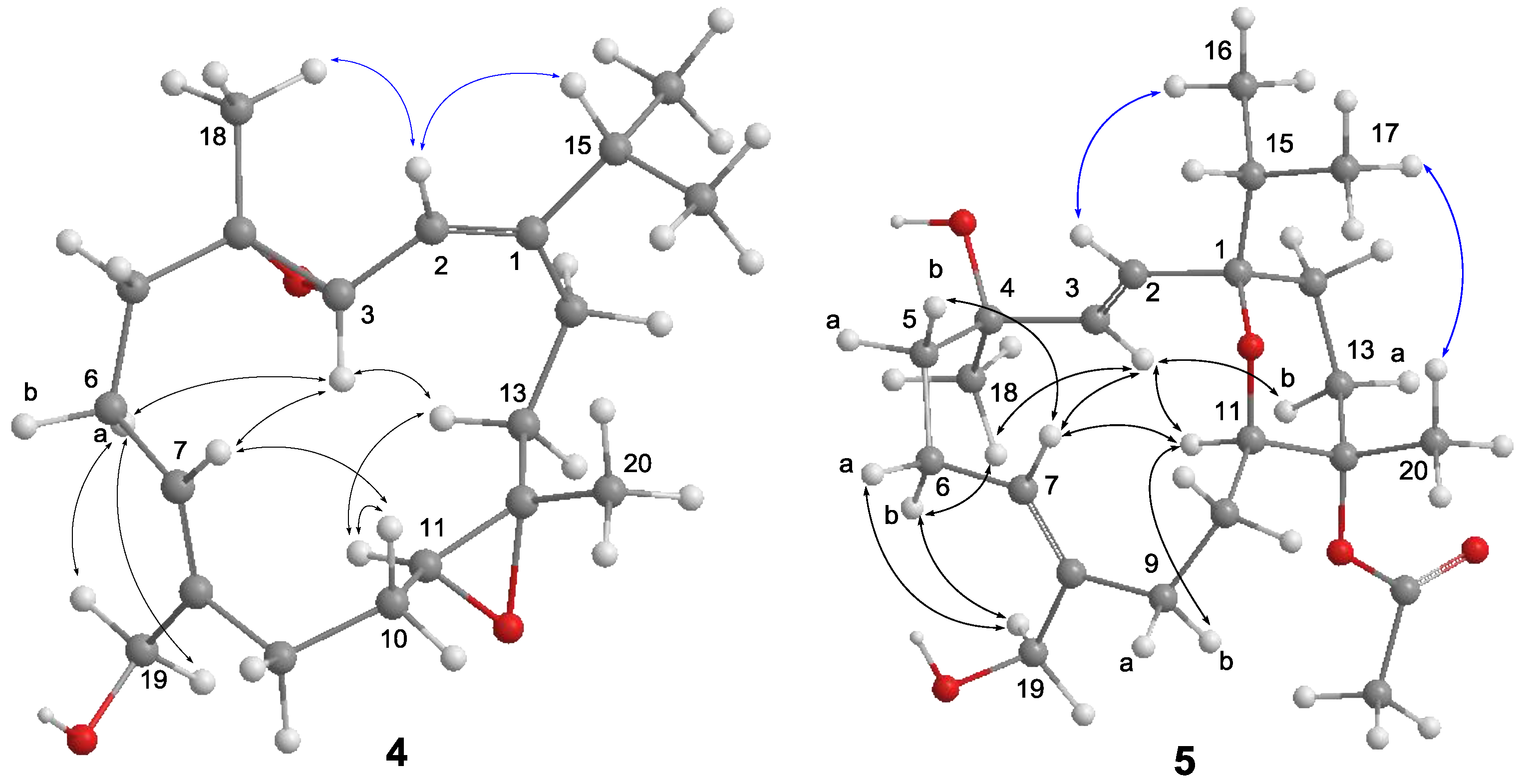
4 to be a 3,4-epoxide analogue of sinulariol L, which was corroborated by the HMBC correlations from H
3-18 to C-3, C-4, and C-5 as well as COSY correlation between H-2 and H-3. The 1
E and 7
Z geometries were inferred according to the NOE correlations of H-2/H-15 and H
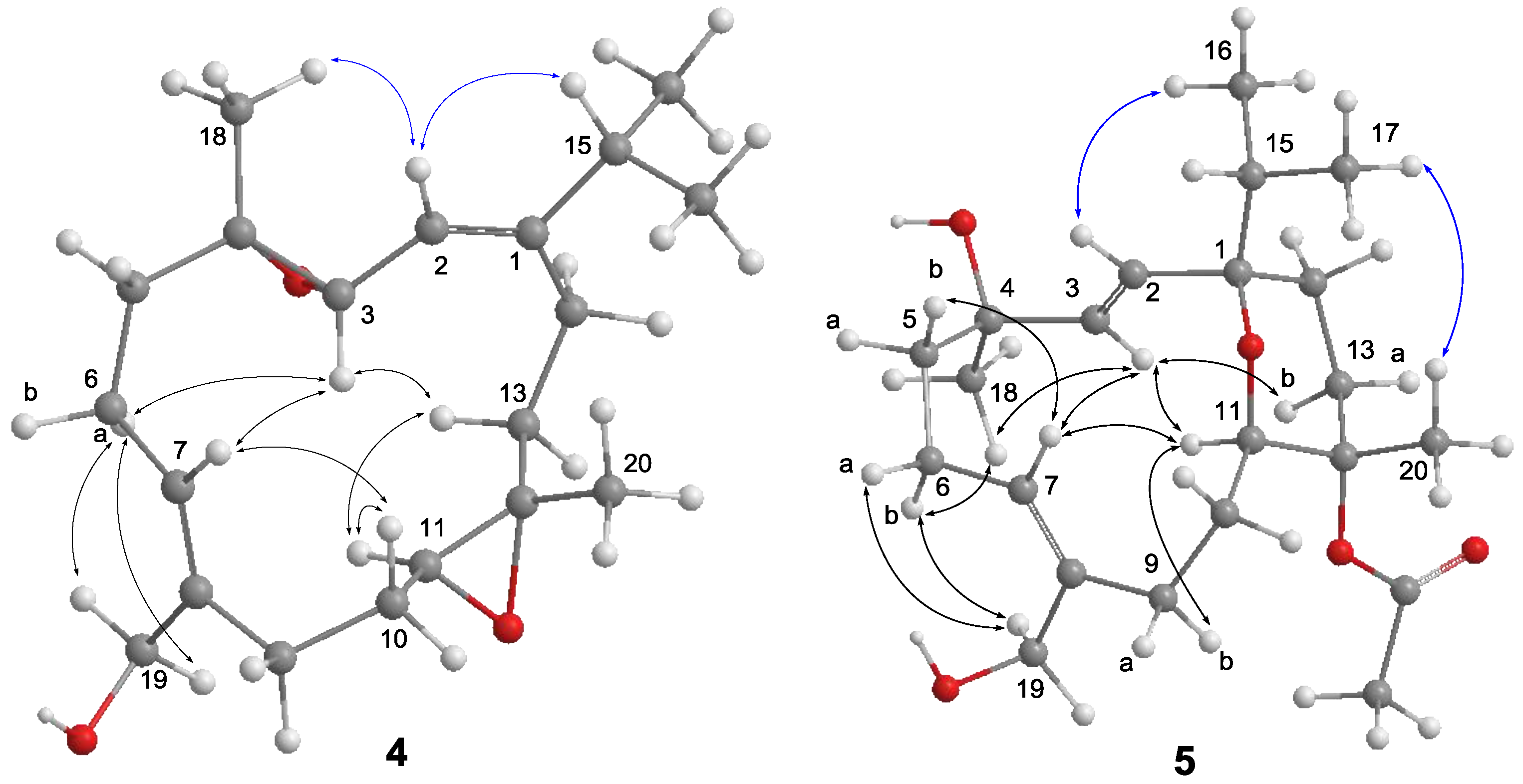
2-19/H-6a, while correlations of H
3-18/H-2 and H-11/H-13b suggested the
trans geometries for both epoxides (
Figure 5). The relative configurations at C-3, C-4, C-11, and C-12 were determined by further analysis of NOE correlations. The NOE correlations of H-3/H-6a, H-3/H-7, H-3/H-13b, and H-11/H-13b indicated the spatial proximity among these protons and, thus, a 3
S*, 4
S*, 11
S*, 12
S*-configuration was determined.
