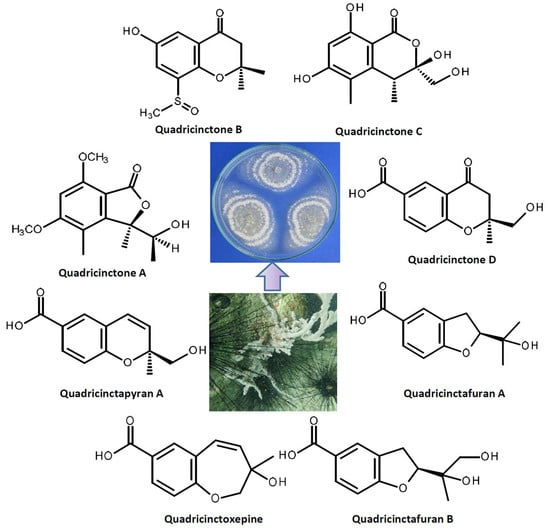
2. Results and Discussion
Compound
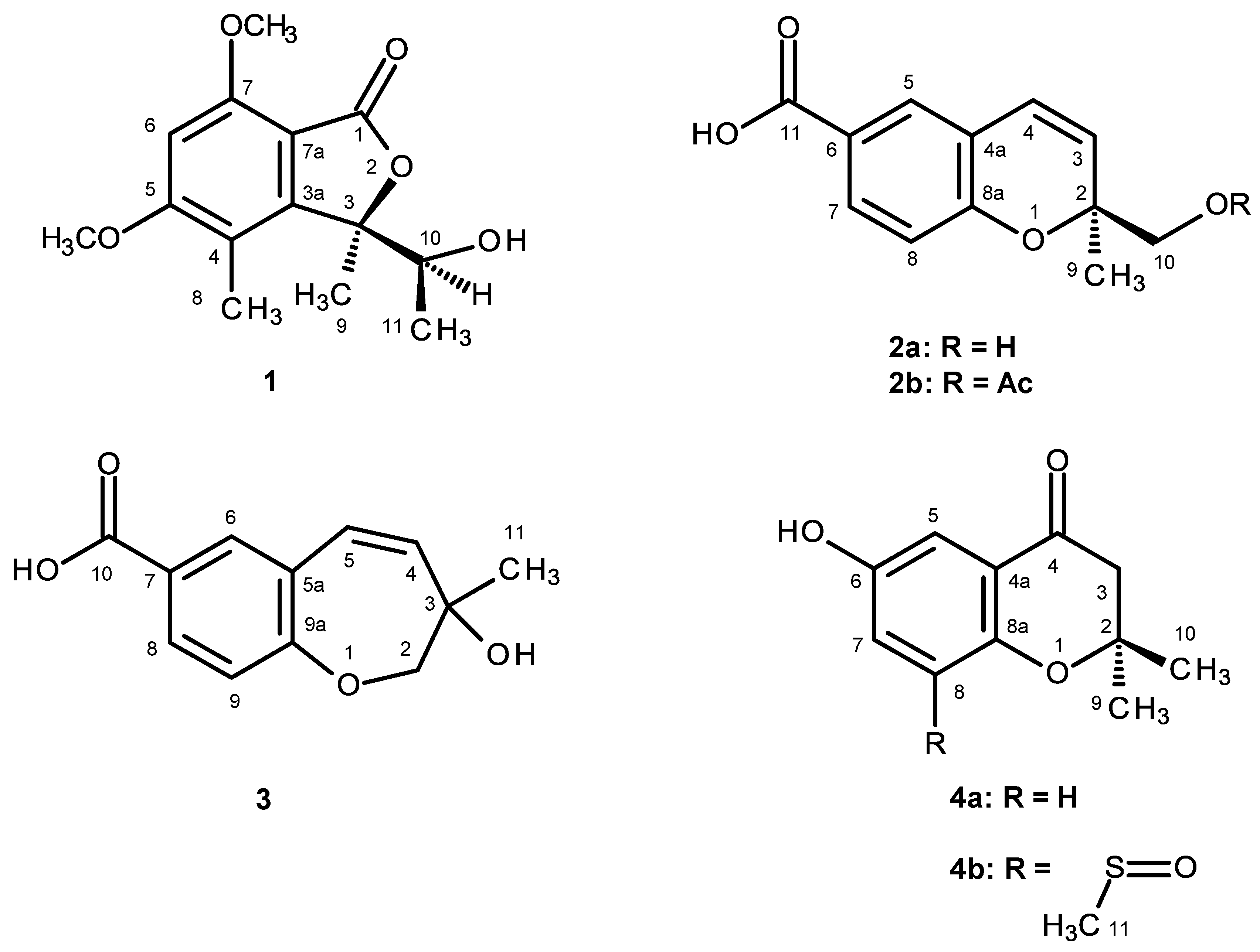
1 was isolated as white crystals (mp, 176–177 °C), and its molecular formula C
14H
18O
5 was established on the basis of the (+)-HRESIMS
m/z 267.1243 [M + H]
+ (calculated 267.1332), indicating six degrees of unsaturation. The IR spectrum showed absorption bands for hydroxyl (3455 cm
−1), conjugated ester carbonyl (1723 cm
−1) and aromatic (1612, 1596 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 1,
Supplementary Information, Figures S2 and S4) exhibited the signals of one conjugated ester carbonyl (δ
C 168.2), five quaternary sp
2 (δ
C 164.6, 158.3, 152.8, 111.7, 105.5), one methine sp
2 (δ
C 94.5), one oxygen bearing quaternary sp
3 (δ
C 88.8), one oxygen bearing methine sp
3 (δ
C 70.8), two methoxyl (δ
C 56.1 and 56.0) and three methyl (δ
C 21.5, 17.8 and 11.2) groups. The
1H NMR spectrum (
Table 1,
Supplementary Information, Figure S1) revealed the presence of, besides a singlet of one aromatic proton at δ
H 6.41, a quartet of the oxymethine proton at δ
H 4.22 (
J = 6.4 Hz), two singlets of the methoxyl groups at δ
H 3.97 s and 3.92 s, two methyl singlets at δ
H 2.12 s and 1.76 s, a methyl doublet at δ
H 0.87 (
J = 6.4 Hz) and a broad band of the hydroxyl proton at δ
H 2.15. The
1H and
13C data (
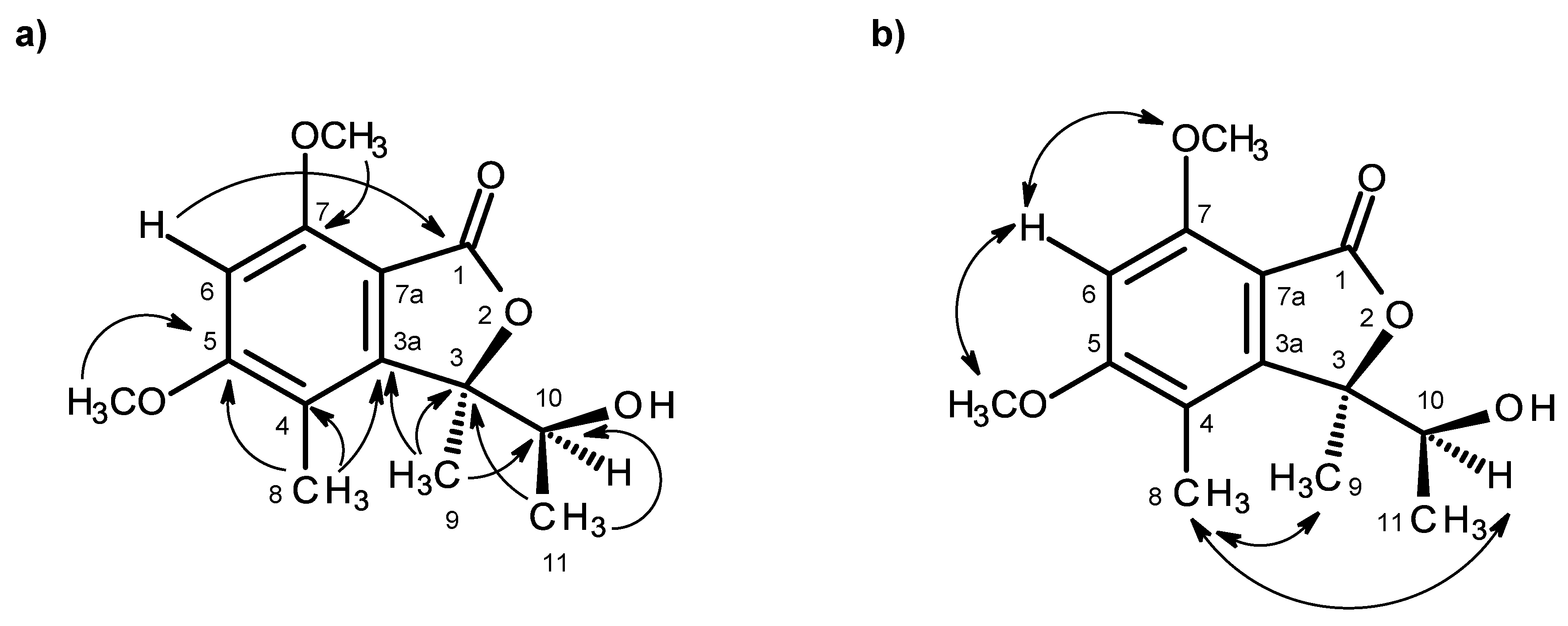
Table 1) revealed the presence of a pentasubstituted benzene ring. That this pentasubstituted benzene ring was part of the 5,7-dimethoxy-3,4-dimethyl-2-benzofuran-1(3
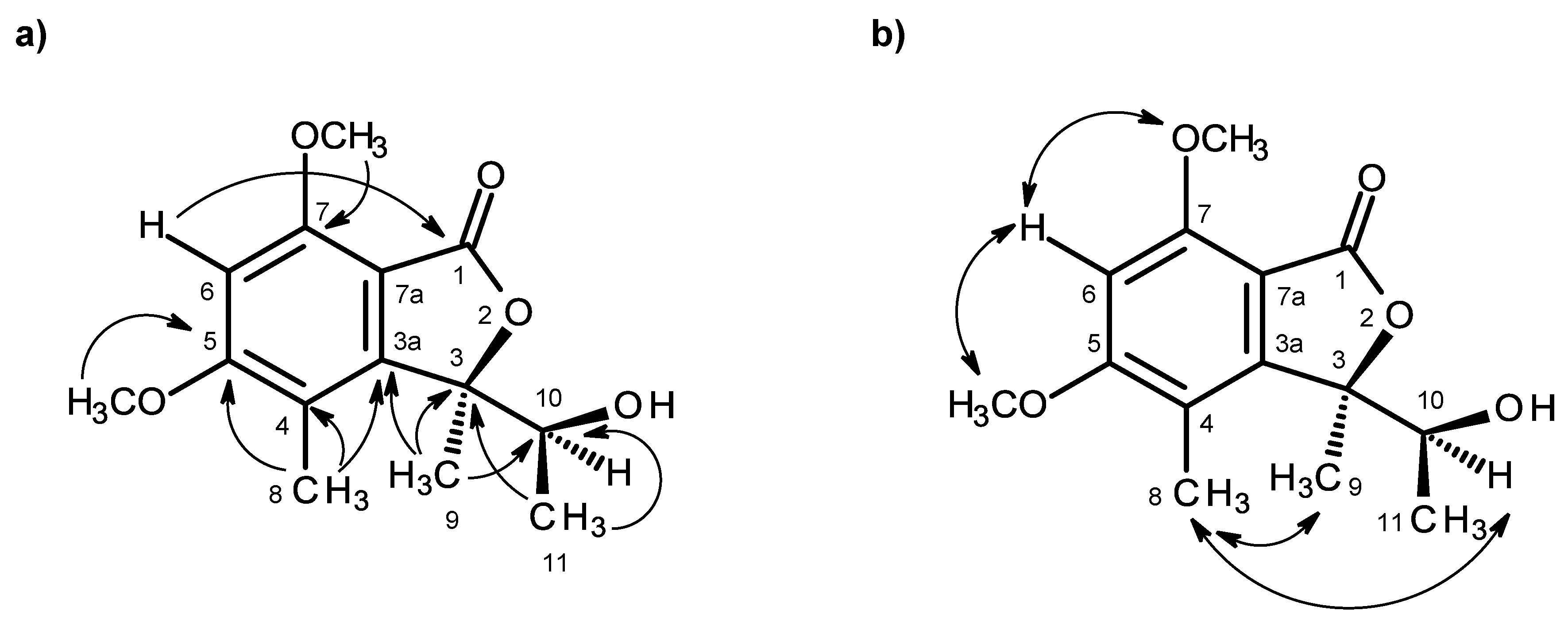
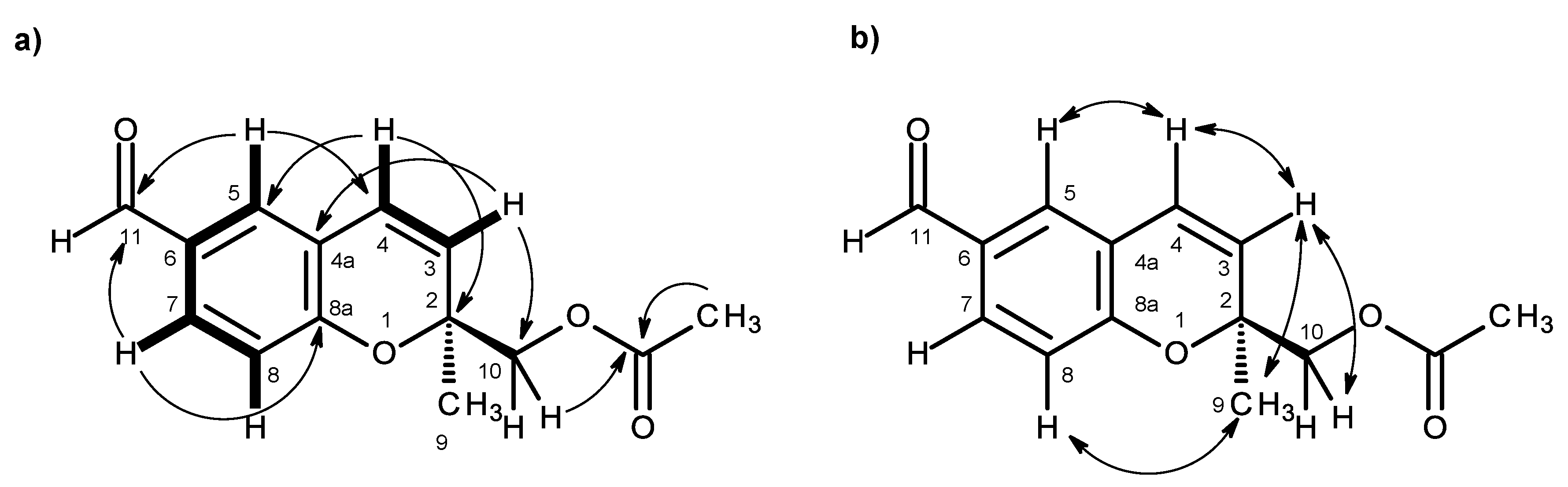
H)-one ring system was corroborated by the HMBC correlations (
Table 1,
Figure 2a,
Supplementary Information, Figure S5) of H-6 (δ
H 6.41, s) to C-7a (δ
C 105.3), C-4 (δ
C 111.7), C-7 (δ
C 158.3), C-5 (δ
C 164.6) and C-1 (δ
C 168.2), of OMe-5 (δ
H 3.92, s) to C-5, of OMe-7 (δ
H 3.97, s) to C-7, of H
3-8 (δ
H 2.12, s) to C-4, C-3a (δ
C 152.8) and C-5, of H
3-9 (δ
H 1.76, s) to C-3a and of the NOESY correlations of H-6 to OMe-5 and OMe-7. That another substituent of C-3 was a 1-hydroxyethyl group was supported by the COSY correlations of H-10 (δ
H 4.22, q,
J = 6.4 Hz) to H
3-11 (δ
H 0.87, d,
J = 6.4 Hz), by the HMBC correlations of H
3-9 to C-10 (δ
C 70.8), C-3 (δ
C 88.8) and C-3a and of H
3-11 (δ
H 0.87, d,
J = 6.4 Hz) to C-3 and C-10 (
Table 1,
Figure 2a), as well as by the NOESY correlations of H
3-8 to H-10, H
3-9, of H
3-11 to H-10, OH-10 and of H
3-9 to H-10 (
Table 1,
Figure 2b,
Supplementary Information, Figure S6). Final proof of the structure and the stereochemistry assigned to compound
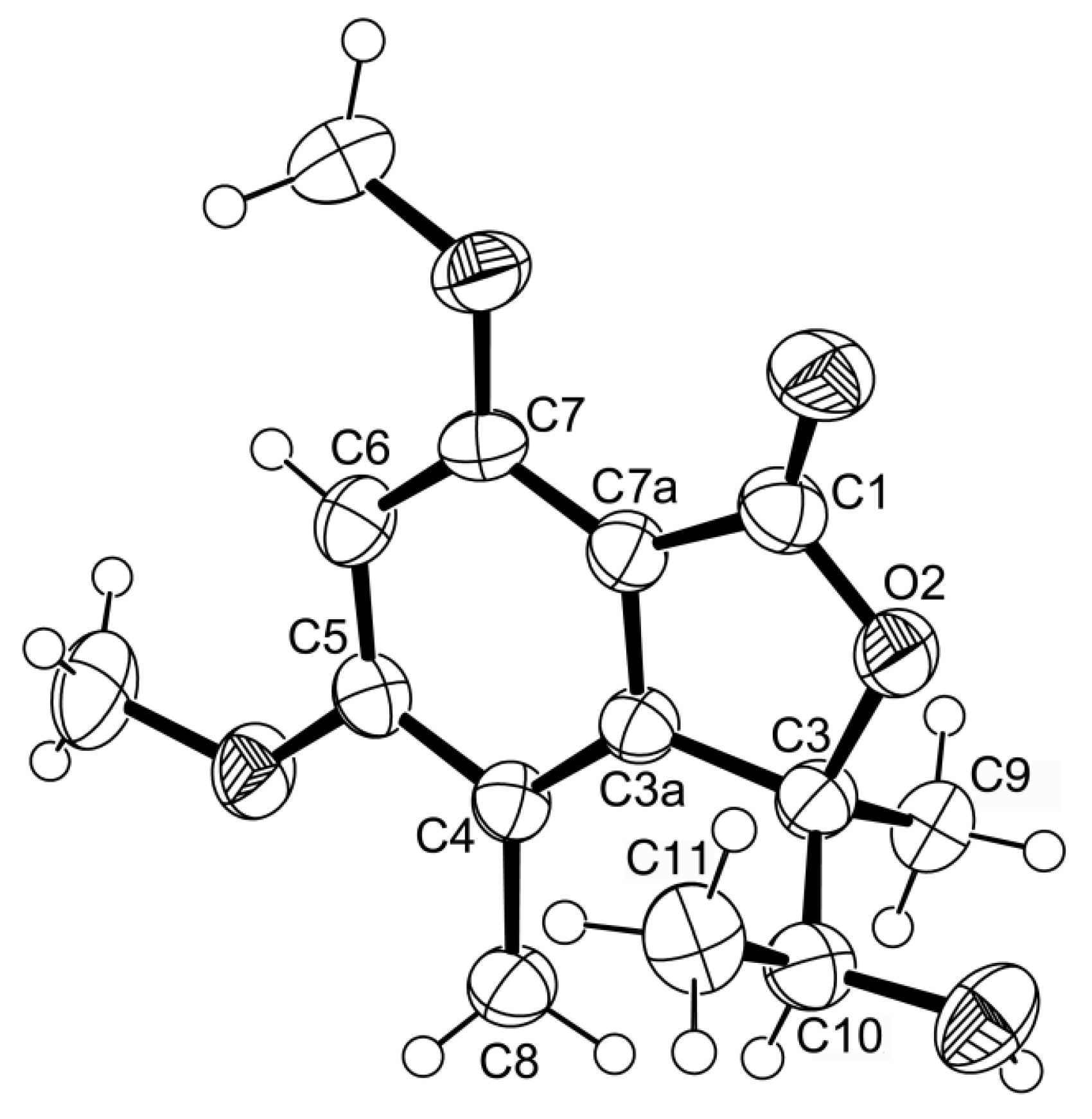
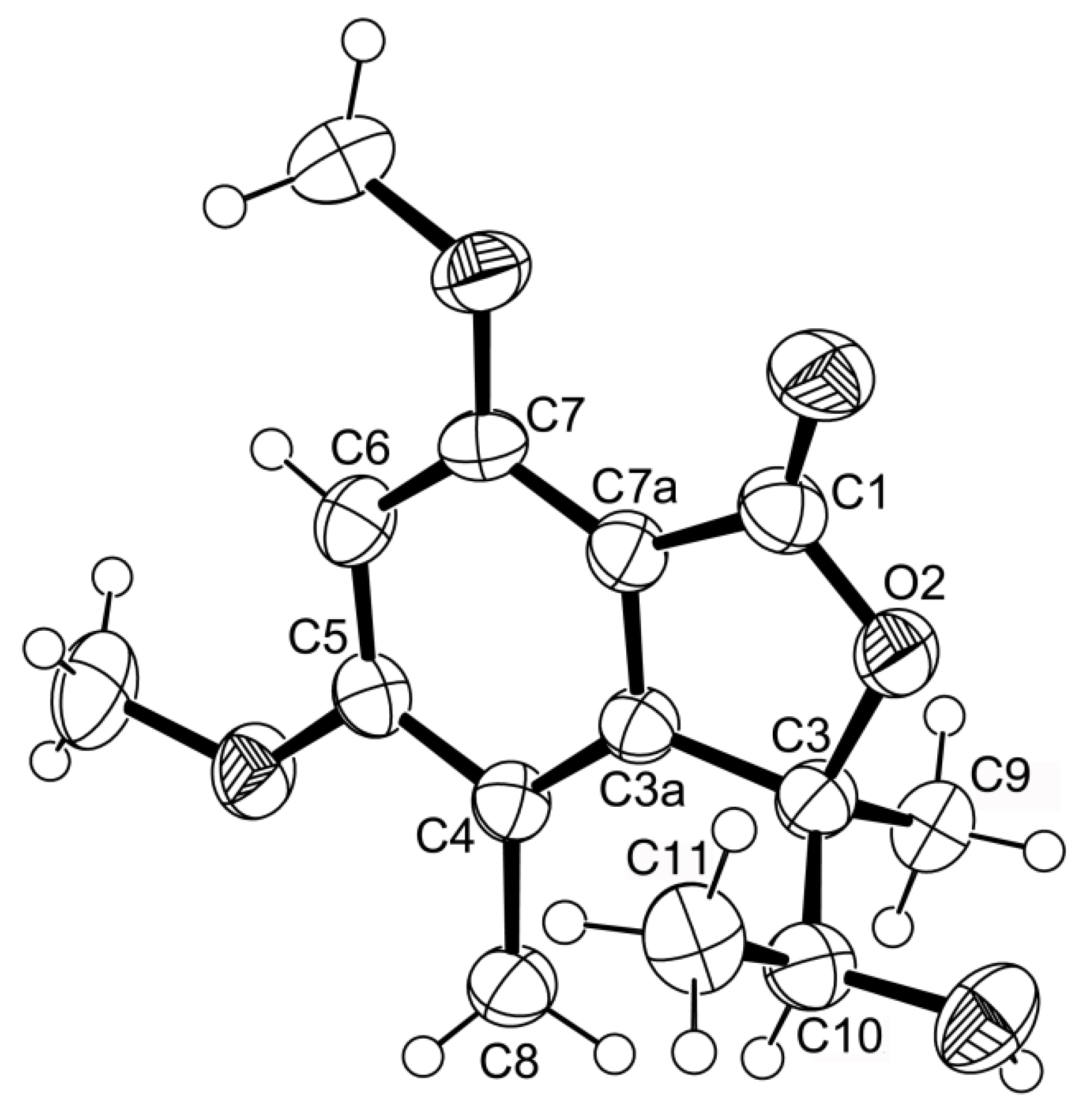