Compound
5 has a molecular formula of C
22H
36O
5 as determined by HRESIMS and
13C NMR spectrum (
Figures S13 and S15). The NMR data of
5 (
Table 2) substantially resemble those of sinulariol J [
13], except for the presence of an additional acetyl group featured resonances at δ
C 170.2 (C), δ
C 22.4 (CH
3), and δ
H 1.98 (3H, s), suggesting
5 to be an
O-acetyl analogue of sinulariol J. The downfield-shifted C-12 (δ
C 81.9 (C) and H
3-20 (δ
H 1.44) and the HMBC correlations from H
3-20 to C-11, C-12, and C-13 evidenced that this acetoxy group should be attached at C-12. The 2
E and 7
Z geometries were determined to be the same as those of sinulariol J according to the large coupling constant (
J = 16.0 Hz) between H-2 and H-3, as well as NOE correlations of H
2-19/H
2-6 (
Figure 5). In addition, NOE correlations of H-3/H-11 and H-3/H
3-18 suggested they were oriented on the same face, and arbitrarily assigned as α-orientation. The isopropyl group and H
3-20 were, thus, assigned as β-orientation based on the correlations of H-2/H
3-16 and H
3-17/H
3-20. Consequently, an 1
R*,4
R*,11
R*,12
S*-configuration was assigned for
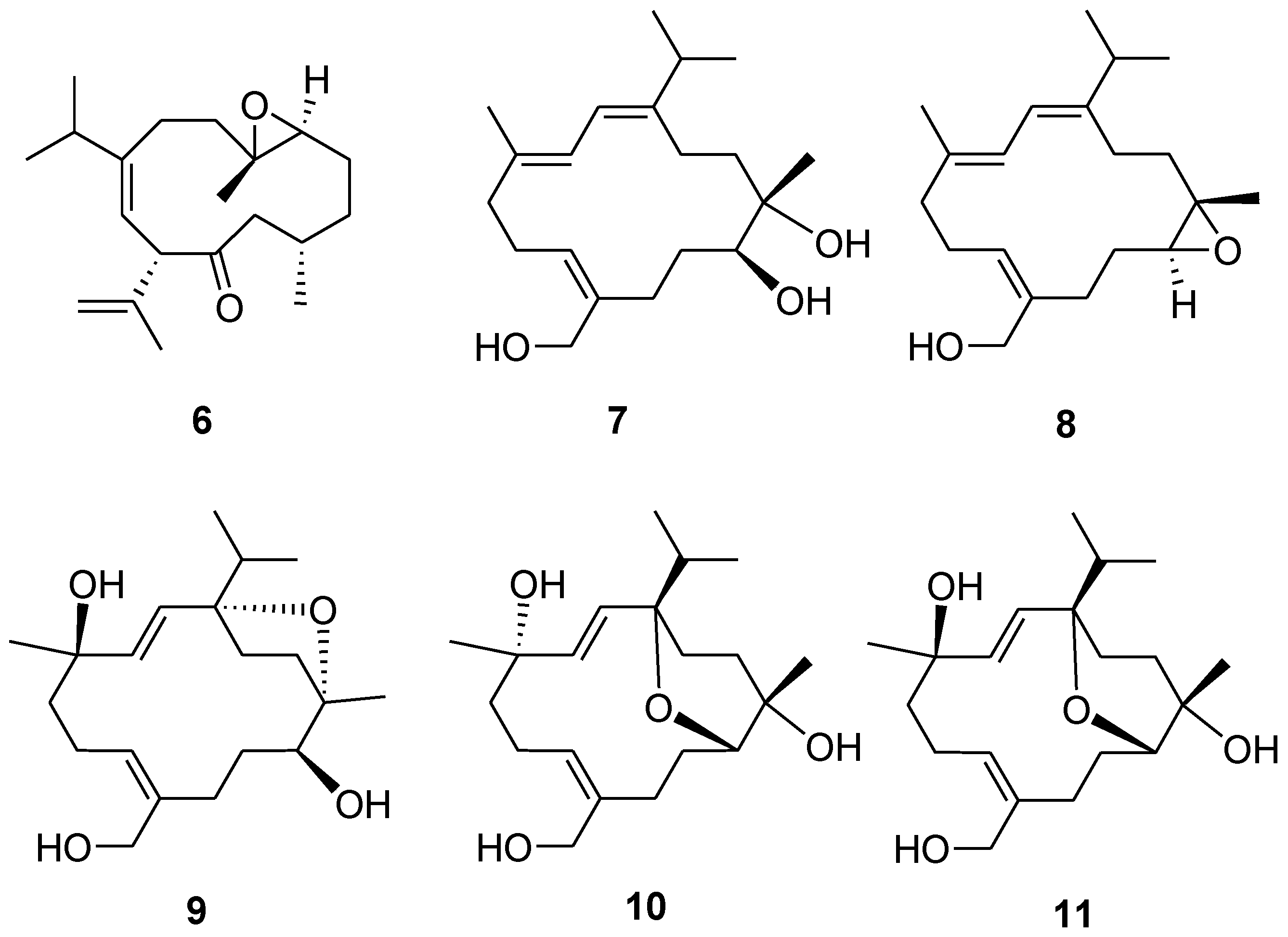
5. Moreover, the NMR spectroscopic data of six known compounds
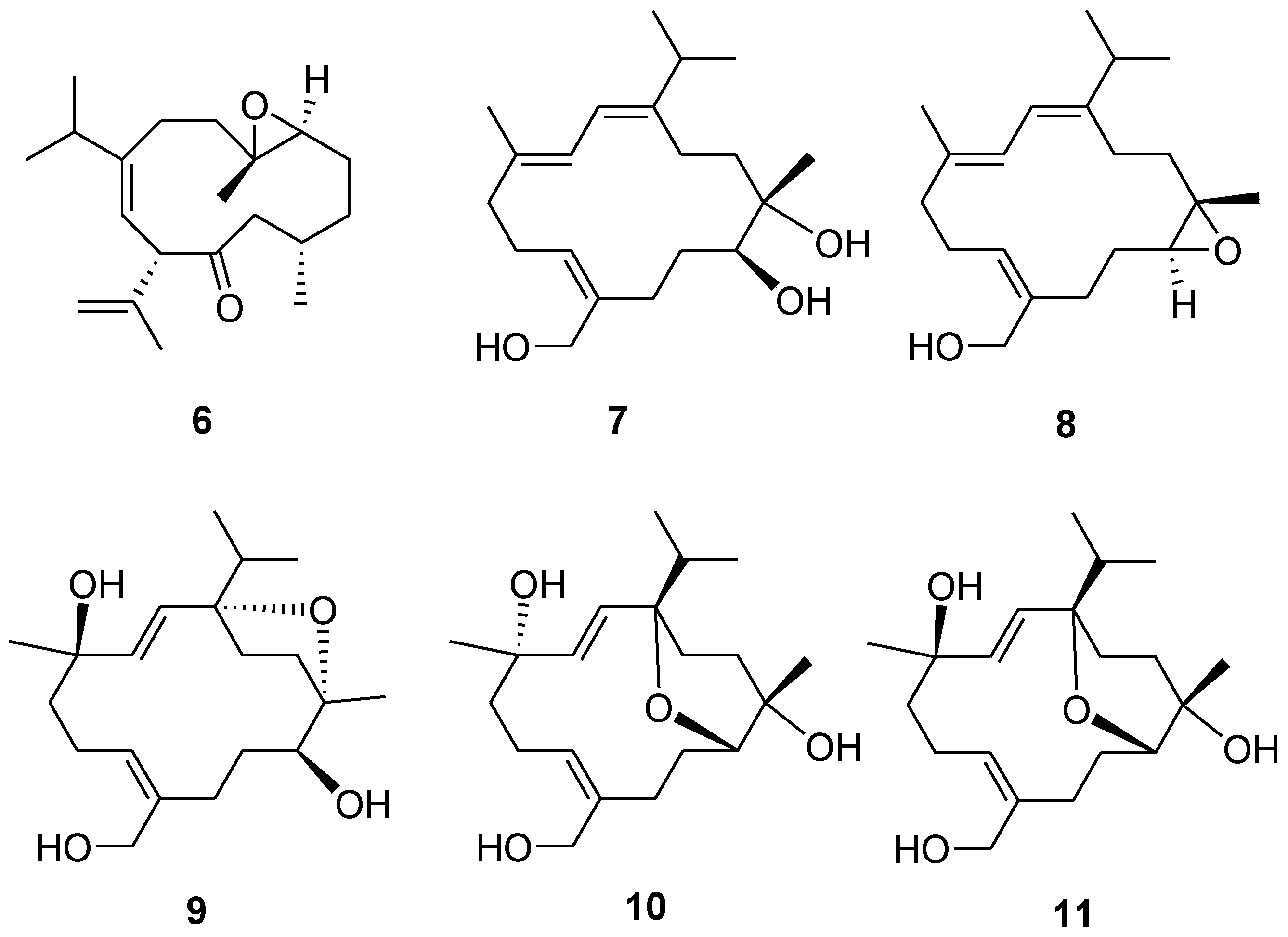
6–
11 (
Figure 6) were found to be identical to those of known compounds based on the comparison of their physical and spectroscopic data with those reported in the literature [
5,
13].
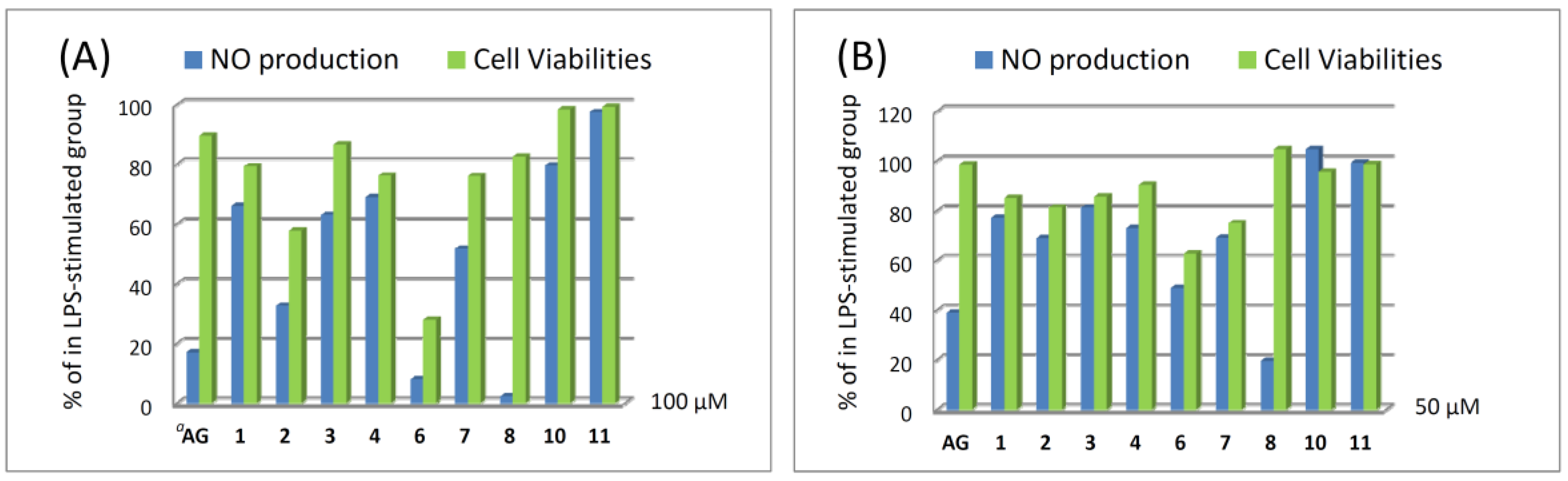
Compounds
1–
11 were evaluated for cytotoxicity activities against P388, K562, and HT-29 cell lines using the Alamar Blue assay. The result showed that they are not cytotoxic against the above three cancer cell lines. In addition, the nitric oxide (NO) inhibitory activities of compounds
1–
4,
6–
8,
10, and
11 were further evaluated by assay of LPS-stimulated NO production in activated RAW264.7 cells, as shown in
Figure 7. The results indicated that compounds
2,
6, and
8 could effectively reduce the levels of NO to 32.6%, 8.0%, and 2.3%, respectively, at a concentration of 100 μM. Moreover, compound
8 at a concentration of 50 μM exhibited good inhibitory activity compared to the positive control aminoguanidine (AG), and the level of NO was also reduced significantly to 19.6%, while giving a 104.6% retention of cell viability. Thus, compound
8 is a promising metabolite which may become a lead compound in the future anti-inflammatory drug development.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO P-1020 digital polarimeter (JASCO Corporation, Tokyo, Japan). IR spectra were recorded on a JASCO FT/IR-4100 infrared spectrophotometer (JASCO Corporation). The 1H NMR and 13C NMR spectra were recorded on a Varian 400MR NMR (400 MHz for 1H and 100 MHz for 13C) and a Varian Unity INOVA500 FT-NMR (500 MHz for 1H and 125 MHz for 13C) instruments (Varian Inc., Palo Alto, CA, USA). The chemical shifts were referenced to the solvent residue of CDCl3 (δH 7.265 ppm and δC 77.0 ppm). The high-resolution mass spectra were acquired via a Bruker APEX II mass spectrometer with an ESI ionization source (Bruker, Bremen, Germany). Silica gel 60 (40−63 μm, Merck, Darmstadt, Germany), C18 gel (LiChroprep RP-18, 40−63 μm, Merck), and Sephadex LH-20 (GE Healthcare, Uppsala, Sweden) were used for column chromatography. Thin-layer chromatography (TLC) analysis was performed on a precoated silica gel plates (Kieselgel 60 F254, 0.25 mm, Merck). High-performance liquid chromatography (HPLC) was performed using a Shimadzu LC-10ATVP series pump equipped with a UV detector (Shimadzu, Milan, Italy) and a semipreparative RP-18 column (5 µm, 250 × 10 mm, Hibar Purospher RP-18e, Merck).
3.2. Animal Material
The soft coral Sinularia nanolobata was collected off the coast of Jihui Fishing Port, Taitung county, Taiwan (latitude: 23°07′ N; longitude: 121°23′ E) in March 2013. The material was frozen at −20 °C until extraction in the laboratory. Species identification of this coral was performed by Prof. C.-F. Dai (National Taiwan University, Taiwan), while the voucher specimen (JiH-201303) was deposited at the Department of Marine Biotechnology and Resources, National Sun Yat-sen University.