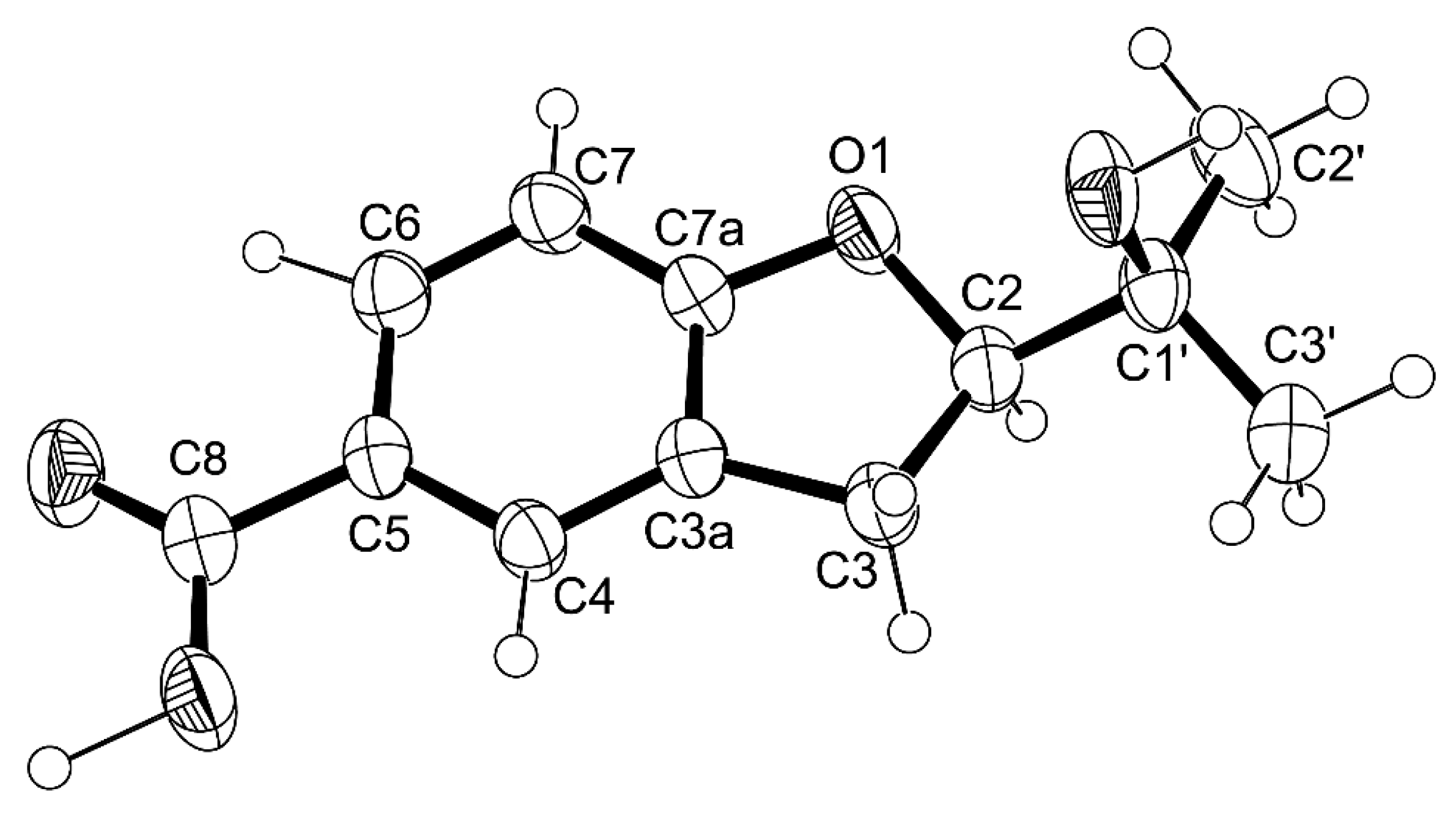
1 was provided by its X-ray analysis (
Figure 3), and since the diffraction data were collected with a Gemini PX Ultra equipped with CuKα radiation, it was possible to establish the absolute configurations of C-3 and C-10, respectively, as 3
R and 10
S. Since
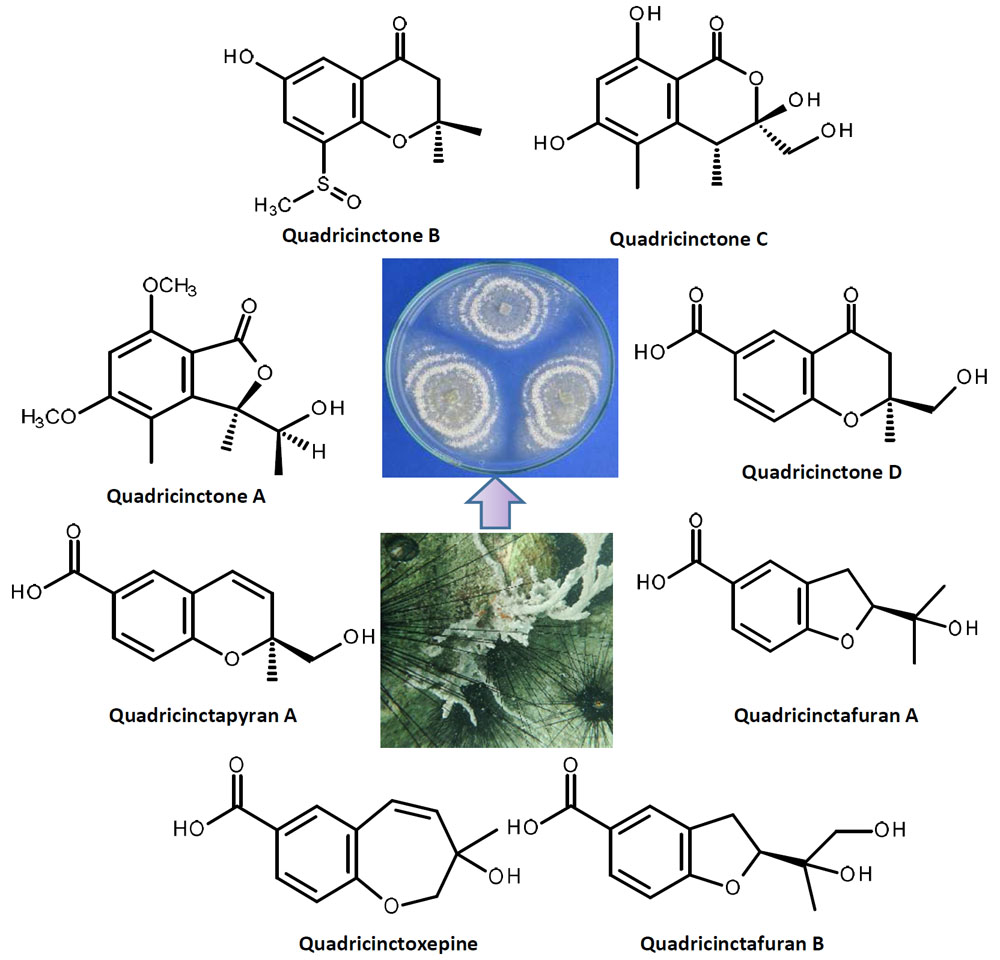
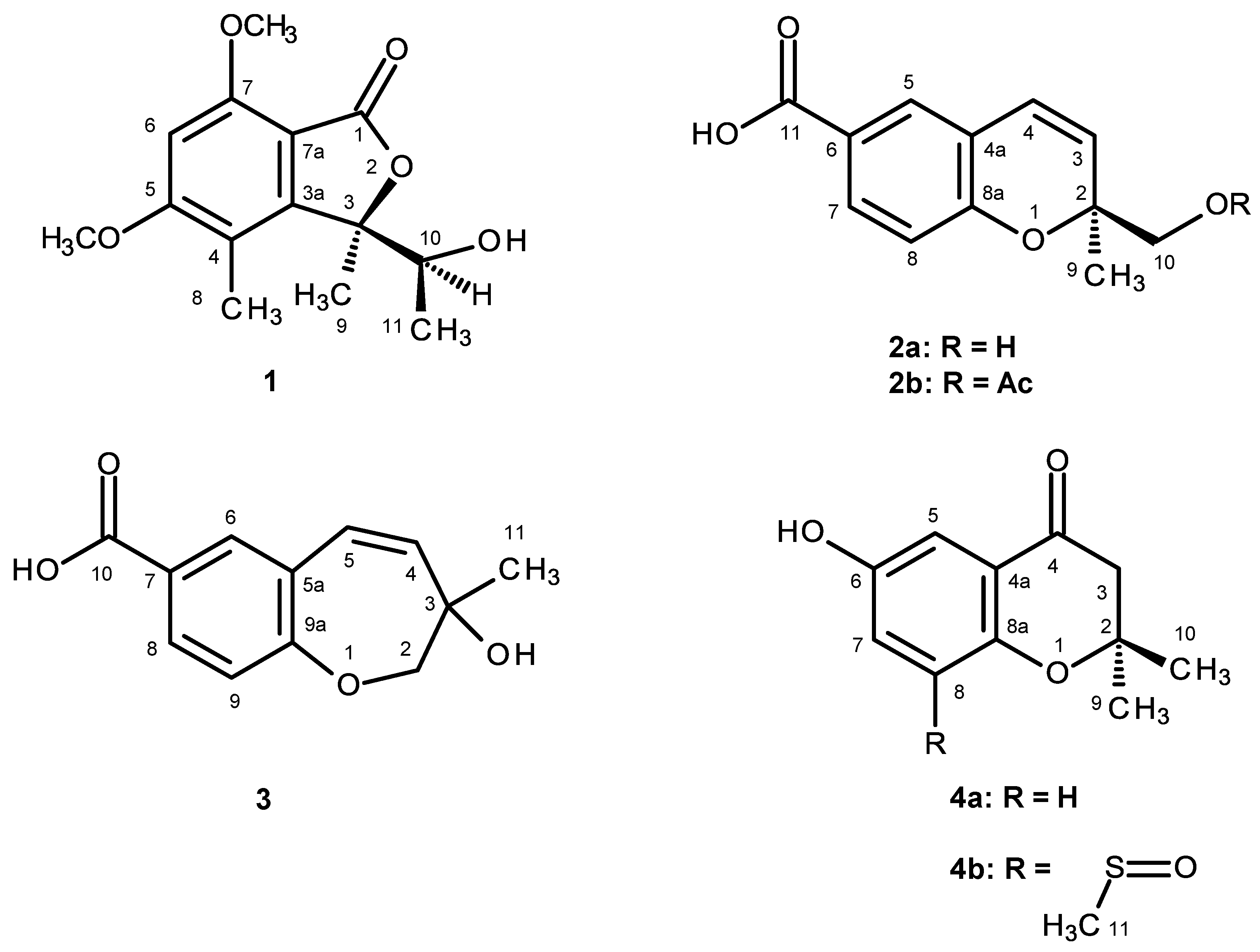
1 is a new compound, we have named it quadricinctone A.
Compound
2a was also isolated as white crystals (mp. 147–148 °C), and its molecular formula C
12H
12O
4 was determined based on the (+)-HRESIMS
m/z 221.0820 [M + H]
+ (calculated 221.0814), indicating seven degrees of unsaturation. The IR spectrum showed absorption bands for hydroxyl (3447 cm
−1), a conjugated carbonyl (1696 cm
−1), aromatic (1609 cm
−1) and olefin (1647 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 2,
Supplementary Information, Figures S8 and S10) exhibited the signals of one conjugated carboxyl carbonyl (δ
C 167.0), three quaternary sp
2 (δ
C 156.8, 123.0, 120.5), five methine sp
2 (δ
C 130.8, 128.6, 127.9, 122.8, 115.7), one oxy-quaternary sp
3 (δ
C 80.5), one oxymethylene sp
3 (δ
C 67.1) and one methyl (δ
C 23.3) groups. The
1H NMR spectrum (
Table 2,
Supplementary Information, Figures S7) revealed, besides the presence of three aromatic protons of the 1,2,4 trisubstituted benzene ring at δ
H 6.79, d (
J = 8.4 Hz), 7.65, d (
J = 2.1 Hz) and 7.69, dd (
J = 8.4, 2.1 Hz), two doublets of the protons of a
cis-double bond at δ
H 6.56, d (
J = 10.0 Hz) and 5.74, d (
J = 10.0 Hz), a methyl singlet at δ
H 1.31, a singlet of two protons at δ
H 3.46 and two broad signals of the hydroxyl protons at δ
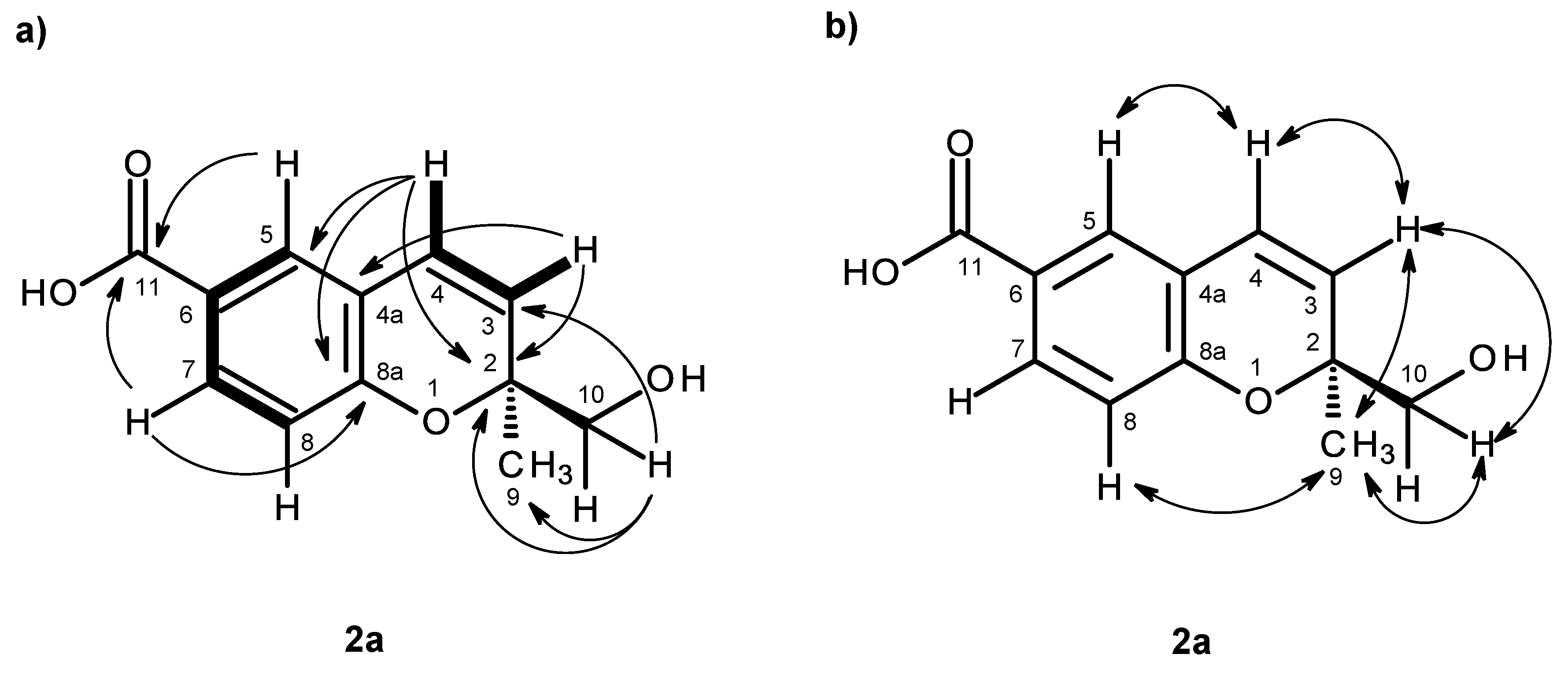
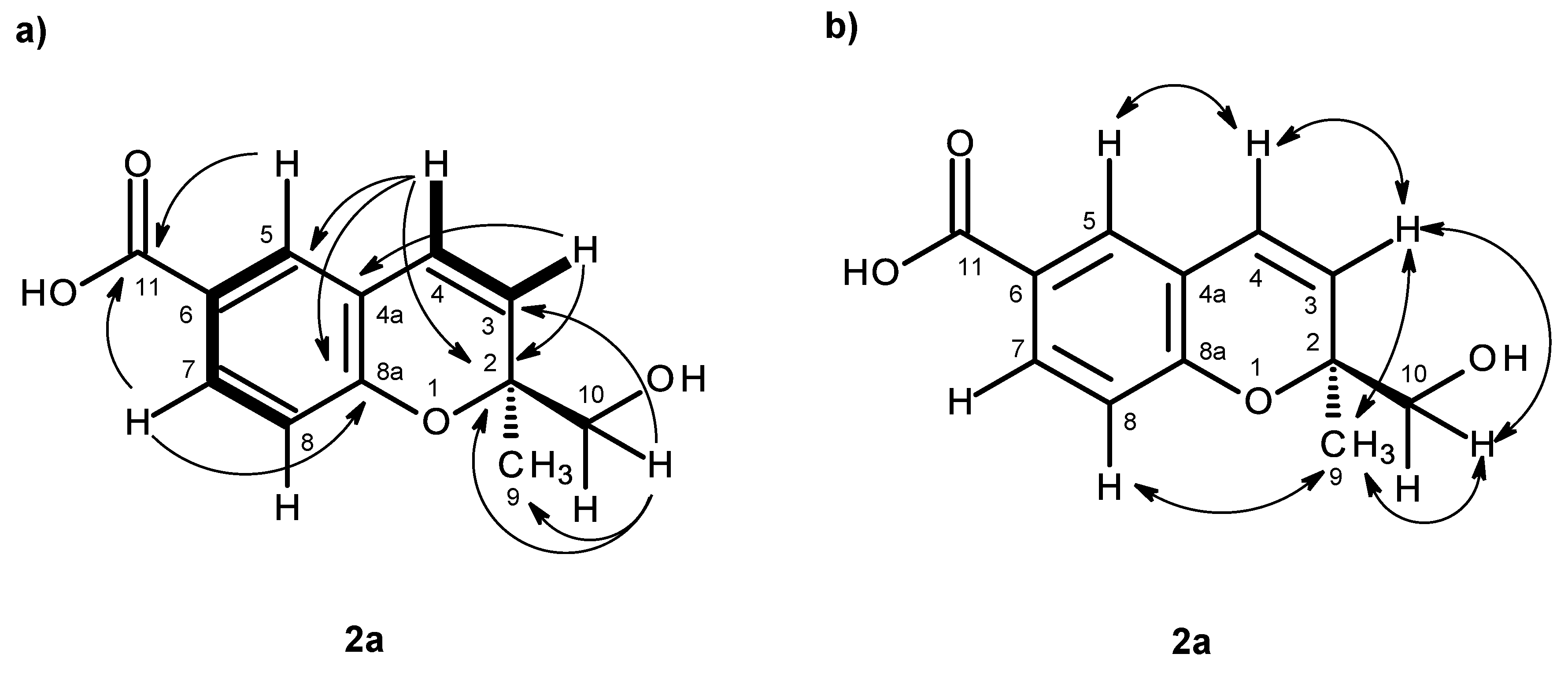
H 12.59 and 5.07, respectively. The COSY spectrum (
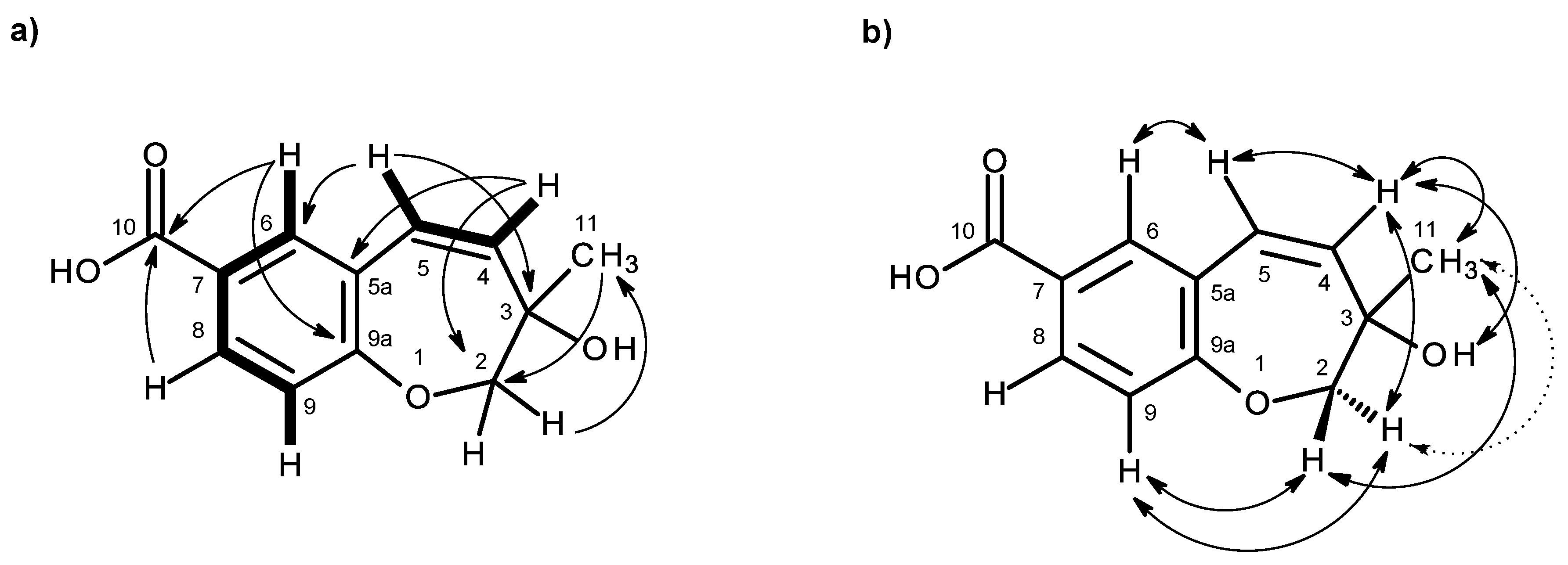
Table 2,
Figure 4a,
Supplementary Information, Figures S9) exhibited cross-peaks of H-4 (δ
H 6.56, d,
J = 10.0 Hz) to H-3 (δ
H 5.74, d,
J = 10.0 Hz), of H-7 (δ
H 7.69, dd,
J = 8.4, 2.1 Hz) to H-5 (δ
H 7.65, d,
J = 2.1 Hz) and H-8 (δ
H 6.79, d,
J = 8.4 Hz), confirming the presence of the 1,2,4 trisubstituted benzene ring and the
cis-double bond. That the 1,2,4-trisubsituted benzene ring and the
cis-double bond were part of the 2
H-chromene-6-carboxylic acid moiety was corroborated by the HMBC correlations (
Figure 4a,
Supplementary Information, Figures S11) of H-5 to C-4 (δ
C 122.8), C-7 (δ
C 130.8), C-8a (δ
C 156.8) and C-11 (δ
C 167.0), of H-7 to C-5 (δ
C 127.9), C-8a, C-11, of H-8 to C-4a (δ
C 120.5), C-6 (δ
C 123.0) and C-8a, as well as of H-3 to C-4a and of H-4 to C-4a, C-5 and C-8a. As the HMBC spectrum also exhibited correlations of the methyl singlet at δ
H 1.31 (H
3-9) to C-3, C-2 (δ
C 80.5) and C-10 (δ
C 67.1) and of the singlet at δ
H 3.46 (H-10) to C-2, C-3 and CH
3-9 (δ
C 23.3), the methyl and hydroxymethyl groups were placed on C-2. The NOESY correlations (
Figure 4b,
Supplementary Information, Figures S12) of H-4 to H-3 and H-5, of H-3 to H-4, H
3-9 and H
2-10, of H-8 to H-7, H
2-10 and H
3-9 and of H
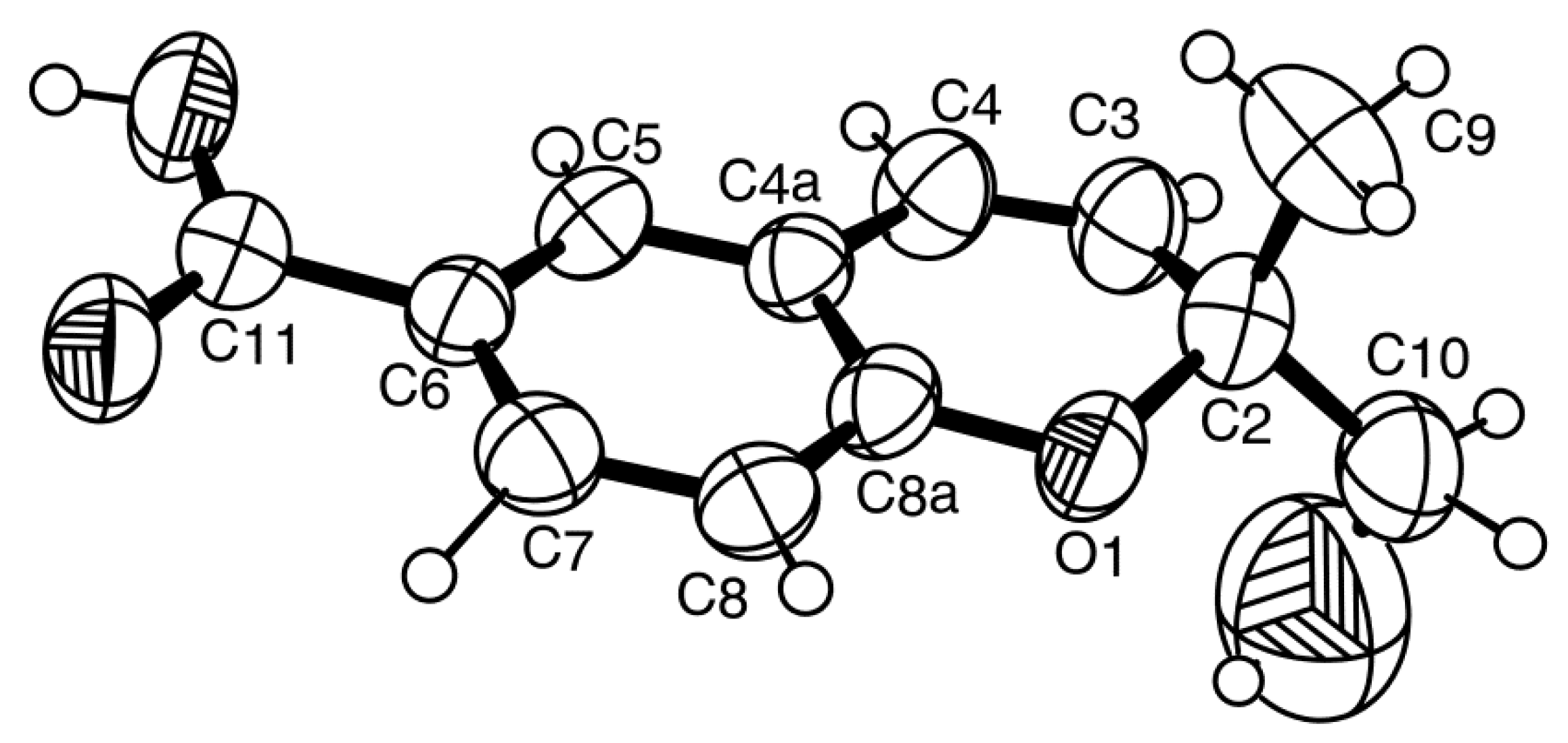
3-9 to H-8, H-10 also confirmed this hypothesis. Since compound
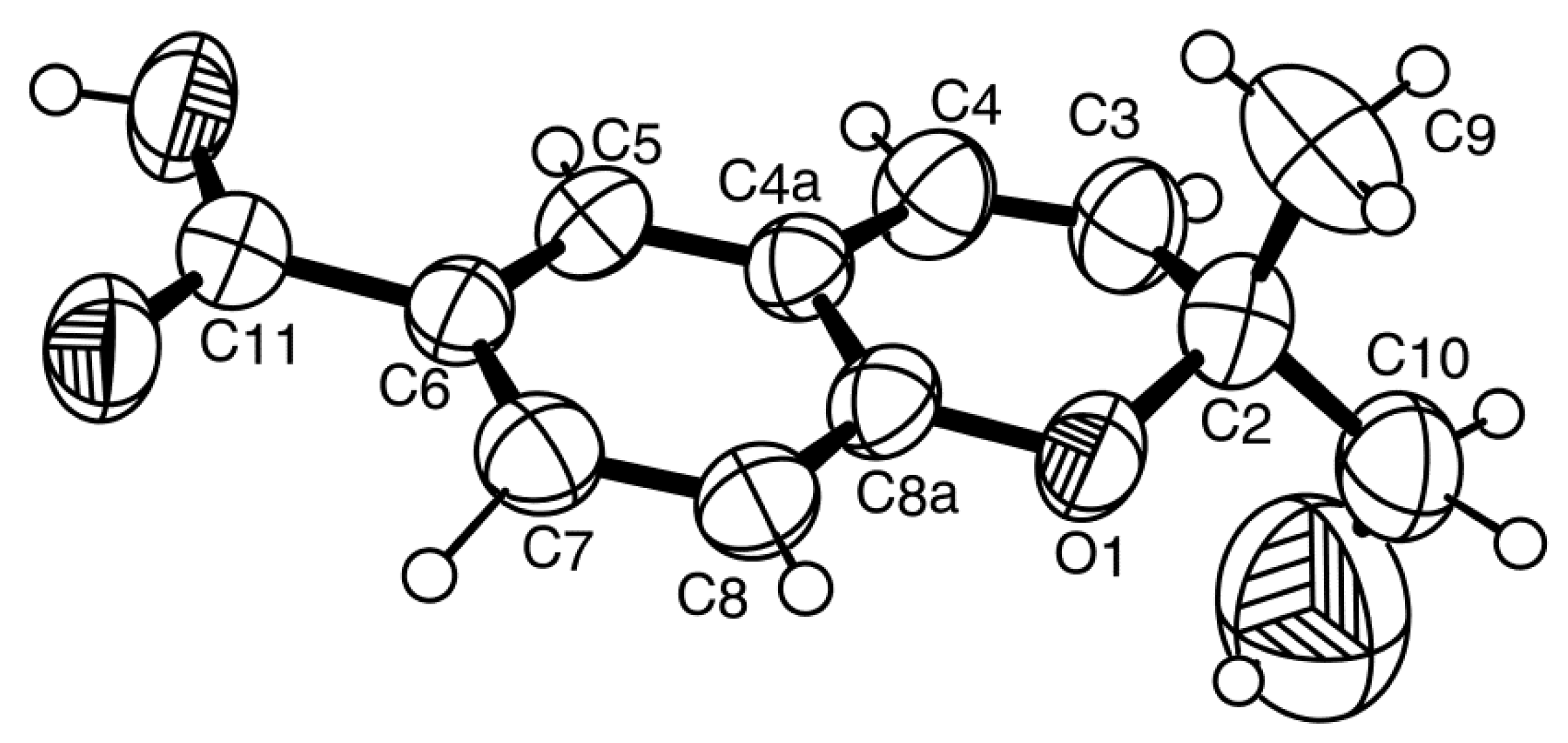
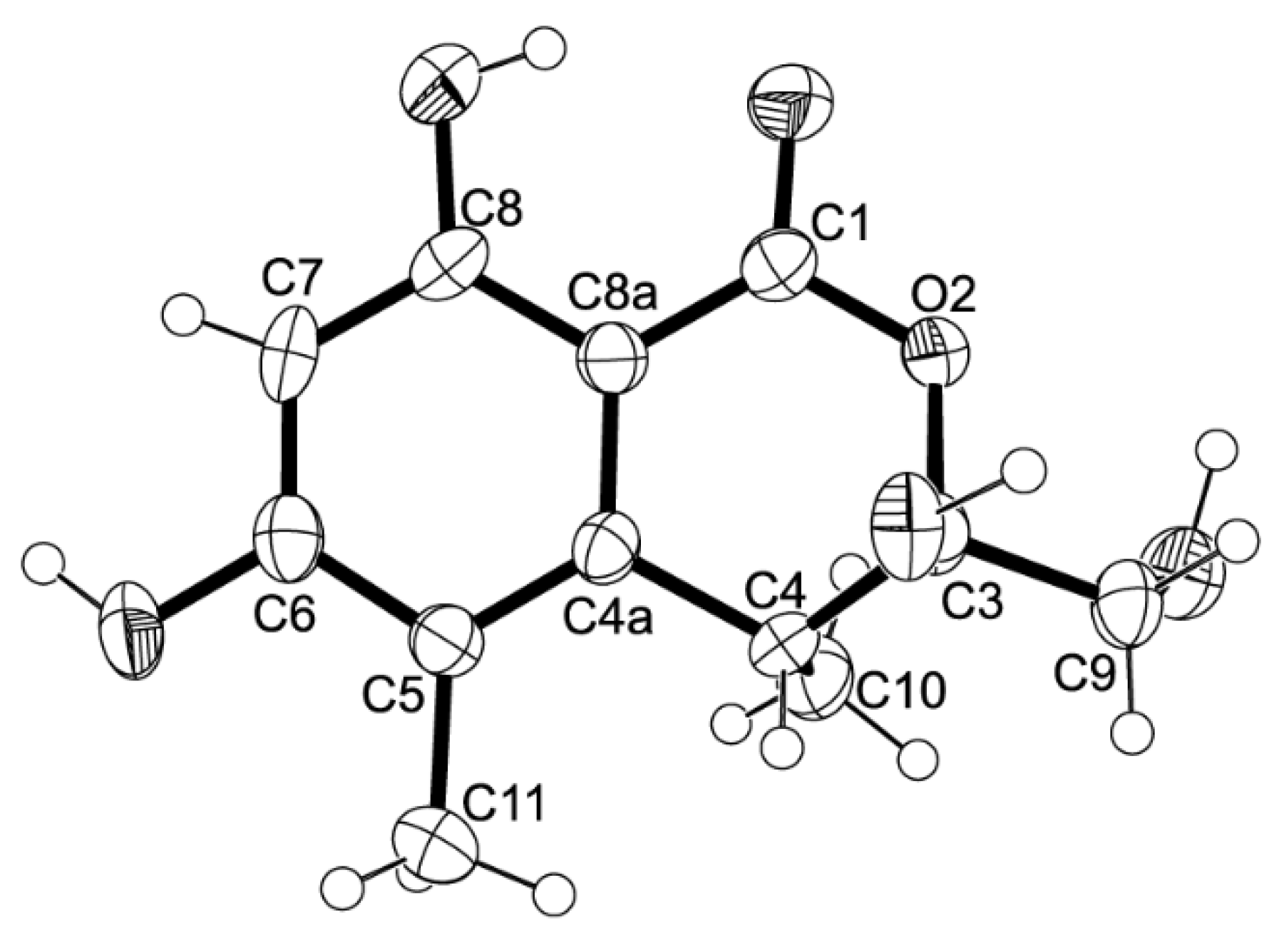
2a was obtained in a suitable crystal, X-ray analysis was carried out, and the ORTEP view shown in
Figure 5 revealed that the absolute configuration of C-2 is
S. A literature search indicated that
2a has never been previously reported; therefore, it was named quadricinctapyran A.
Compound
2b was isolated as a white solid (mp. 118–119 °C), and its molecular formula C
14H
14O
5 was established on the basis of the (+)-HRESIMS
m/z 263.0971 [M + H]
+ (calculated 263.0919), indicating eight degrees of unsaturation. The
1H and
13C NMR spectral features (
Supplementary Information, Figures S13 and S14) of compound
2b resembled those of compound
2a, except for an additional carbonyl carbon at δ
C 170.8 and a methyl group at δ
C 20.7 (δ
H 1.98, s), characteristic of the acetoxyl group (
Table 2). Moreover, since the signals of the oxymethylene protons (H
2-10) of compound
2b appeared as two doublets at δ
H 4.13 (
J = 11.7 Hz) and 4.24 (
J = 11.7 Hz), ca. 0.7 ppm higher than that of H
2-10 in compound
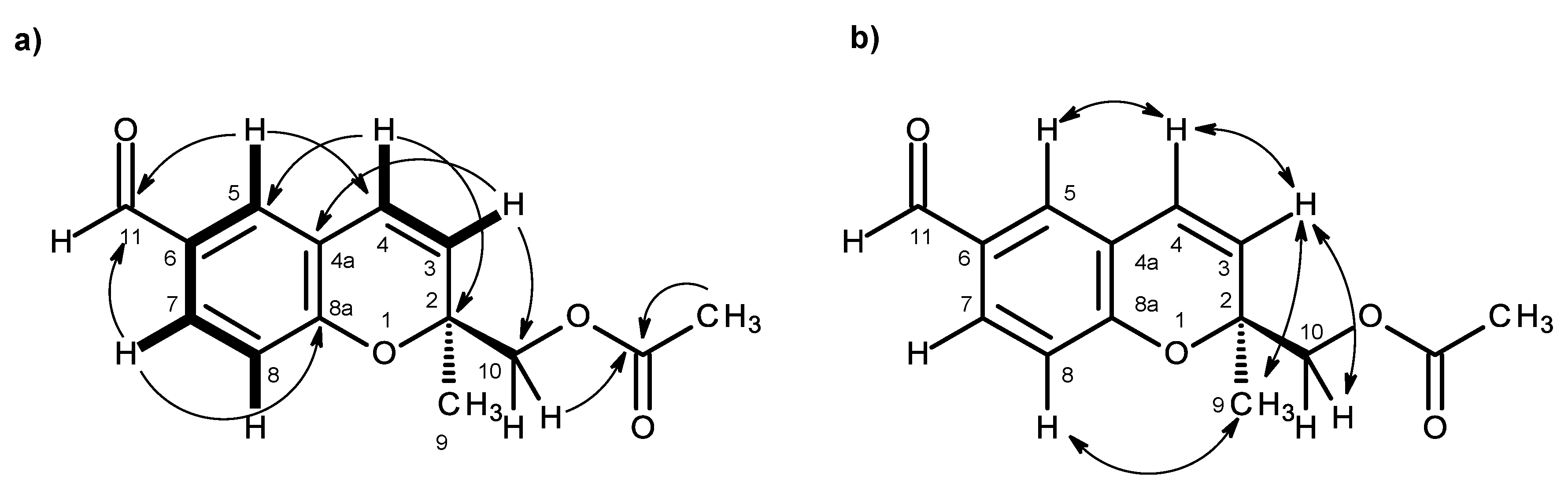
2a, it was clear that the acetoxyl group was on C-10. This was also corroborated by the HMBC correlations (
Table 2,
Figure 6a,
Supplementary Information, Figure S17) of H
2-10 and the methyl singlet at δ
H 1.98 to the carbonyl of the acetoxyl group (δ
C 170.8). Since Compound
2b could not be obtained as a suitable crystal for X-ray analysis, the absolute configuration of its C-3 could not be determined with certainty. However, as compound
2b is the acetate derivative of compound
2a, it was speculated that the stereochemistry of its C-2 should be the same as that of C-2 of compound
2a, i.e., 2
S. In order to confirm this hypothesis, the NOESY experiments were carried out. The NOESY spectrum of compound
2b showed a weak correlation of H-8 to H
3-9 and not to H
2-10 (
Figure 6b,
Supplementary Information, Figure S18), similar to what has been observed for compound
2a. Acid hydrolysis of
2b gave the product whose structure was confirmed as
2a by
1H and
13C NMR data, as well as the optical rotation. Therefore, the absolute configuration of C-2 of
2b is assigned as 2
S. Compound
2b is also a new compound, thus we named it quadricinctapyran B.