3.3. Extraction and Isolation
The minced, wet tissues of S. nanolobata (3.1 kg) were extracted with EtOH (3L × 2). The extract was concentrated under reduced pressure and the residue was partitioned between EtOAc and H2O. The EtOAc layer was dried over Na2SO4 and the resulting residue (27.9 g) was subjected to a silica gel column with a gradient of acetone and hexane in an increasing polarity (acetone-hexane, 2% to 100%), and then a gradient of MeOH and acetone (MeOH-acetone, 2% to 100%) to yield 18 fractions. Fraction 7 was chromatographed on a silica gel column with acetone/hexane (6%) to yield two subfractions 7a and 7b. Subfraction 7a was purified by a C18 gel column (MeOH-H2O, 90%), followed by semipreparative HPLC (MeOH-H2O, 78%) to yield compounds 1 (34.3 mg), 2 (12.0 mg) and 6 (12.6 mg). Fraction 10 was purified by a Sephadex LH-20 column using acetone as eluent to obtain a mixture, which was further separated by silica gel column chromatography (acetone-hexane, 10% to 100%), C18 column chromatography (MeOH-H2O, 90%), and semipreparative HPLC (MeOH-H2O, 83%) to yield compounds 4 (2.6 mg) and 8 (5.5 mg). Fraction 14 was purified by a Sephadex LH-20 column (acetone, 100%) and semipreparative HPLC (MeOH-H2O, 80%) to yield compounds 3 (5.2 mg) and 5 (1.2 mg), and (MeOH-H2O, 67%) to yield compounds 7 (3.8 mg) and 9 (6.4 mg). Fraction 15 was purified by a Sephadex LH-20 column using acetone as eluent to obtain a mixture, which was further separated by silica gel column chromatography (acetone-hexane, 20% to 100%), C18 column chromatography (MeOH-H2O, 90%), and semipreparative HPLC (MeOH-H2O, 66%) to yield compounds 10 (2.9 mg) and 11 (2.6 mg).
Nanoculone A (
1): colorless oil;
+6 (
c 1.20, CHCl
3); IR (neat) ν
max 2962, 1710, 1644, 1457, 1379, and 895 cm
−1;
13C and
1H NMR data, see
Table 1; ESIMS
m/z 327 [M + Na]
+; HRESIMS
m/z 327.2298 [M + Na]
+ (calcd. for C
20H
32O
2Na, 327.2300).
Nanoculone B (
2): colorless oil;
+175 (
c 0.49, CHCl
3); IR (neat) ν
max 2957, 2866, 1705, 1455, 1105, and 895 cm
−1;
13C and
1H NMR data, see
Table 1; ESIMS
m/z 327 [M + Na]
+; HRESIMS
m/z 327.2297 [M + Na]
+ (calcd. for C
20H
32O
2Na, 327.2300).
Nanolobol A (
3): colorless oil;
+3 (
c 1.30, CHCl
3); IR (neat) ν
max 3437, 2957, 2960, 1732, 1459, 1372, 1246, 1020, and 756 cm
−1;
13C and
1H NMR data, see
Table 2; ESIMS
m/z 403 [M + Na]
+; HRESIMS
m/z 403.2456 [M + Na]
+ (calcd. for C
22H
36O
5Na, 403.2455).
Nanolobol B (
4): colorless oil;
−67 (
c 0.65, CHCl
3); IR (neat) ν
max 3441, 2923, 2960, 1731, 1462, 1024, and 856 cm
−1;
13C and
1H NMR data, see
Table 2; ESIMS
m/z 343 [M + Na]
+; HRESIMS
m/z 343.2244 [M + Na]
+ (calcd. for C
20H
32O
3Na, 343.2244).
Nanolobol C (
5): colorless oil;
−86 (
c 0.20, CHCl
3); IR (neat) ν
max 3384, 2959, 2874, 1724, 1601, 1438, 1367, 1256, and 1095 cm
−1;
13C and
1H NMR data, see
Table 2; ESIMS
m/z 403 [M + Na]
+; HRESIMS
m/z 403.2456 [M + Na]
+ (calcd. for C
22H
36O
5Na, 403.2455).
Calyculone I (
6): colorless needles;
+41 (
c 1.09, CHCl
3); lit.
+52.1 (c 1.0, CHCl
3) [
5]; IR (neat) ν
max 2959, 1708, 1463, 1385 and 897 cm
−1; ESIMS
m/z 455 [M + Na]
+ (calcd. for C
20H
32O
2Na).