The molecular formula C
12H
12O
4 of compound
3, a white solid (mp 189–191 °C), was established based on the (+)-HRESIMS
m/z 221.0819 [M + H]
+ (calculated 221.0814), and thus, it is an isomer of compound
2a. Furthermore, the general features of its
1H and
13C NMR spectra resembled those of compound
2a. However, the chemical shift values of some of the proton and carbon signals were slightly different from those observed in compound
2a. The
13C NMR, DEPT and HSQC spectra (
Table 3,
Supplementary Information, Figures S20 and S22) exhibited the signals of one conjugated carboxyl carbonyl (δ
C 166.7), three quaternary sp
2 (δ
C 161.2, 125.3, 125.0), five methine sp
2 (δ
C 139.5, 134.6, 129.8, 123.5, 119.8), one oxy-quaternary sp
3 (δ
C 70.7), one oxymethylene sp
3 (δ
C 77.0) and one methyl (δ
C 26.1) groups. The
1H NMR spectrum (
Table 3,
Supplementary Information, Figure S19) revealed the existence of three aromatic protons of the 1,2,4-trisubstituted benzene ring, similar to that of compound
2a, at δ
H 7.05, d (
J = 8.4 Hz), 7.74, dd (
J = 8.4, 2.1 Hz), 7.89, d (
J = 2.1 Hz), two olefinic protons of the
cis-double bond at δ
H 5.95, dd (
J = 12.0, 1.2 Hz) and 6.31, d (
J = 12.0 Hz), two oxymethylene protons at δ
H 3.84, d (
J = 11.1 Hz) and 4.02, dd (
J = 11.1, 1.6 Hz), and a methyl singlet at δ
H 1.26. That the carboxylic acid functionality was on C-7, and the substituent with a
cis-double bond was on C-5a was substantiated by the HMBC correlations of H-6 (7.89, d,
J = 2.1 Hz) to C-10 (δ
C 166.7), C-9a (δ
C 161.2), C-8 (δ
C 129.8) and C-5 (δ
C 123.5), of H-5 (δ
H 6.31, d,
J = 12.0 Hz) to C-6 (δ
C 134.5) and C-9a and of H-4 (δ
H 5.95, dd, d,
J = 12.0, 1.2 Hz) to C-5a (δ
C 125.0) (
Table 3 and
Figure 7a,
Supplementary Information, Figure S23). However, contrary to compound
2a, compound
3 showed the HMBC correlations of H
2-2 (δ
H 3.84, d,
J = 11.1 Hz and 4.02, dd,
J = 11.1, 1.6 Hz) to not only C-4 (δ
C 139.5), but also to C-9a (
Table 3 and
Figure 7a). Consequently, the benzoic acid moiety was fused with the 2,3,6,7-tetrahydro-oxepine ring through C-5a and C-9a. That the methyl group and the hydroxyl group were on C-3 of the oxepin ring was confirmed by the HMBC correlations of the methyl singlet at δ
H 1.26 (H-11) to C-3 (δ
C 70.7), C-2 (δ
C 77.0) and C-4, as well as of H
2-2 to C-11 (δ
C 26.1) and C-3 (
Table 3 and
Figure 7). This was also corroborated by the NOESY correlations of H
3-11 to OH-3 (δ
H 3.39, br), H
2-2 and H-4 (
Table 3 and
Figure 7b,
Supplementary Information, Figure S24). Therefore, Compound
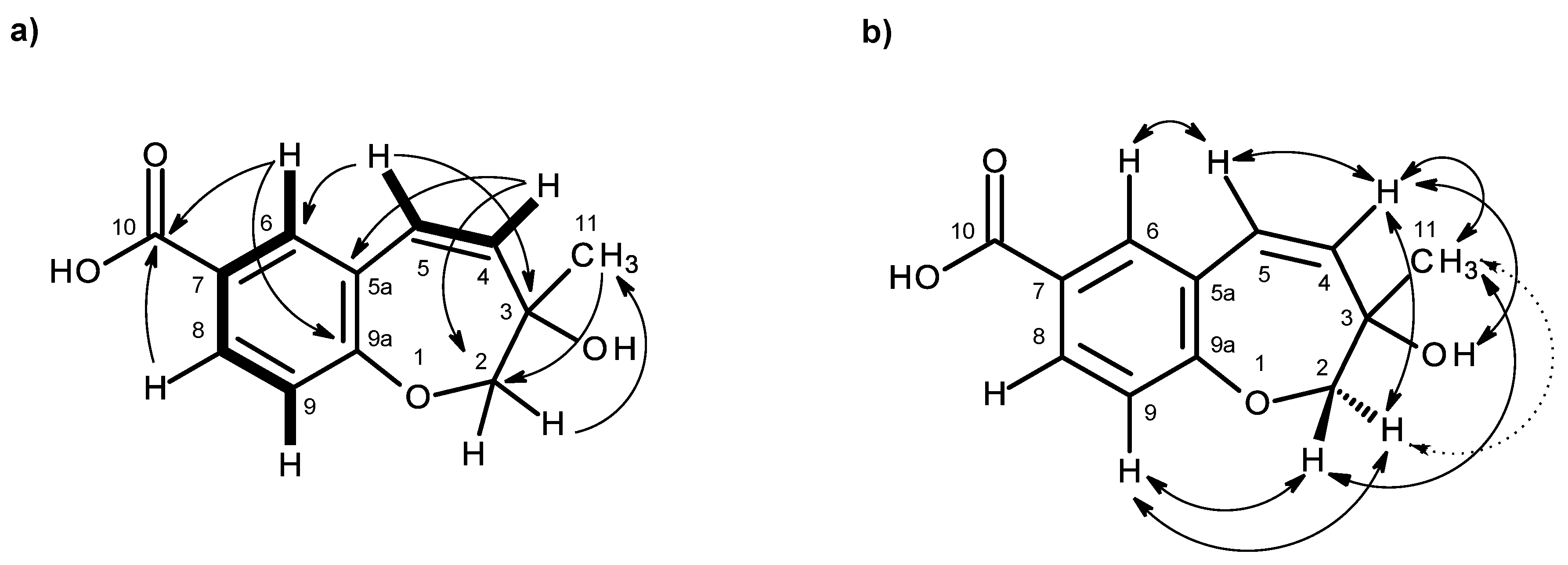
3 was identified as 3-hydroxy-3-methyl-2,3-dihydro-1-benzoxepine-7-carboxylic acid.
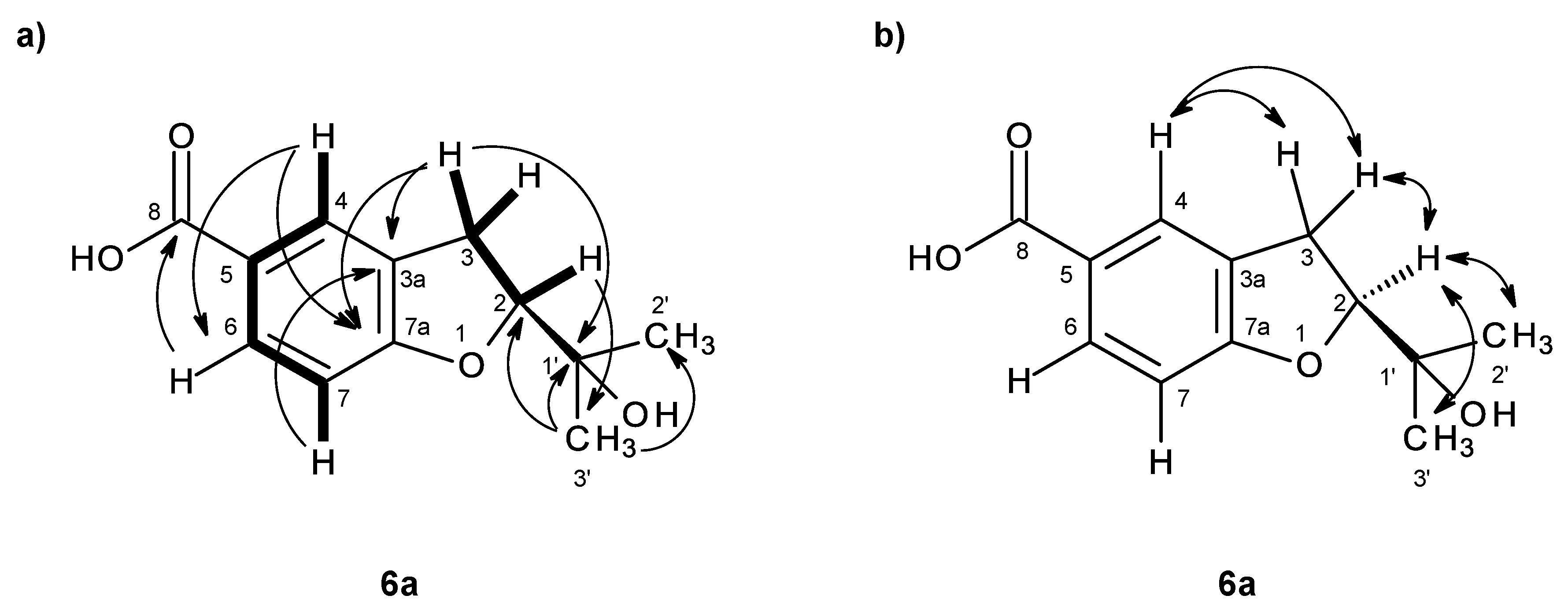
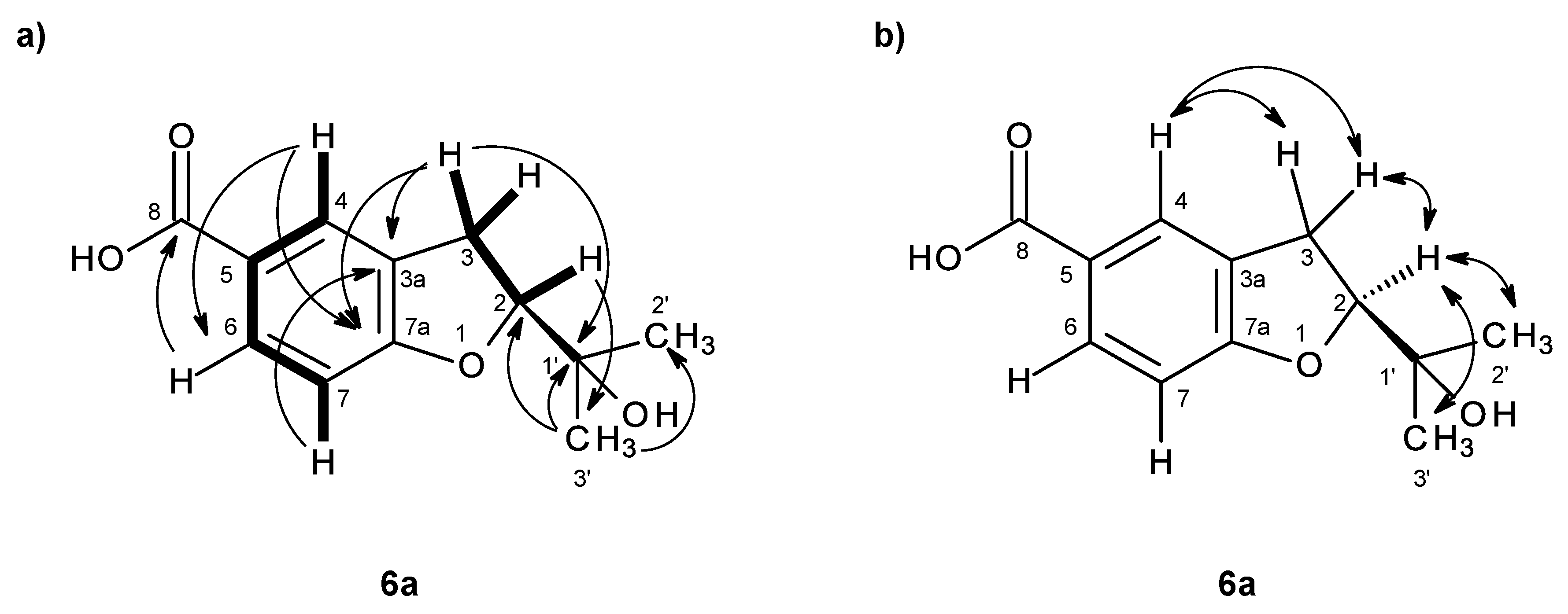
As compound
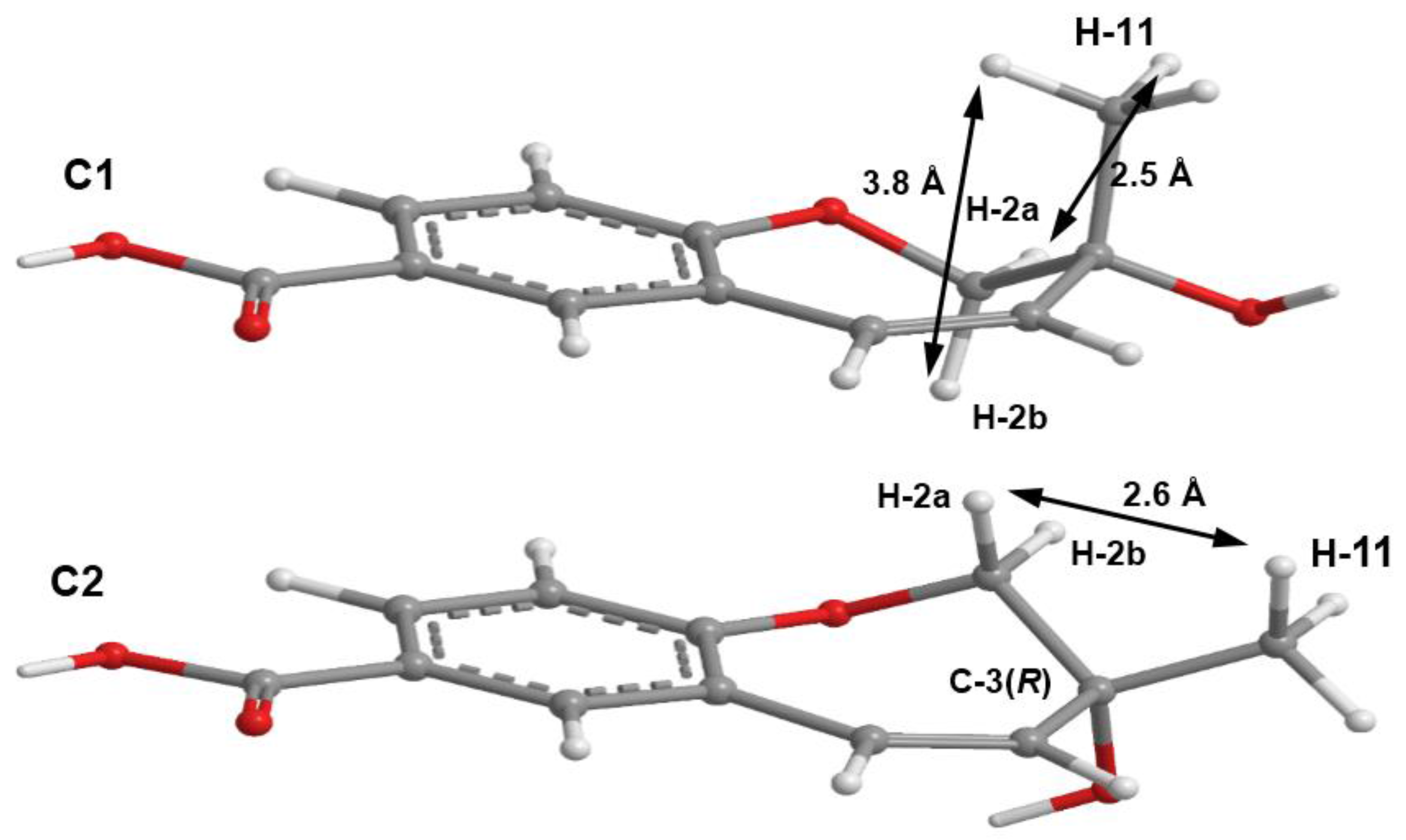
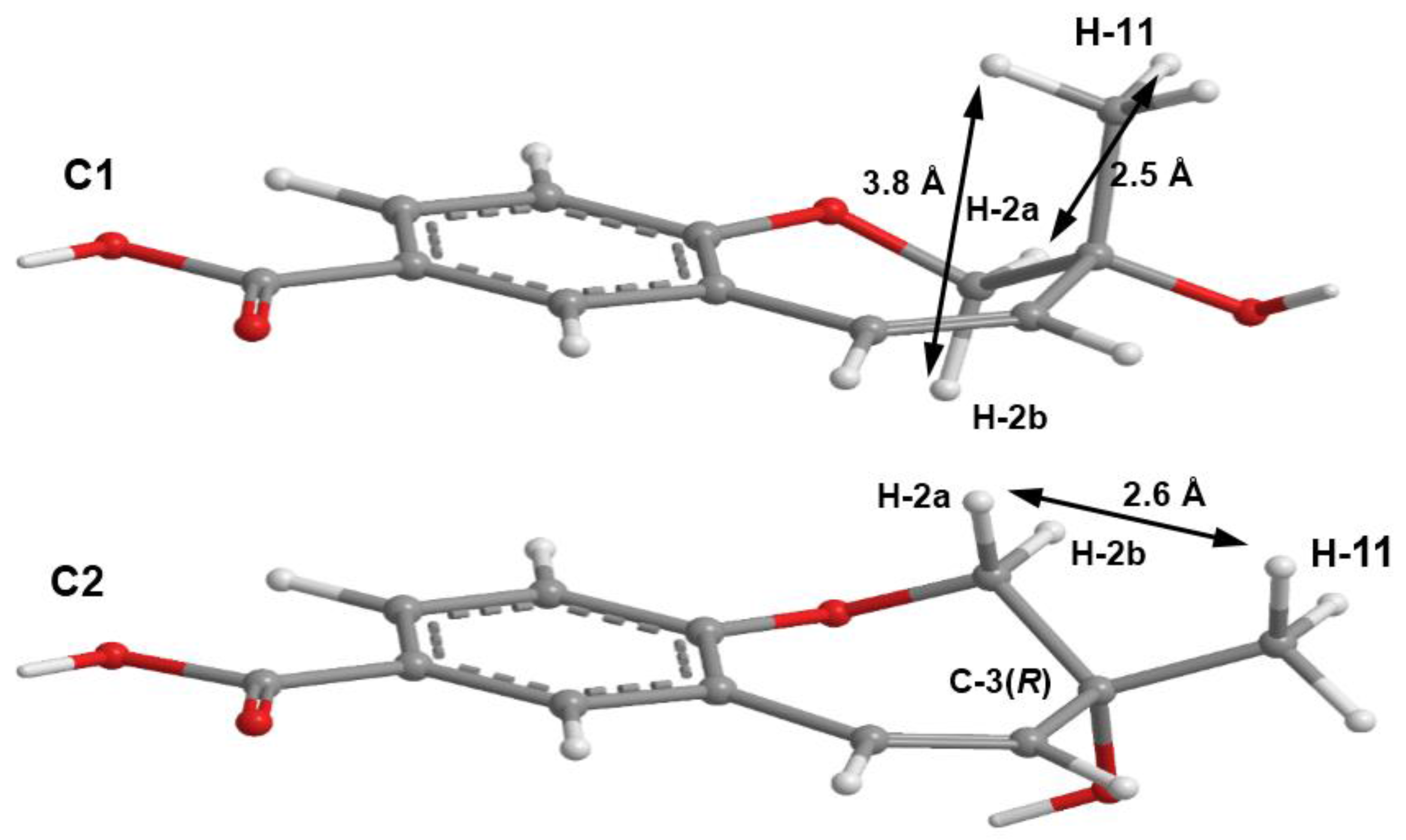
3 could not be obtained as a suitable crystal for X-ray analysis, an effort to tentatively determine the relative configuration of the stereogenic carbon (C-3) by molecular mechanics conformation analysis and the NOESY experiments was carried out. Stochastic conformational search on the computational models of the structure of
3 with C-3 in
R configuration, followed by energy minimization, converged to two half-chair conformations for the seven-membered ring C1 and C2, as depicted in
Figure 8, regardless of the modelling level of the theory used (MP2/6-311G, PM3, MMFF and MM2). All methods, except for PM3, also agree that conformation C2, with the methyl group in the equatorial position, is more stable by ca. 2 kcal/mol. However, this difference can be attributed to a weak intramolecular hydrogen bond in conformation C2, between HO-3 and O-1, which is not possible in conformation C1. The semi-empirical PM3 method gives less weight to non-ideal intramolecular hydrogen bonds, as compared to the other methods, and assigns virtually the same energy to both conformations of
3, while still orienting HO-3 towards the seven-membered ring. Since DMSO solvent molecules compete for HO-3 hydrogen bonding, it is more likely that the intramolecular bond is not an important feature of ring conformation C2 and that, in reality, both conformations have approximately the same energy.