Sinulariol A (
7): colorless oil;
+47 (
c 0.95, CHCl
3); lit.
+17.1 (c 0.79, CHCl
3) [
13]; IR (neat) ν
max 3369, 2959, 2871, 1447, 1379 and 756 cm
−1; ESIMS
m/z 345 [M + Na]
+ (calcd. for C
20H
34O
3Na).
Sinulariol C (
8): colorless oil;
−30 (
c 0.31, CHCl
3); lit.
−8.8 (c 0.15, CHCl
3) [
13]; IR (neat) ν
max 3421, 2960, 1671, 1458, 1384, 1021, 857, 755 and 669 cm
−1; ESIMS
m/z 327 [M + Na]
+ (calcd. for C
20H
32O
2Na).
Sinulariol D (
9): colorless oil;
+13 (
c 0.46, CHCl
3); lit.
+7.5 (c 1.35, CHCl
3) [
13]; IR (neat) ν
max 3355, 2963, 2873, 1653, 1455, 1367, 1077 and 756 cm
−1; ESIMS
m/z 361 [M + Na]
+ (calcd. for C
20H
34O
4Na).
Sinulariol H (
10): colorless oil;
−78 (
c 0.73, CHCl
3); lit.
−71.3 (c 0.28, CHCl
3) [
13]; IR (neat) ν
max 3371, 2963, 2933, 1444, 1374 and 756 cm
−1; ESIMS
m/z 361 [M + Na]
+ (calcd. for C
20H
34O
4Na).
Sinulariol J (
11): colorless oil;
−77 (
c 0.65, CHCl
3); lit.
−40.5 (c 0.15, CHCl
3) [
13]; IR (neat) ν
max 3350, 2959, 2871, 1447, 1379 and 756 cm
−1; ESIMS
m/z 361 [M + Na]
+ (calcd. for C
20H
34O
4Na).
3.4. Crystallographic Data of 6
The colorless crystal size of 0.15 × 0.10 × 0.10 mm was obtained at 4 °C in a refrigerator by slow evaporation in an acetone solution. Diffraction intensity data were acquired with a CCD area detector with graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Crystal data for 6: C20H32O2, M = 304.45, monoclinic, a = 8.4794 (6) Å, b = 11.0386 (8) Å, c = 10.2704 (7) Å, α = 90°, β = 104.094 (3)°, γ = 90°, V = 932.38 (11) Å3, T = 100 (2) K, space group P21, Z = 2, μ (CuKα) = 0.521 mm−1, 6301 reflections collected, 3100 independent reflections (Rint = 0.0201). The final R1 values were 0.0275 (I > 2σ(I)). The final wR (F2) values were 0.0733 (I > 2σ(I)). The final R1 values were 0.0276 (all data). The final wR (F2) values were 0.0734 (all data). The goodness of fit on F2 was 1.050. Flack parameter = 0.1 (2). Crystallographic data for 6 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1482769). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK.
3.5. Cytotoxicity Assay
The Alamar Blue assay were performed as previous reported [
14,
15]. After the cell lines (P388, K562, and HT-29) were cultured for 15 h according to the published procedure [
16], the tested compounds in DMSO solutions were added and cultured for 72 h. The attached cells were incubated with Alamar Blue (10 μL/well, 4 h) and the absorbance was measured at wavelength of 595 nm using a microplate reader.
3.6. Nitric Oxide Inhibitory Activity
The nitrite concentration in the culture medium was measured as an indicator of NO production according to the Griess reaction [
17]. Briefly, 80 μL of cell culture supernatant was reacted with 100 μL of Griess reagent (1:1 mixture of 0.1%
N-(1-naphthyl)ethylenediamine dihydrochloride in water and 1% sulfanilamide in 5% phosphoric acid) in a 96-well plate and incubated at room temperature for 10 min. The absorbance at 550 nm was recorded using the ELISA reader [
18,
19]. Fresh medium was used as the blank. The results are expressed as the percentage of inhibition calculated relative to the cells treated with vehicle and LPS.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}