Both model conformations of
3 predict hydrogen-hydrogen distances that are similar to within 0.2 Å, with the exception of some distances to the methyl group (H
3-11). The most notable are to the diastereotopic hydrogens (H
2-2), partially presented in
Figure 8. While both conformations show almost the same distance between H-2a and H-11, a difference is predicted between H-2b and H-11 if the conformation C1 predominates, which should be apparent in the build-up rate of NOESY cross-peaks for small mixing times. Alternatively, the predominance of the conformation C2 would be indicated by two equal strength cross-peaks for H-2a and H-2b in cross-relaxation with H-11. It is observed that the H-2b (δ
H 3.84, d,
J = 11.1 Hz)/H-11 NOESY cross-peak is weak while the H-2a (δ
H 4.02, dd,
J = 11.1, 1.6 Hz)/H-11 is medium, suggesting that the conformation C1 predominates. NOE effective distances, r
eff, are calculated by [
13] :
The average effective positions of the three methyl H-11
i protons are relative to H-2a or H-2b. The predicted ratio
reff (H-2a/H-11)/
reff (H-2b/H-11) is 1.40 (1.36, if
r−3 averages are used instead of
r−6). Assuming that the cross-relaxation rate is similar in both cases, the NOE intensities should also have a similar ratio [
14]. Since the observed intensities ratio is actually closer to two, the evidence points towards the predominance of the conformation C1. Since, as stated previously, both conformations have similar conformational energy, the higher stability of 3
R-C1 of
3 (or of its stereoisomer 3
S-C2) must originate from the ready interaction of the equatorial HO-3 with the hydrogen-bonding solvent and also from a higher entropic rotational freedom of the hydroxyl and methyl groups. The NOESY spectrum also revealed the correlations of both H
2-2 to H-9. However, only one of the H
2-2, i.e., the doublet at δ
H 3.84 (
J = 11.1 Hz), showed a weak cross-peak to H-4, while the double doublet at δ
H 4.02 (
J = 11.1, 1.6 Hz) did not give any cross-peak to H-4. This observation led to the conclusion that the plucked oxepin ring should adopt the conformation in which H-2 at δ
H 3.84 is near H-4, i.e., in the α (axial), while H-2 at δ
H 4.02 is in β (equatorial) positions, which is in agreement with our conformational analysis. A literature search revealed that compound
3 is also a new compound, so we named it quadricinctoxepine.
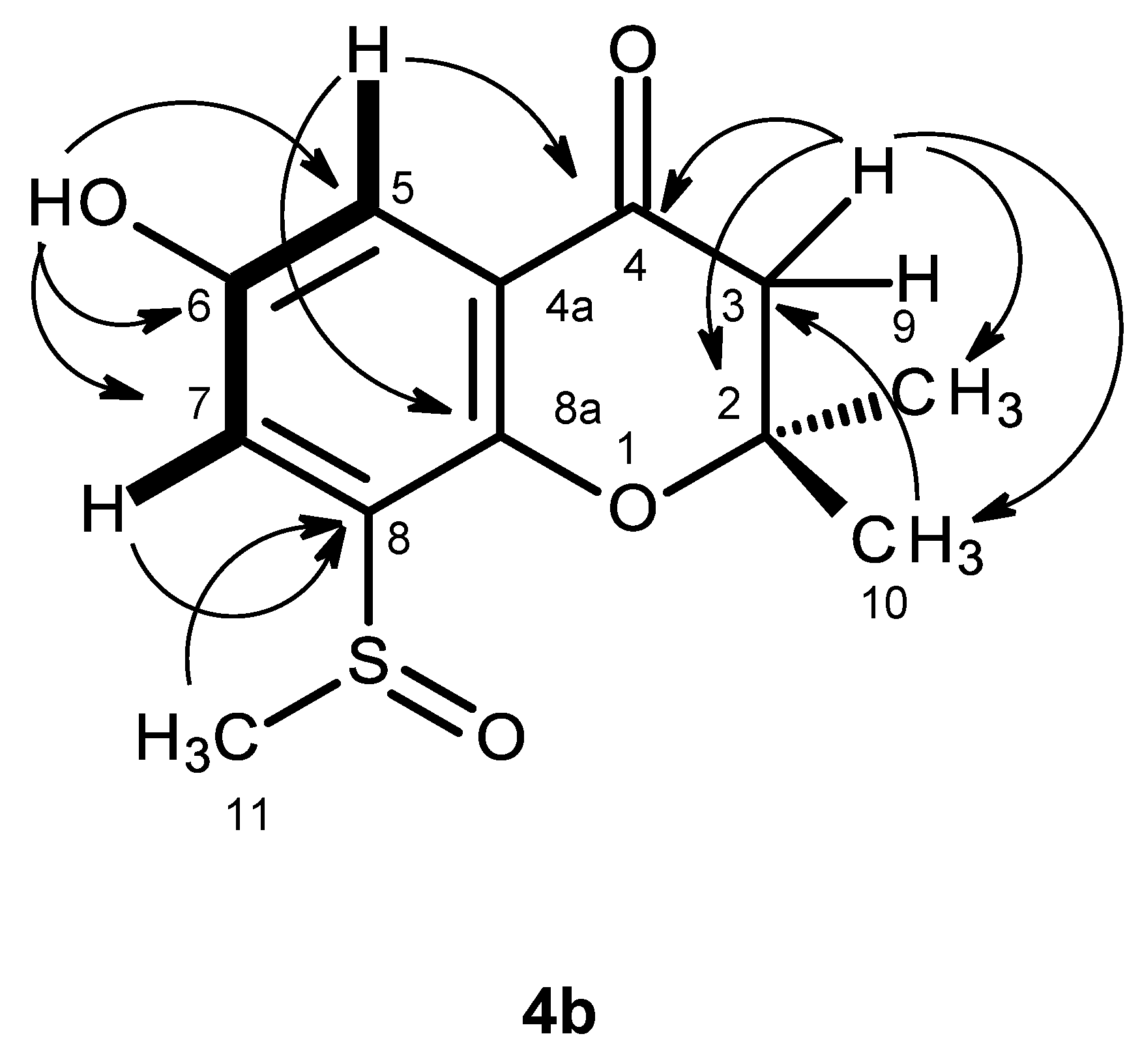
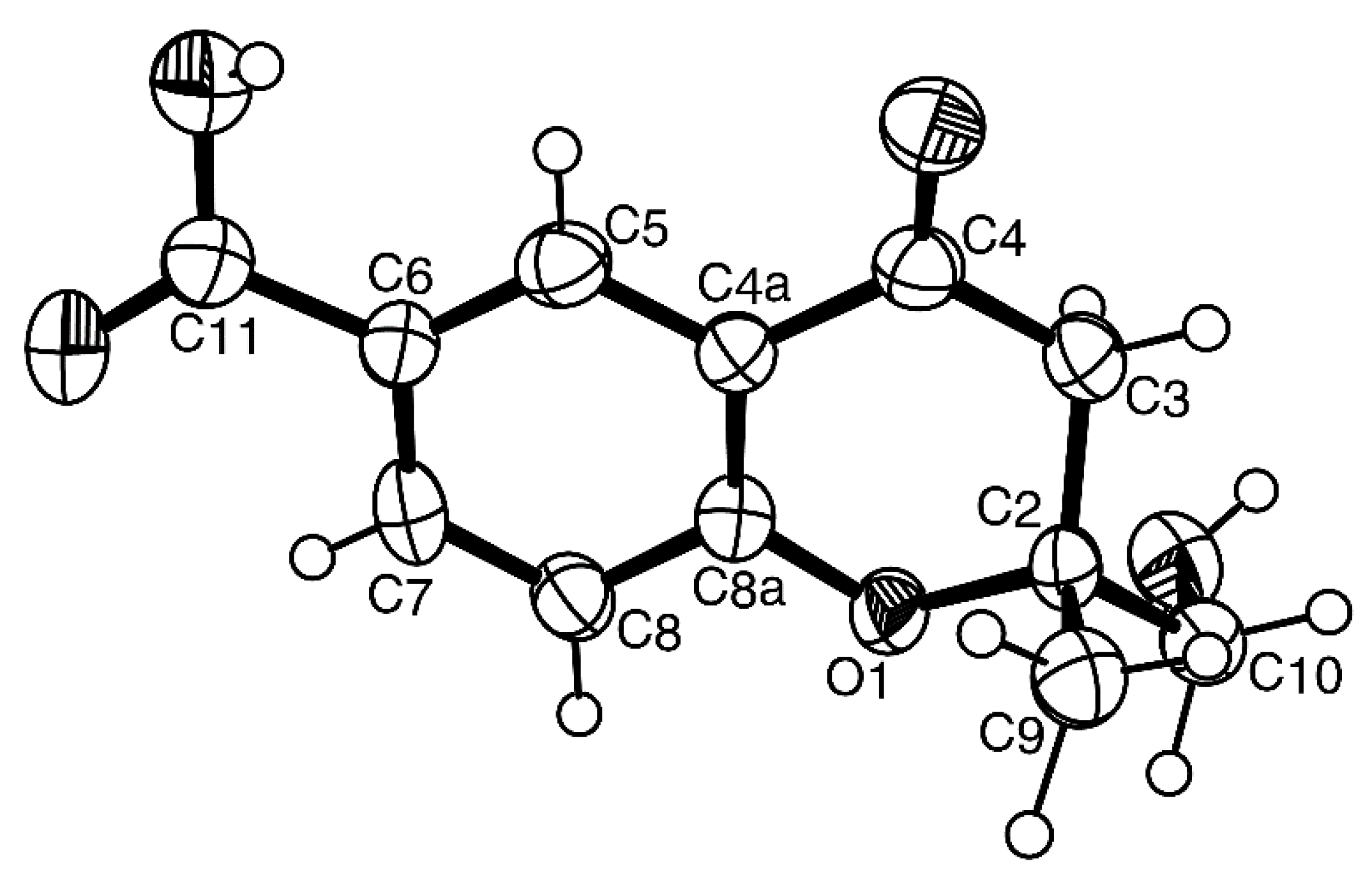
Compound
4b was isolated as white crystals (mp, 227–228 °C), and its molecular formula C
12H
14O
4S was established on the basis of the (+)-HRESIMS
m/z 255.0694 [M + H]
+ (calculated 255.0691). The IR spectrum showed absorption bands for hydroxyl (3442 cm
−1), conjugated ketone carbonyl (1690 cm
−1) and aromatic (1622 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 4,
Supplementary Information, Figures S26 and S28) exhibited the signals of one conjugated ketone carbonyl (δ
C 191.2), four quaternary sp
2 (δ
C 151.6, 147.4, 135.7, 120.8), two methine sp
2 (δ
C 118.0, 112.4), one oxy-quaternary sp
3 (δ
C 80.9), one methylene sp
2 (δ
C 48.0) and three methyl (δ
C 40.8, 26.1 and 25.7) groups. The
1H NMR spectrum (
Table 4,
Supplementary Information, Figure S25) exhibited, besides a broad singlet of the phenolic hydroxyl group at δ
H 9.85, the signals of two
meta-coupled aromatic protons at δ
H 7.17, d (
J = 3.1 Hz) and δ
H 7.35, d (
J = 3.1 Hz), two geminally-coupled methylene protons at δ
H 2.80, d (
J = 16.6 Hz) and 2.82, d (
J = 16.6 Hz) and three methyl singlets at δ
H 1.38, s, 1.39, s, and 2.77, s. The chemical shift value of the methyl singlet at δ
H 2.77, s (δ
C 40.8), indicated that it was on the electron-withdrawing moiety. That compound
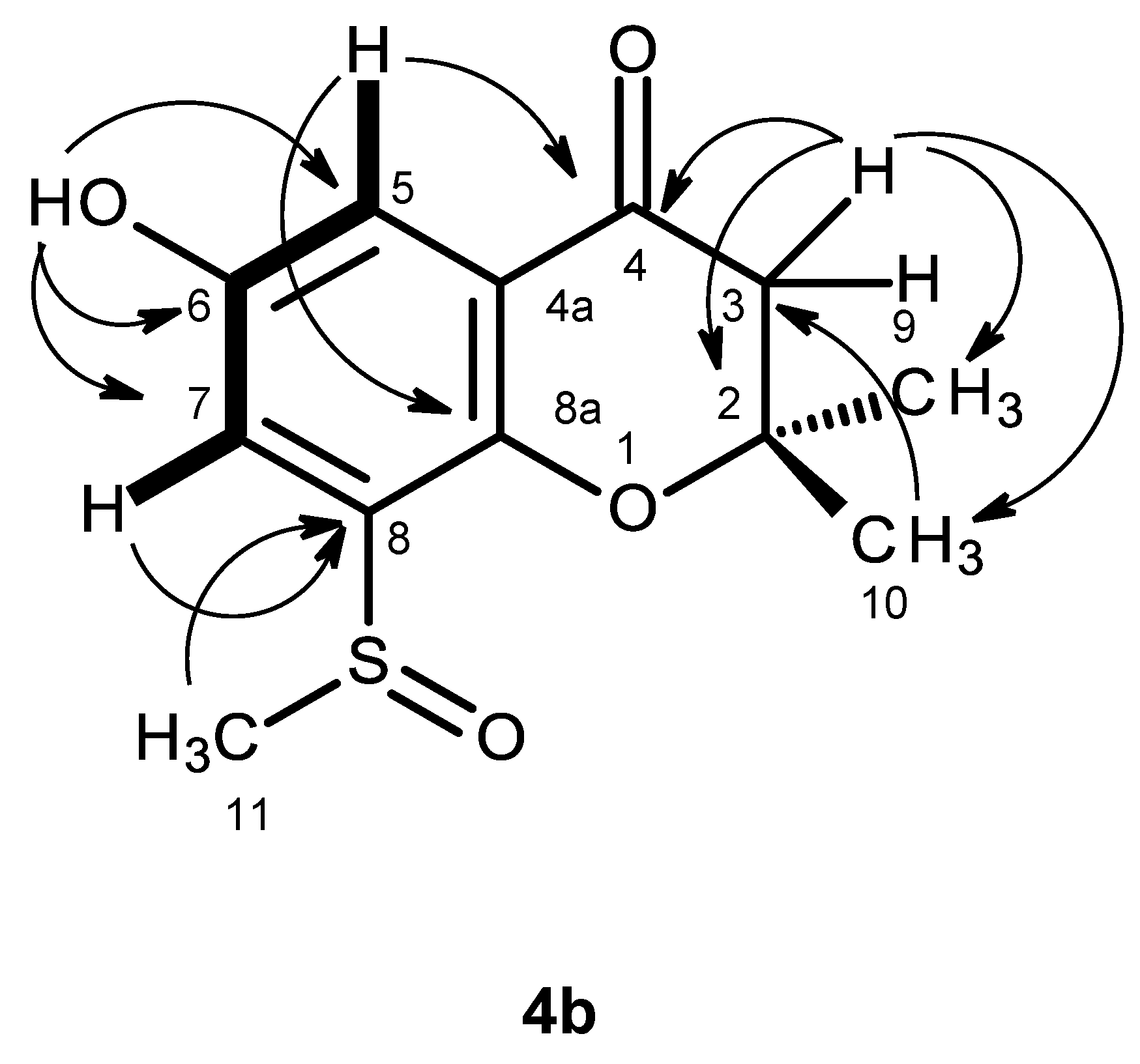
4b was a 2,2,6,8-tetrasubstituted 2,3-dihydro-4
H-chromen-4-one derivative was supported by the HMBC correlations of H-5 (δ
H 7.17, d,
J = 3.1 Hz) to C-4 (δ
C 191.2), C-7 (δ
C 118.0) and C-8a (δ
C 147.4), of H-7 (δ
H 7.35, d,
J = 3.1 Hz) to C-5 (δ
C 112.4), C-8 (δ
C135.7), C-8a, as well as of H-2 (δ
H 2.80, d,
J = 16.6 Hz and 2.82, d,
J = 16.6 Hz) to C-3 (δ
C 80.9) and C-4 (
Table 4,
Figure 9,
Supplementary Information, Figure S28). As the HMBC spectrum also exhibited cross-peaks of a broad singlet of the phenolic hydroxyl proton at δ
H 9.85 to C-5, C-6 (δ
C 151.6) and C-7 and of the singlets of the methyl groups at δ
H 1.38 and 1.39 to C-2 (δ
C 48.0) and C-3, the hydroxyl group was placed on C-6, and the two methyl groups were placed on C-2. Since this partial structure accounted for only C
11H
11O
3, another portion of the molecule must contain CH
3SO. That the methyl sulfoxide group was on C-8 was supported by the presence of the deshielded methyl group at δ
H 2.77, s (δ
C 40.8), as well as by the HBMC cross-peak of H
3-11 (δ
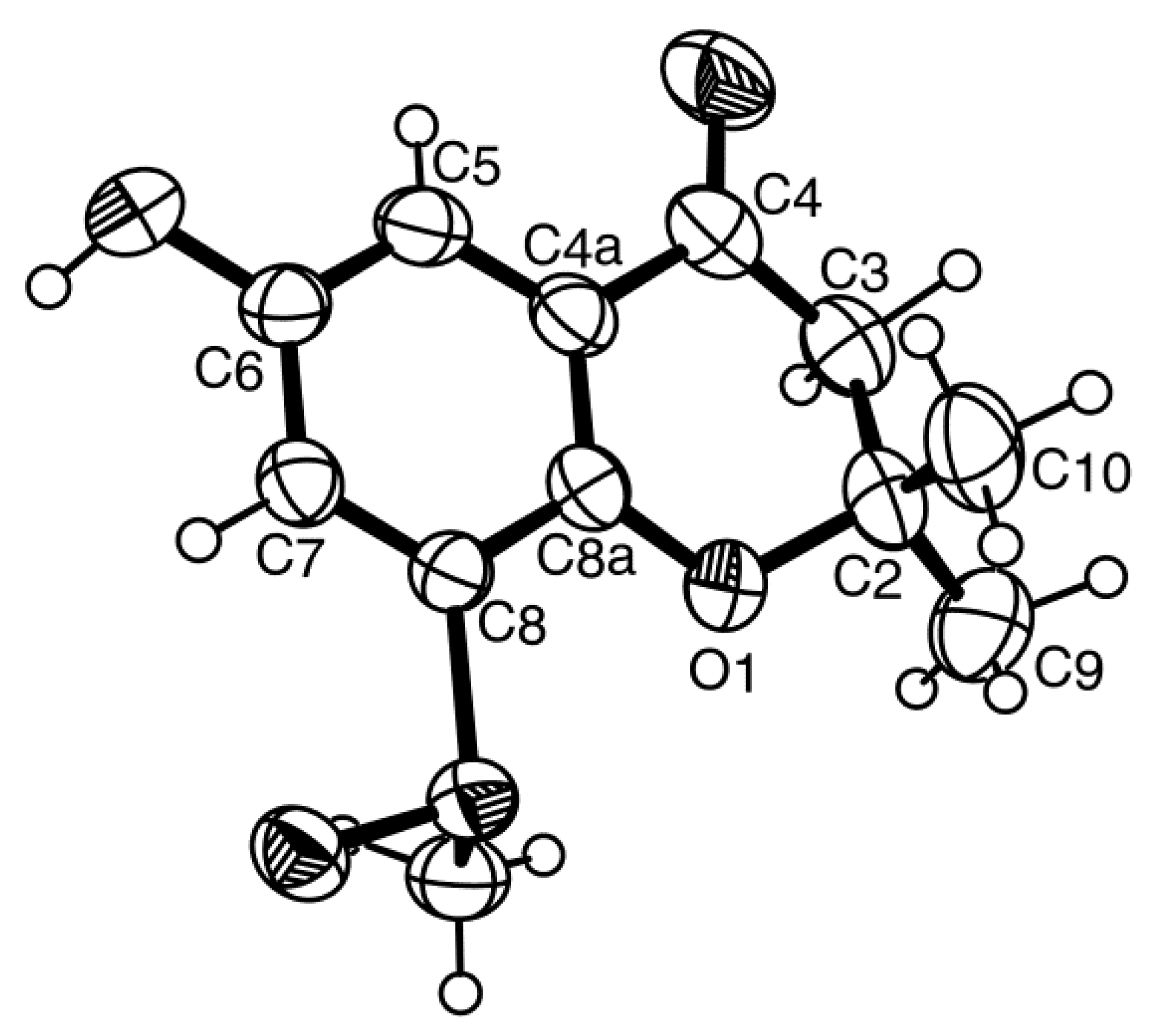
H 2.77, s) to C-8. Final proof of the structure of compound
4b was provided by its X-ray analysis, and its ORTEP view is shown in
Figure 10. Moreover, the ORTEP view also revealed that the absolute configuration of the sulfoxide group in compound
4b is
R. A literature search showed that compound
4b is a new compound, and thus, we have named it quadricinctone B.
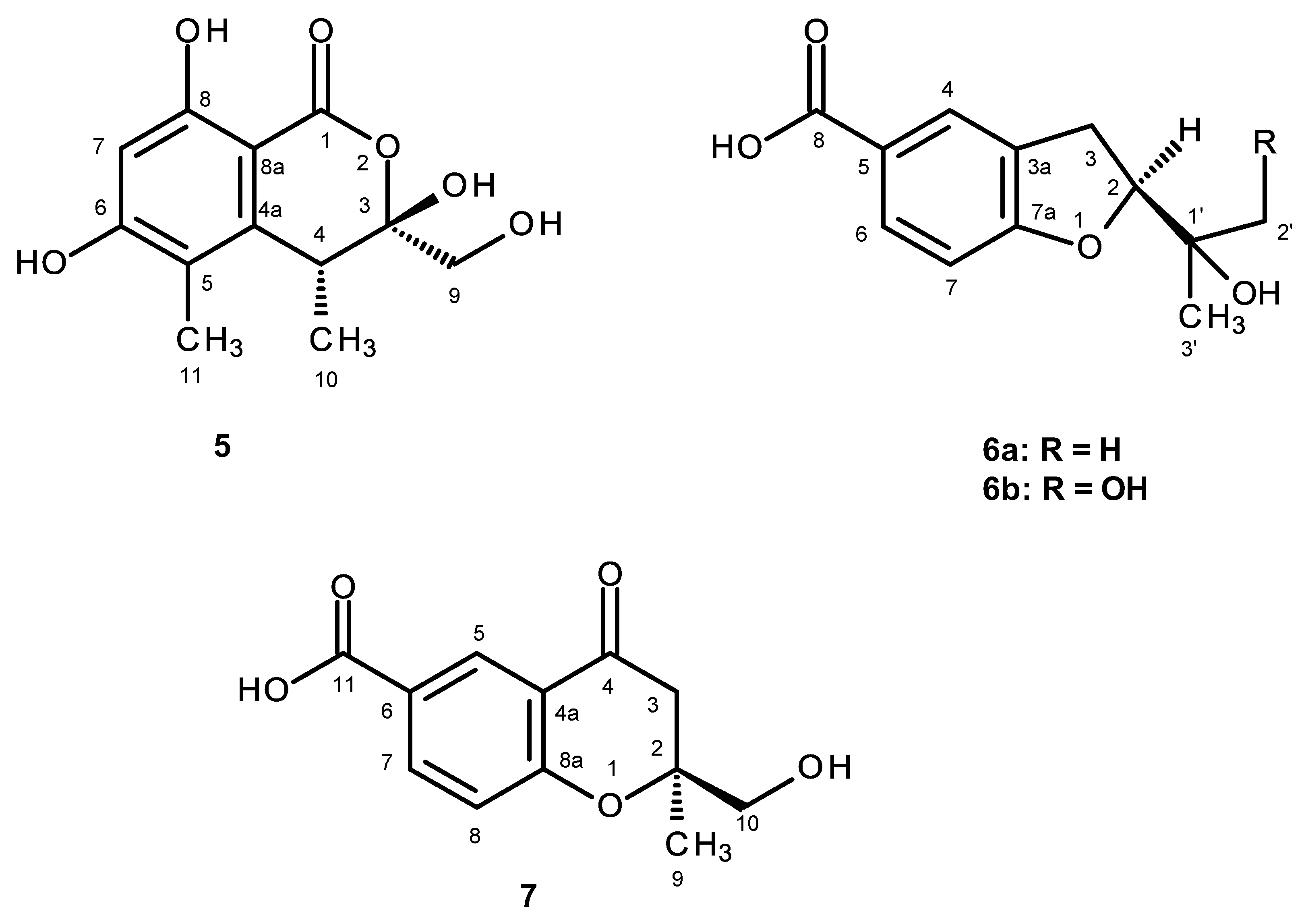
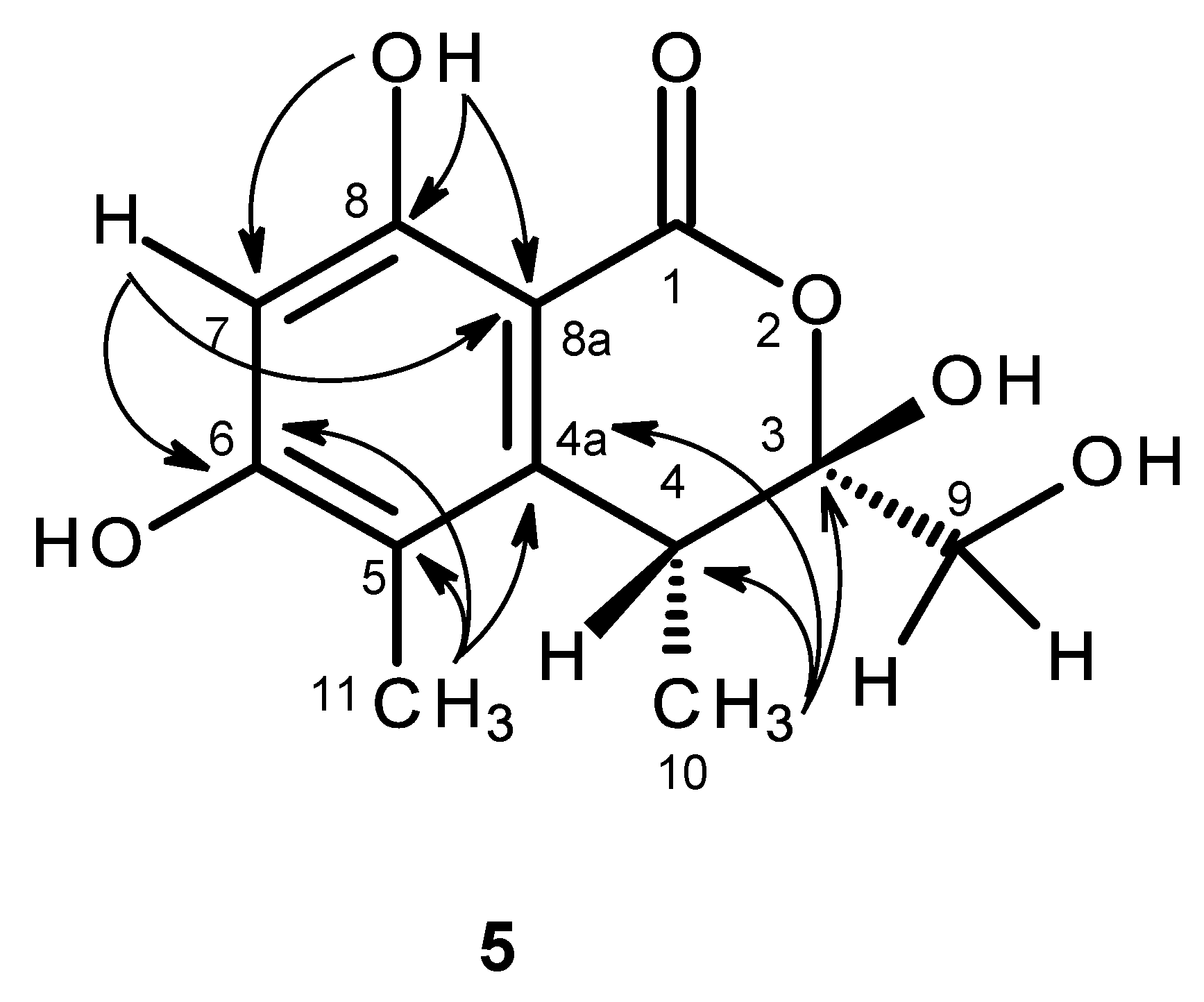
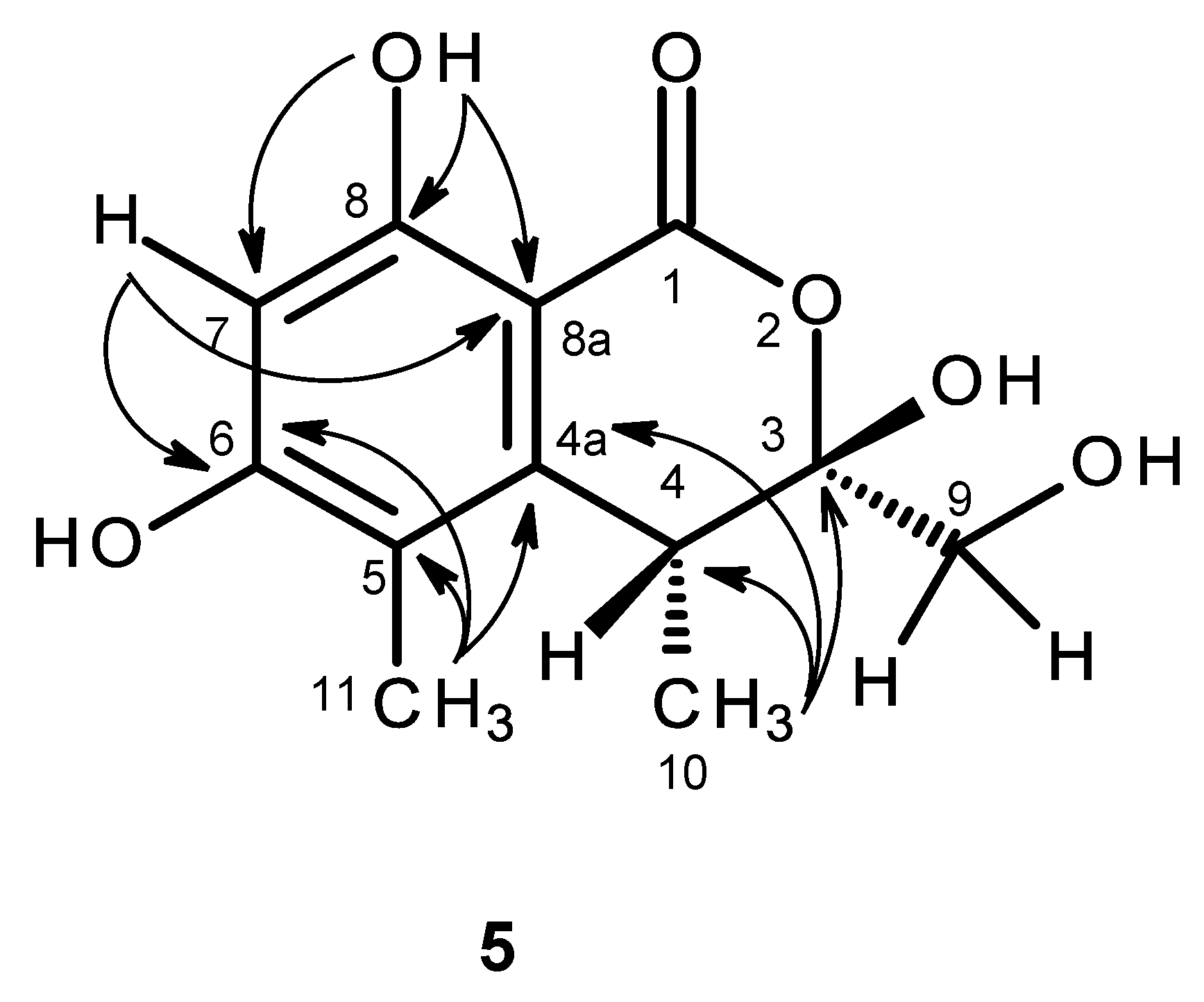
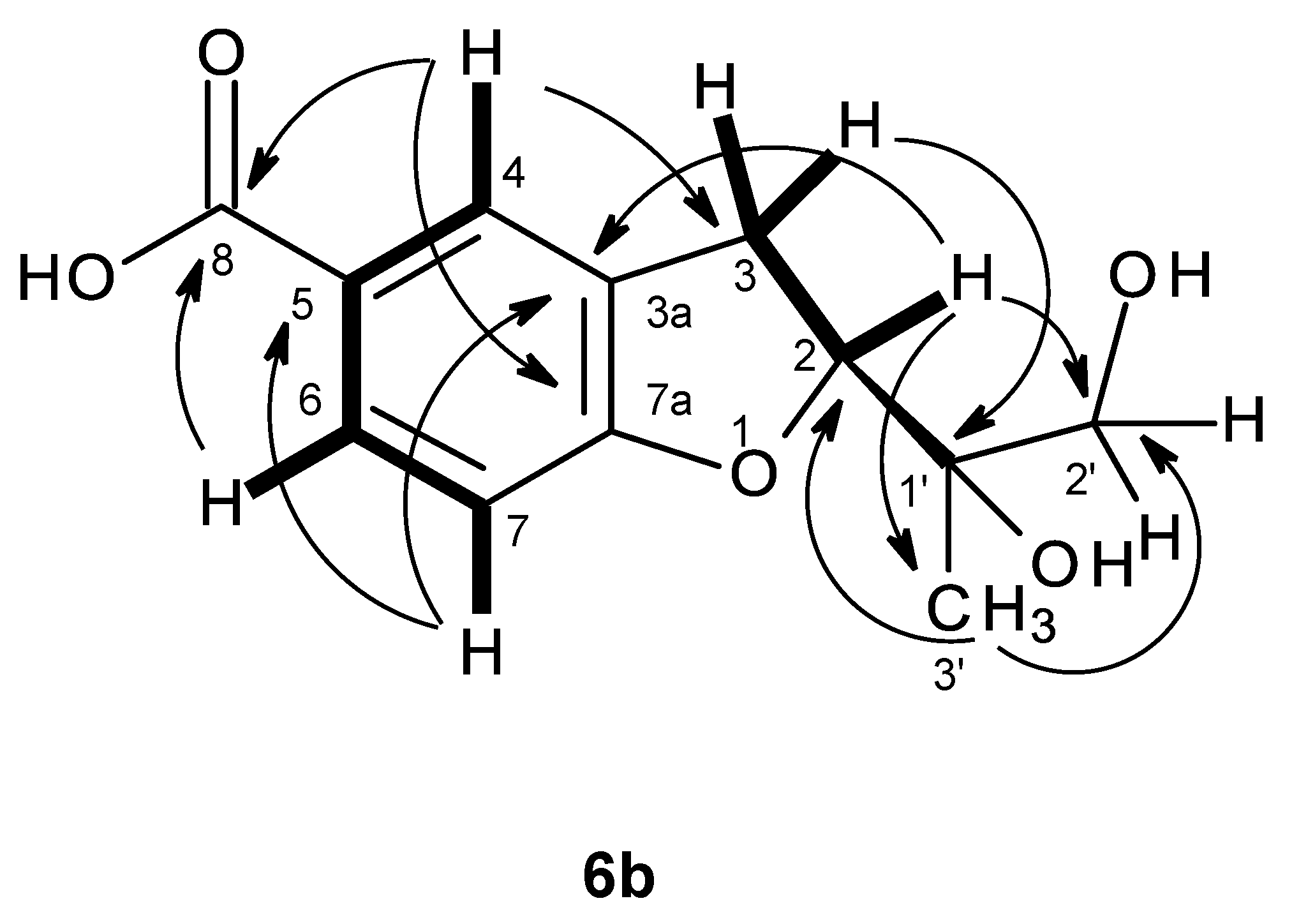
The molecular formula C
12H
14O
6 of compound
5 was determined based on the (+)-HRESIMS
m/z 255.0875 (calculated 255.0869), indicating six degrees of unsaturation. The IR spectrum exhibited absorption bands for hydroxyl (3439 cm
−1), conjugated ester carbonyl (1660 cm
−1) and aromatic (1643 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 5,
Supplementary Information, Figures S31 and S33) exhibited the signals of one conjugated ester carbonyl (δ
C 168.8), five quaternary sp
2 (δ
C 162.9, 161.3, 144.6, 113.5, 98.9), one methine sp
2 (δ
C 99.9), one quaternary sp
3 of a hemiketal (δ
C 104.8), one methine sp
3 (δ
C 34.9), one oxymethylene sp
3 (δ
C 63.7) and two methyl (δ
C 16.3 and 9.8) carbons. The
1H NMR spectrum (
Table 5,
Supplementary Information, Figure S30) exhibited, besides a singlet of one aromatic proton at δ
H 6.27, a singlet of a hydrogen-bonded phenolic hydroxyl at δ
H 11.24 and a broad signal of another phenolic hydroxyl at δ
H 10.62, two broad signals of the hydroxyl groups at δ
H 5.26 and 7.07, one broad doublet of two methylene protons at δ
H 3.65 (
J = 16.6 Hz), a broad signal of one methine proton at δ
H 3.26, one methyl doublet at δ
H 1.06 (
J = 7.2 Hz) and one methyl singlet at δ
H 1.98. That compound
5 was a 3,3,4,5,6,8-hexasubstituted 3,4-dihydro-1
H-isochromen-1-one was supported by the HBMC correlations of OH-8 (δ
H 11.24, s) to C-8 (δ
C 161.3), C-7 (δ
C 99.9), C-8a (δ
C 98.9) and of H-7 (δ
H 6.27, s) to C-5 (δ
C 113.5), C-6 (δ
C 162.9), C-8 and C-8a (
Table 5,
Figure 11,
Supplementary Information, Figure S34). Since the methyl singlet at δ
H 1.98 gave HMBC cross-peaks to C-5 and the carbons at δ
C 144.6 and 162.9 (
Table 5,
Figure 11), the methyl group (δ
H 1.98; δ
C 9.8) and another hydroxyl group (δ
H 10.62, br) were placed on C-5 and C-6, respectively, and the carbon signals at δ
C 144.6 and 162.9 were assigned for C-4a and C-6, respectively. On the other hand, the COSY spectrum (
Table 5,
Supplementary Information, Figure S32) showed cross-peaks of the broad signal at δ
H 3.26 to the methyl doublet at δ
H 1.06 (
J = 7.2 Hz) and of the broad signal at δ
H 5.26 to the broad doublet at δ
H 3.65 (
J = 16.6 Hz), while the HMBC spectrum (
Table 5,
Figure 11) gave cross-peaks of the methyl doublet at δ
H 1.06 (
J = 7.2 Hz) to C-4a, C-4 (δ
C 34.9), and the signal of the quaternary carbon at δ
C 104.8 (C-3), the methyl and the hydroxymethyl substituents were placed on C-4 and C-3, respectively. Therefore, another hydroxyl group (δ
H 7.07, br) was on C-3. This was supported by the chemical shift value of C-3, which is typical for a hemiketal carbon. Since compound
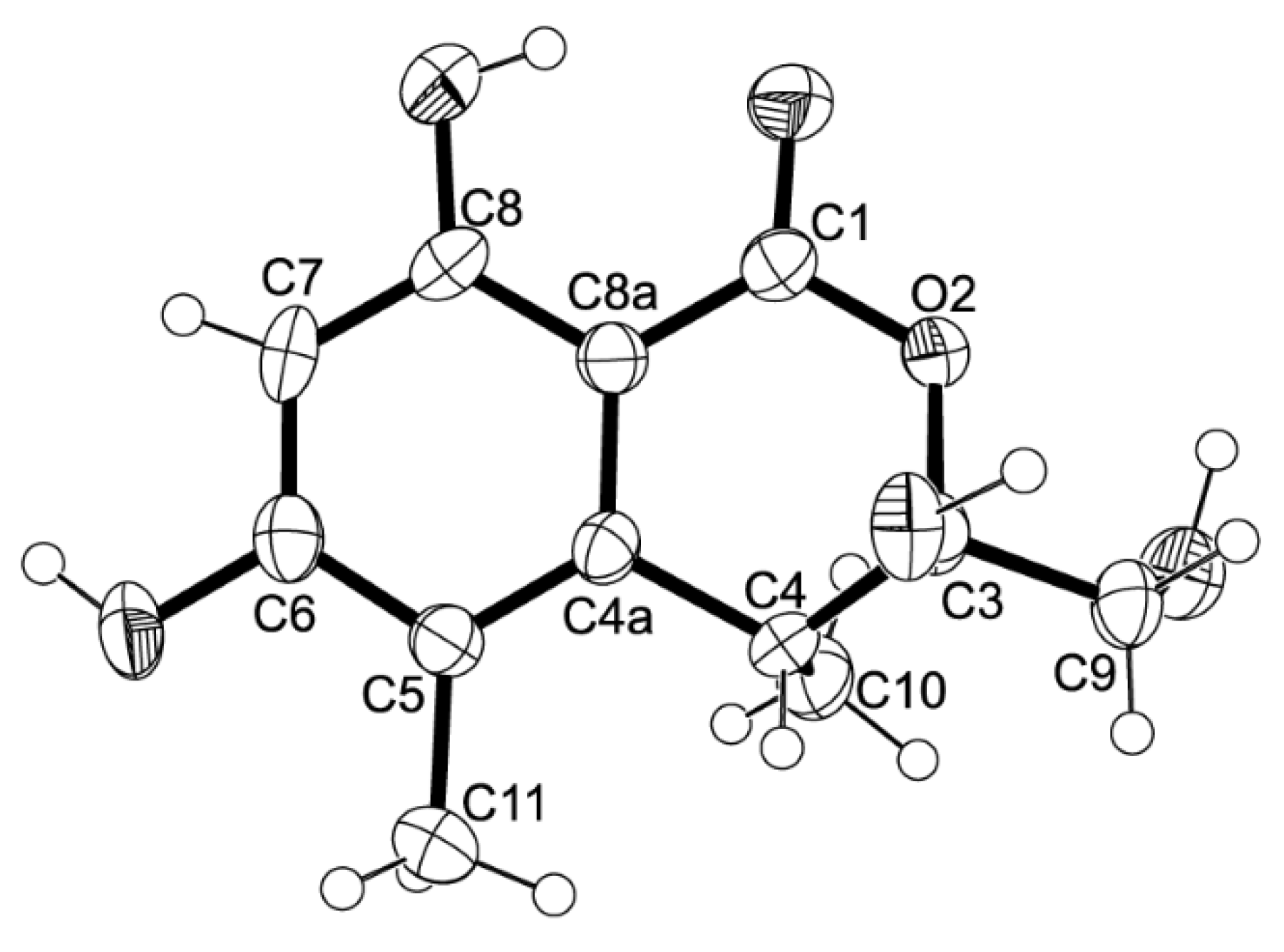
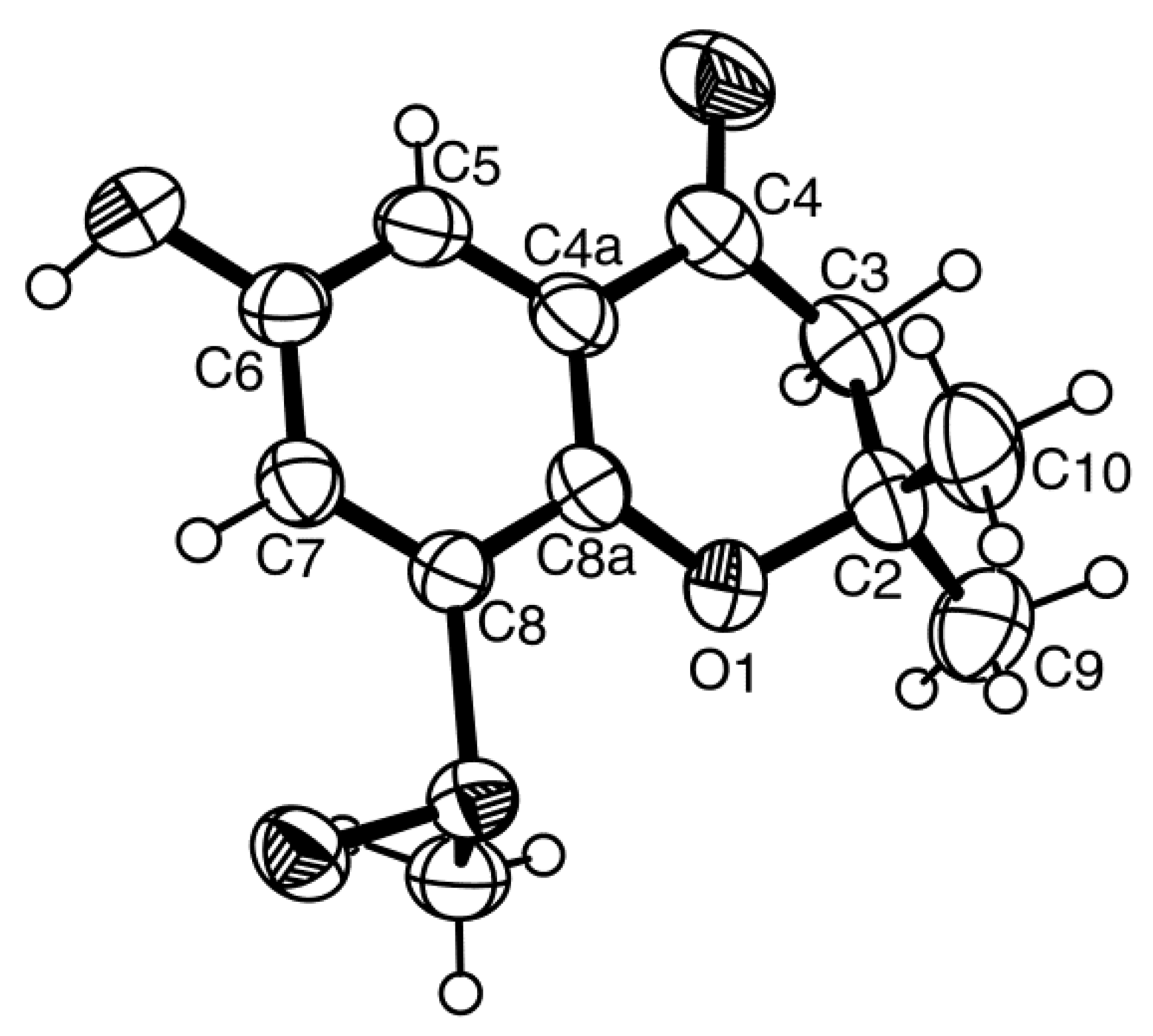
5 could be obtained as a suitable crystal (mp. 223–224 °C), its X-ray analysis was performed. The ORTEP view of compound
5, shown in
Figure 12, not only confirmed the proposed structure, but also determined the absolute configuration of C-3 and C-4 as 3
S, 4
R. Since compound
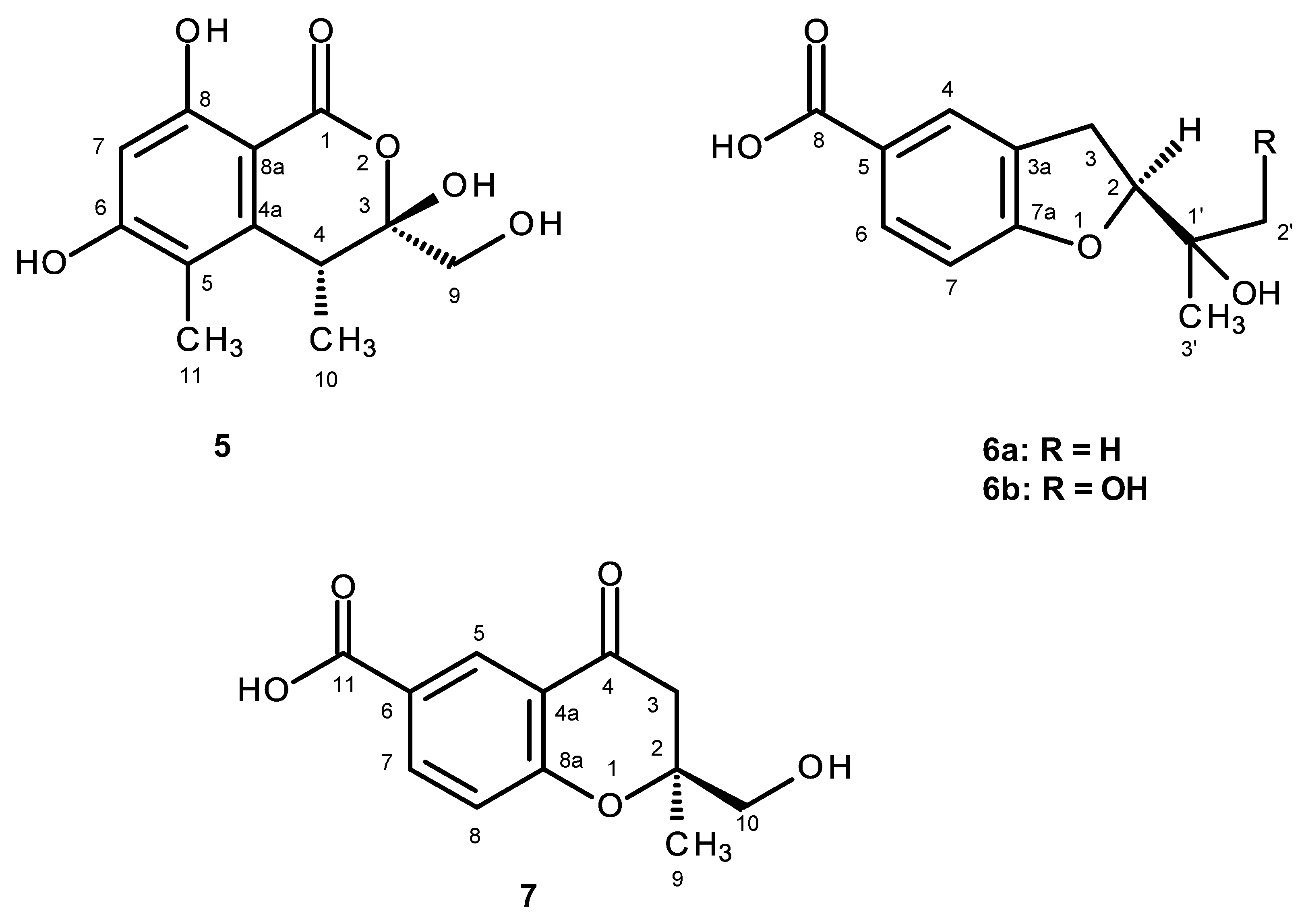
5 is a new compound, it was named quadricinctone C.
Compound
6a was also isolated as white crystals (mp. 149–150 °C), and its molecular formula C
12H
14O
4 was determined based on the (+)-HRESIMS
m/z 223.9067 [M + H]
+ (calculated 223.9070), indicating six degrees of unsaturation. The IR spectrum showed absorption bands for hydroxyl (3417 cm
−1), a conjugated carbonyl (1681 cm
−1) and aromatic (1634 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 6,
Supplementary Information, Figures S36 and S38) exhibited the signals of one conjugated carboxyl carbonyl (δ
C 167.2), two quaternary sp
2 (δ
C 128.2, 122.6), one oxy-quaternary sp
2 (δ
C 163.7), three methine sp
2 (δ
C 130.4, 126.4, 108.4), one oxy-quaternary sp
3 (δ
C 70.0), one oxymethine sp
3 (δ
C 90.2), one methylene sp
2 (δ
C 29.3) and two methyl (δ
C 25.9 and 24.9) carbons. The
1H NMR spectrum (
Table 6,
Supplementary Information, Figure S35) showed signals of three aromatic protons of the 1,2,4-trisubstituted benzene ring at δ
H 7.75, d (
J = 1.8 Hz), 7.72, dd (
J = 8.3, 1.8 Hz), 6.80, d (
J = 8.3 Hz), in addition to a broad signal of the hydroxyl proton at δ
H 12.49, a triplet of one proton at δ
H 4.64 (
J = 8.9 Hz), a doublet of two protons at δ
H 3.17 (
J = 8.9 Hz) and two methyl singlets at δ
H 1.13 and 1.14. That the 1,2,4-trisubstituted benzene ring was part of 2,5-disubstituted 2,3-dihydro-1-benzofuran was supported by the COSY correlations of H-6 (δ
H 7.72, dd,
J = 8.3, 1.8 Hz) to H-4 (δ
H 7.75, d,
J = 1.8 Hz) and H-7 (δ
H 6.80, d,
J = 8.3 Hz) and of H-2 (δ
H 3.17,
J = 8.9 Hz) to H-3 (δ
H 4.64,
J = 8.9 Hz)(
Table 6,
Figure 13a,
Supplementary Information, Figure S37), as well as of the HMBC correlations of H-4 to C-6 (δ
C 130.4) and C-7a (δ
C 163.7), of H-6 to C-4 (δ
C 126.4), of H-3 to C-2 (δ
C 90.2), C-3a (δ
C 128.2) and C-7a and of H-7 to C-3a, C-7a and C-5 (δ
C 122.6) (
Table 6,
Figure 13a,
Supplementary Information, Figure S39). That the carboxyl group was on C-5 was supported by the HMBC correlations of H-4 and H-6 to the conjugated carbonyl at δ
C 167.2 (
Table 6,
Figure 13a), along with the presence of a broad signal at δ
H 12.49, which is characteristic of a hydroxyl group of carboxylic acid. In the same manner, that the 2-hydroxypropyl substituent was placed on C-2 was evidenced by the HMBC correlations of H-2 (δ
H 4.64,
J = 8.9 Hz) to C-3 (δ
C 29.3), C-1′ (δ
C 70.0), C-2′ (δ
C 24.9), C-3′ (δ
C 25.9) (
Table 6,
Figure 13a), as well as by the NOESY correlations of H-2 to H-3, H
3-2′ (δ
H 1.14, s) and H
3-3′ (δ
H 1.13, s)(
Figure 13b). Therefore, compound
6a was identified as 2-(2-hydroxypropan-2-yl)-2,3-dihydro-1-benzofuran-5-carboxylic acid. A literature search revealed that compound
6a has the same flat structure as anodendroic acid (2-(1-hydroxy-1-methylethyl)-2,3-dihydrobenzofuran-5-carboxylic acid), which was first isolated from the stem of
Anodendron affine Durce [
15]. Anodendroic acid was later synthesized by Yamaguchi et al. [
16]; however, its
1H NMR data (DMSO-
d6) were slightly different from those reported by Shima et al. [
15]. Anodendroic acid was also obtained by basic hydrolysis of its methyl ester, a constituent of
Eriodictyon sessilifolium [
17], and also by biotransformation of 3-(γ,γ-dimethylallyl)-
p-coumaric acid [
18]. It is interesting to note that, in all of these reports, the identity of anodendroic acid was achieved by comparing its melting point,
1H NMR and MS data with those reported by Shima et al. [
15], while no stereochemistry of C-2 was indicated. However, when compared to the
1H NMR and other physical data of compound
6a with those reported for anodendroic acid by Shima et al. [
15], it was found that the
1H NMR data of anodendroic acid (in pyridine-
d6) [
15] were slightly different from those of compound
6a (in DMSO-
d6). Therefore, it was not possible to compare these
1H NMR data, since they were obtained in different solvents. Interestingly, the
1H NMR data, obtained in DMSO-
d6 by Yamaguchi et al. [
16], were also slightly different from those of compound
6a. The obvious differences between anodendroic acid [
15] and compound
6a are their melting points and optical rotations. While anodendroic acid (mp. 212–214 °C) is laevorotatory (
−19°,
c 0.7, EtOH) [
15], compound
6a (mp. 149–150 °C) is dextrorotatory (
+74°,
c 0.03, MeOH). Consequently, we concluded that the structure of compound
6a is different from that of anodendroic acid. This fact has prompted us to investigate the stereochemistry of compound
6a. Since Compound
6a was obtained as a suitable crystal for X-ray diffraction, its X-ray analysis was carried out. The ORTEP view of compound
6a shown in
Figure 14 not only confirmed its proposed structure, but also revealed the absolute configuration for C-2 as 2
R. Therefore, compound
6a is a new compound, and we have named it quadricinctafuran A. Since anodendroic acid exhibited the opposite sign of rotation to that of compound
6a, it is probable that the absolute configuration of its C-2 is 2
S. This is not unusual, since fomannoxin, another 2,3-dihydrobenzofuran derivative, also showed the opposite optical rotation and has the opposite absolute configuration to tremetone, even though they have very similar structures [
16].
The (+)-HRESIMS of compound
6b gave the [M + H]
+ at
m/z 239.0919 (calculated 239.0919), indicating its molecular formula C
12H
14O
5 and, therefore, six degrees of unsaturation. The general features of its
1H and
13C NMR spectra resembled those of compound
6a. The
13C NMR, DEPT and HSQC spectra (
Table 6,
Supplementary Information, Figures S42 and S44) exhibited the signals of one conjugated carboxyl carbonyl (δ
C 167.2), two quaternary sp
2 (δ
C 128.4, 122.6), one oxy-quaternary sp
2 (δ
C 163.6), three methine sp
2 (δ
C 130.8, 126.4, 108.4), one oxy-quaternary sp
3 (δ
C 72.5), one oxymethine sp
3 (δ
C 86.5), one methylene sp
2 (δ
C 28.7), one oxymethylene sp
2 (δ
C 66.7) and one methyl (δ
C 20.0) carbons. The
1H NMR and COSY spectra (
Table 6,
Supplementary Information, Figures S41 and S43) showed similar proton signals of the 2,3-dihydro-1-benzofuran-5-carboxylic acid nucleus to those of
6a. However, it exhibited only one methyl singlet at δ
H 1.09 and another singlet of oxymethylene protons at δ
H 3.33, instead of two methyl singlets as in compound
6a. Therefore, the difference between the structures of compounds
6a and
6b resides in the substitutions on C-2. That the substituent on C-2 of compound
6b was 1,2-dihydroxypropyl was evidenced by not only its molecular formula (C
12H
14O
5), which has one more oxygen atom than that of compound
6a (C
12H
14O
4), but also the presence of the oxymethylene carbon at δ
C 66.7 (C-2′; δ
H 3.33, s). Moreover, the chemical shift value of C-2 (δ
C 86.5) was nearly 4 ppm lower than that of compound
6a (δ
C 90.2). Thus, compound
6b was identified as 2-(1,2-dihydroxypropan-2-yl)-2,3-dihydro-1-benzofuran-5-carboxylic acid. This was confirmed by the HMBC correlations of H
3-3′ (δ
H 1.09) to C-1′ (δ
C 72.5) and C-2′ (δ
C 66.7), as well of H
2-2′ (δ
H 3.33, s) to C-1′ (δ
C 72.5) and C-3′ (δ
C 20.0) (
Figure 15,
Supplementary Information, Figure S45). Since compound
6b could not be obtained as suitable crystals for X-ray crystallography, it was not possible to determine the absolute configuration of C-2 by X-ray analysis. Moreover, as compound
6b also has a tertiary hydroxyl group on C-1′ and a primary hydroxyl group on C-2′, it was not possible to determine the absolute configurations of C-2 and C-1′ by Mosher’s method. On the other hand, since the methyl group on C-1 in compound
6a was replaced by the hydroxymethyl group incompound
6b, it is legitimate to postulate that compounds
6b is derived from compound
6a. Consequently, the absolute configuration of C-2 of compound
6b should be the same as that of compound
6a, i.e., 2
R. However, it is not yet possible to determine the absolute configuration of C-1′. Compound
6b is also a new compound, and we have named it quadricinctafuran B.
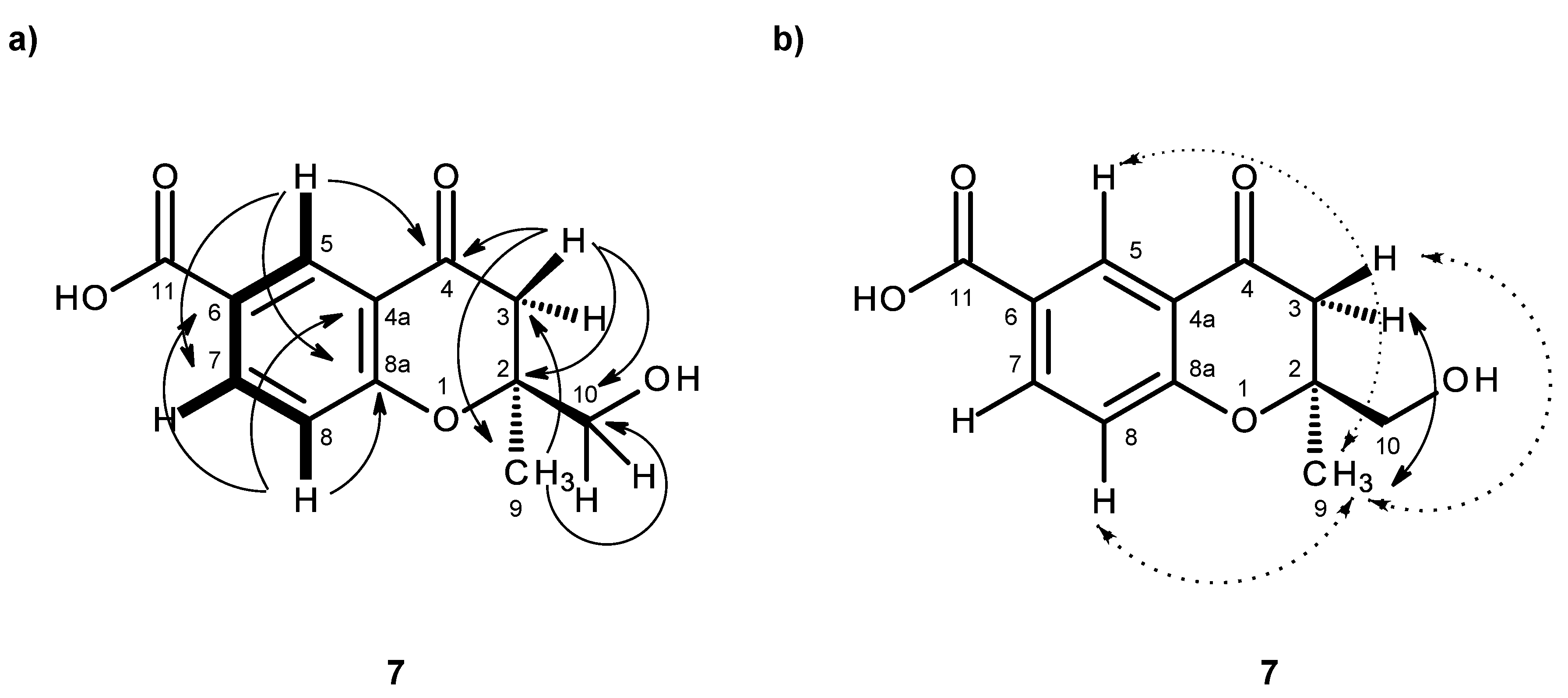
Compound
7 was also isolated as white crystals (mp. 196–197 °C), and its molecular formula C
14H
12O
5 was established on the basis of the (+)-HRESIMS
m/z 237.0792 [M + H]
+ (calculated 237.0763), indicating seven degrees of unsaturation. The IR spectrum showed absorption bands for hydroxyl (3404 cm
−1), conjugated ketone carbonyl (1708 cm
−1), conjugated carboxyl carbonyl (1670 cm
−1) and aromatic (1558, 1540 cm
−1) groups. The
13C NMR, DEPT and HSQC spectra (
Table 7,
Supplementary Information, Figures S48 and S50) exhibited the signals of one conjugated ketone carbonyl (δ
C 191.1), one conjugated carboxyl carbonyl (δ
C 166.4), one oxy-quaternary sp
2 (δ
C 162.9), two quaternary sp
2 (δ
C 123.0, 119.6), three methine sp
2 (δ
C 136.4, 127.5, 118.5), one oxy-quaternary sp
3 (δ
C 83.0), one oxymethylene sp
2 (δ
C 66.8), one methylene sp
2 (δ
C 43.4) and one methyl (δ
C 21.2) carbons. The
1H NMR spectra (
Table 7,
Supplementary Information, Figure S47) exhibited, besides the signals of three aromatic protons of the 1,2,4-trisubstituted benzene ring at δ
H 8.26, d (
J = 2.3 Hz), 8.04, dd (
J = 8.7, 2.3 Hz), 7.07, d (
J = 8.7 Hz), a methyl singlet at δ
H 1.30, two pairs of geminally-coupled methylene protons at δ
H 2.74, d (
J = 16.7 Hz)/3.00, d (
J = 16.7 Hz) and δ
H 3.49, dd (
J = 11.6, 4.5 Hz)/3.59, dd (
J = 11.6, 4.2 Hz); the latter showed COSY correlations with the broad triplet of the hydroxyl proton at δ
H 5.26 (
Table 7,
Figure 16a,
Supplementary Information, Figure S49). As the HMBC spectrum exhibited correlations of the aromatic proton signal at δ
H 8.26, d (
J = 2.3 Hz, H-5) to the oxy-quaternary sp
2 carbon at δ
C 162.9 (C-8a), as well as to the conjugated ketone carbonyl carbon at δ
C 191.1 (C-4) and the methine sp
2 at δ
C 136.4 (C-7) (
Table 7,
Figure 16a,
Supplementary Information, Figure S51), the ketone moiety was placed on C-4a. Additionally, the HMBC spectrum also showed correlations of H-8 (δ
H 7.07, d,
J = 8.7 Hz) to C-8a and the quaternary sp
2 carbons at δ
C 123.0 and 119.6; they were therefore assigned to C-4a and C-6, respectively (
Table 7,
Figure 16a). The presence of the 1-hydroxy-2-methyl-2-oxypropyl moiety was supported by the HMBC correlations of the methyl singlet at δ
H 1.30 (CH
3-9) to the oxy-quaternary sp
3 carbon at δ
C 83.0 (C-2), the oxymethylene sp
3 carbon at δ
C 66.8 (C-10) and the methylene sp
3 carbon at δ
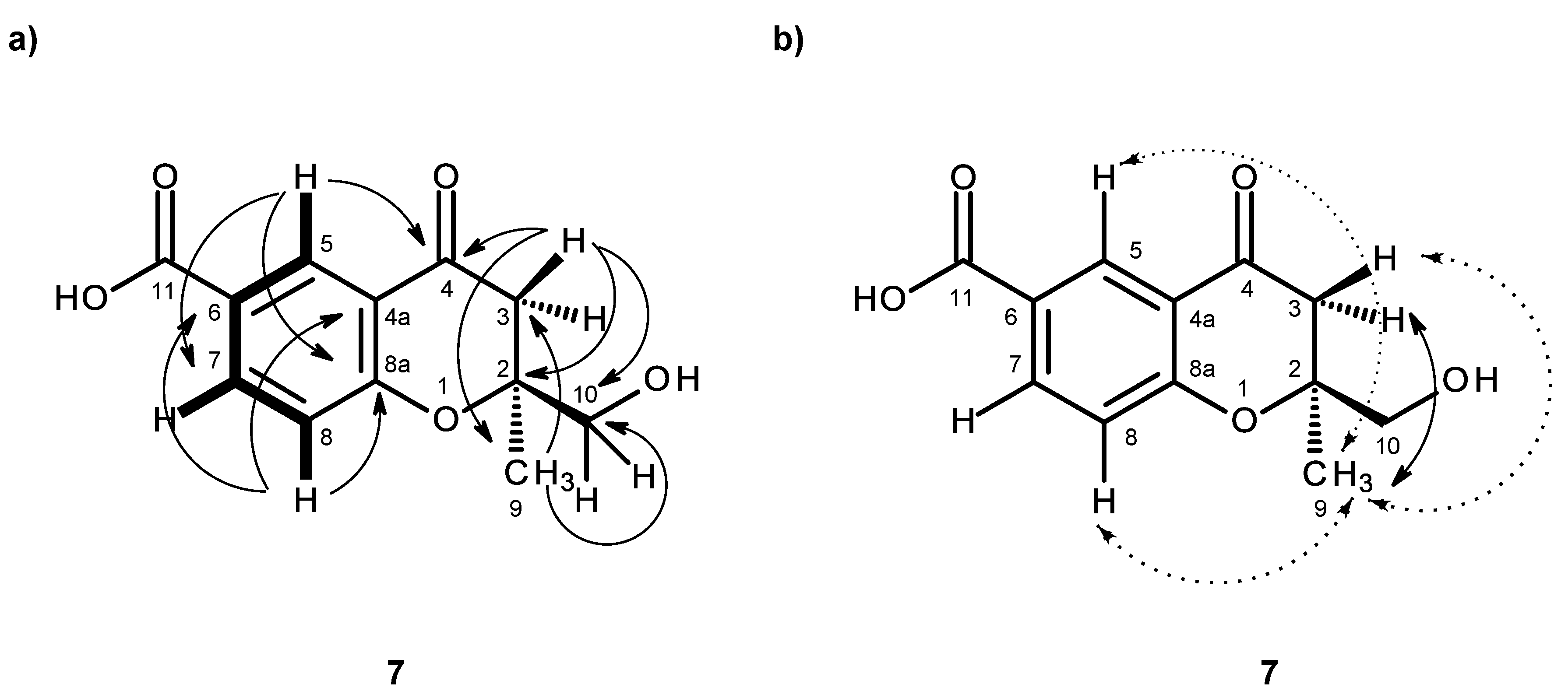
C 43.4 (C-3), as well as of the H
2-3 (δ
H 2.74, d,
J = 16.7 Hz/3.00, d,
J = 16.7 Hz) to C-2, C-9 (δ
C 21.2) and C-10 (
Table 7,
Figure 16a). Since H
2-3 also gave a HMBC cross-peak to C-4, the 1-hydroxy-2-methyl-2-oxypropyl moiety was linked to C-4. Due to the fact that these two moieties accounted for only C
11H
12O
4, it was concluded that the carboxyl group (δ
C 166.4, δ
H 12.69 br) was on C-6. Therefore, compound
7 was identified as 2-(hydroxymethyl)-2-methyl-4-oxo-3,4-dihydro-2
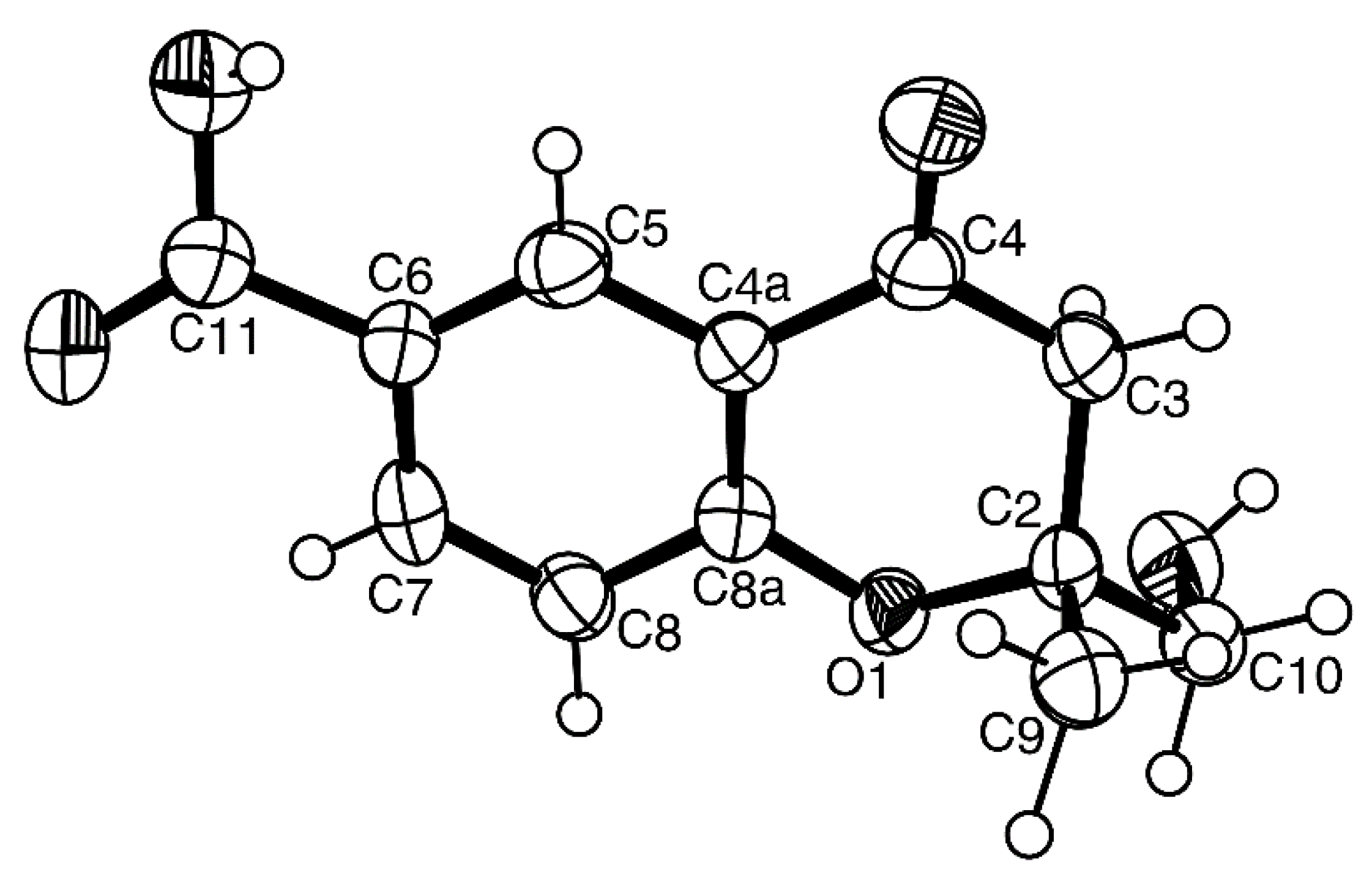
H-chromene-6-carboxylic acid. As compound
7 was obtained as a suitable crystal for an X-ray diffraction, its X-ray analysis was carried out. The ORTEP view of compound
7, shown in
Figure 17, revealed the absolute configuration for C-2 as 2
S. A literature search revealed that compound
7 is also a new compound, so we named it quadricinctone D.
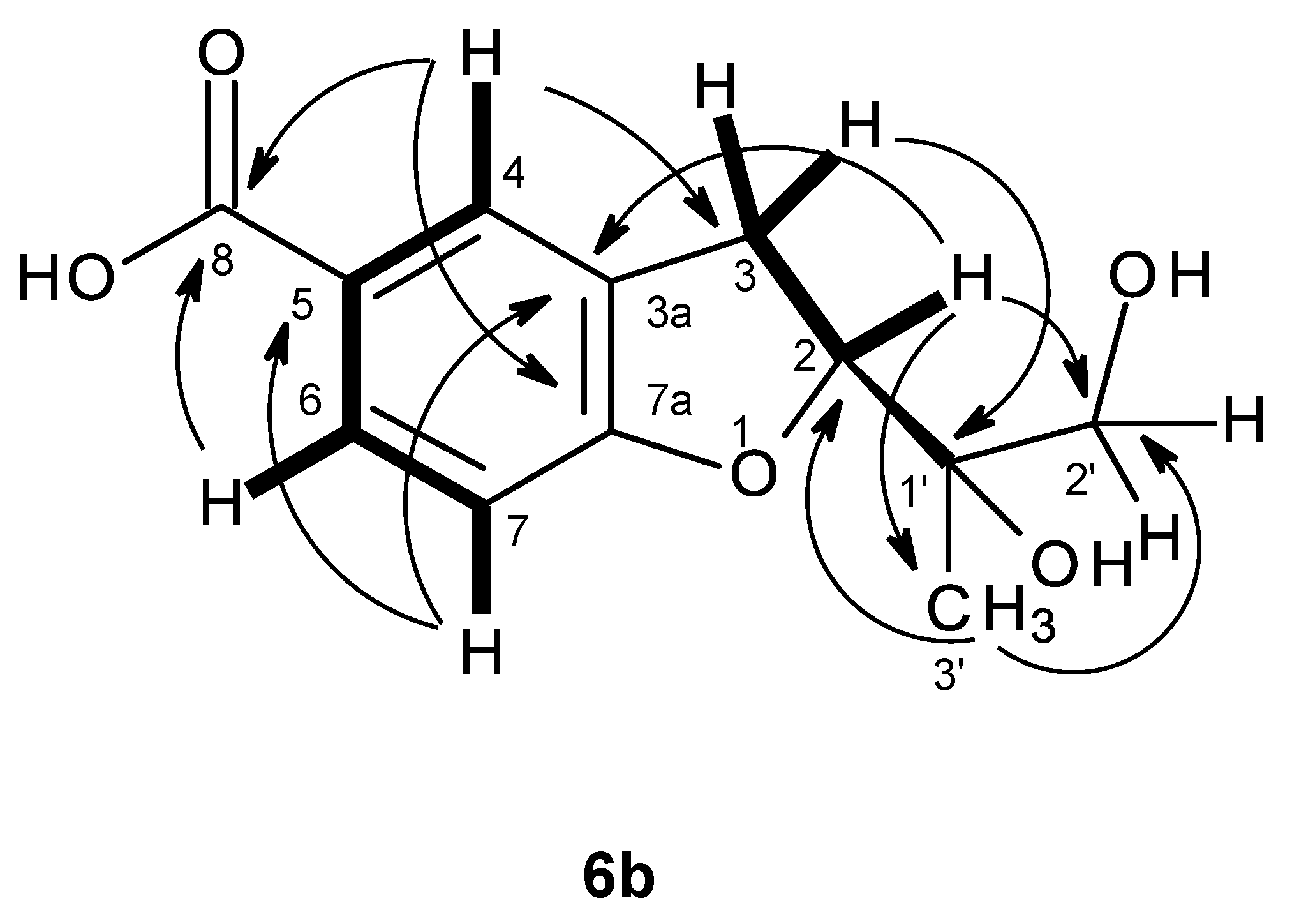
In order to establish the conformation of the 2,3-dihydro-
4H-pyran-4-one, analysis of the NOESY correlations was carried out. The NOESY spectrum (
Table 7,
Figure 16b,
Supplementary Information, Figure S52) exhibited not only a strong correlation of H-7 to H-8, but also week correlations of H
3-9 to H-5 and H-8. Therefore, CH
3-9 is in the α-axial position. Additionally, since H
3-9 also exhibited a strong cross-peak with the signal of the methylene proton at δ
H 2.74, d (
J = 16.7 Hz), and a weak cross-peak with the proton signal at δ
H 3.00, d (
J = 16.7 Hz), the former was assigned to H-3α and the latter to H-3β. It is interesting to observe that the structure of compound
7 is analogous to the structure of compound
3 in that it also exhibits the same methyl-methylene bridge structural feature (at C-9 and C-3), with the same relative intensity of NOESY cross-peaks between the protons of the two groups and with similar conformational energies for the two half-chair conformations of the non-aromatic ring. Therefore, the conclusions drawn for compound
3 are also valid for compound
7, whose stereochemistry is unequivocally defined by X-ray analysis.
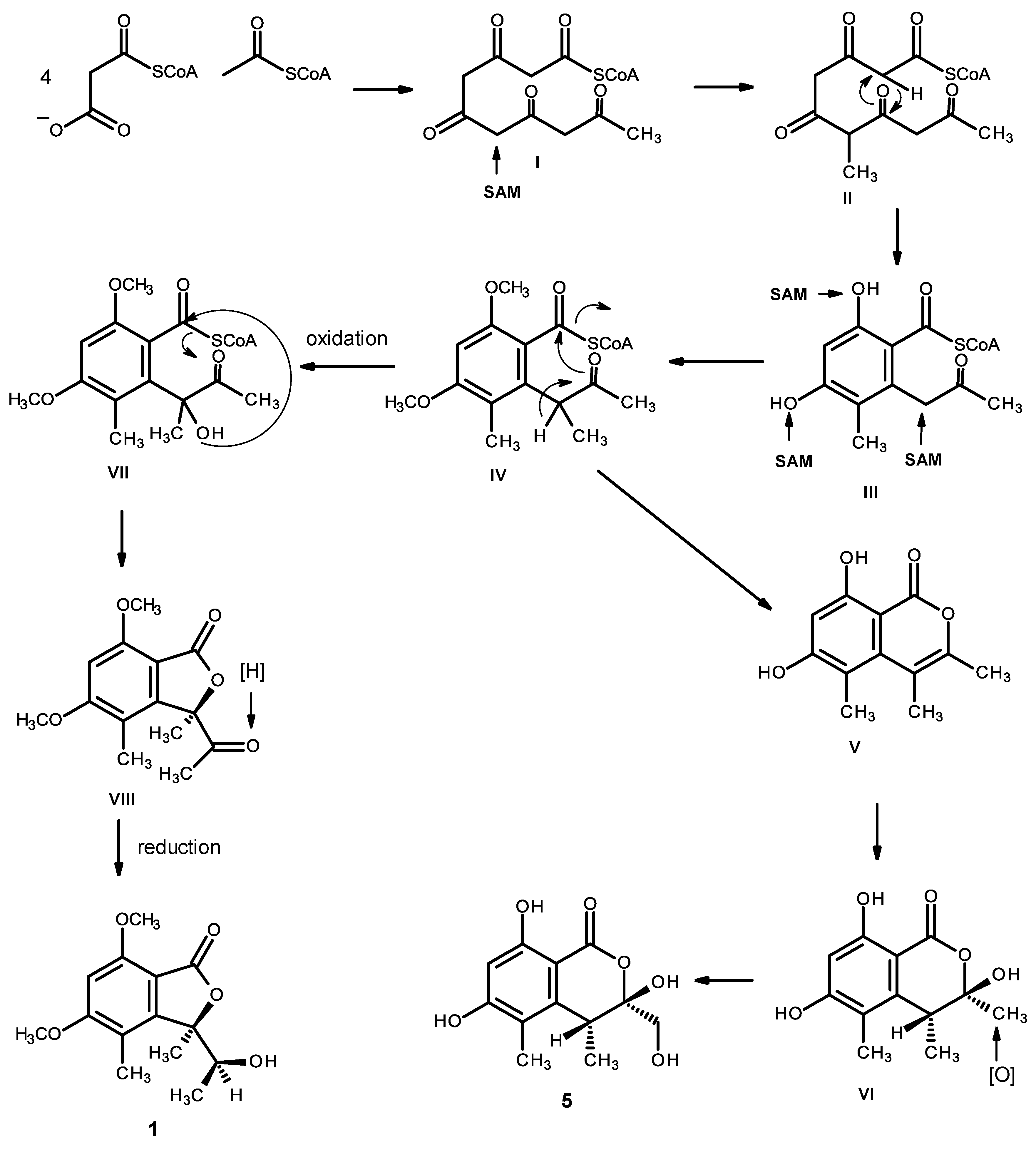
Compounds
1 and
5 can be hypothesized as originating from the pentaketide intermediate (I). Methylation (by SAM) gives II, which undergoes cyclization and enolization to give the intermediate III. Methylation of the phenolic hydroxyl groups and the α-carbon of the carbonyl ketone leads to the intermediate IV. Enolization, followed by a lactonization, originates V, which undergoes hydration to give VI, and oxidation of one of the methyl groups gives rise to compound
5. Alternatively, oxidation of the α-carbon of the side chain of IV leads to the intermediate VII, which, after lactonization and reduction of the ketone carbonyl, gives rise to compound
1 (
Figure 18).
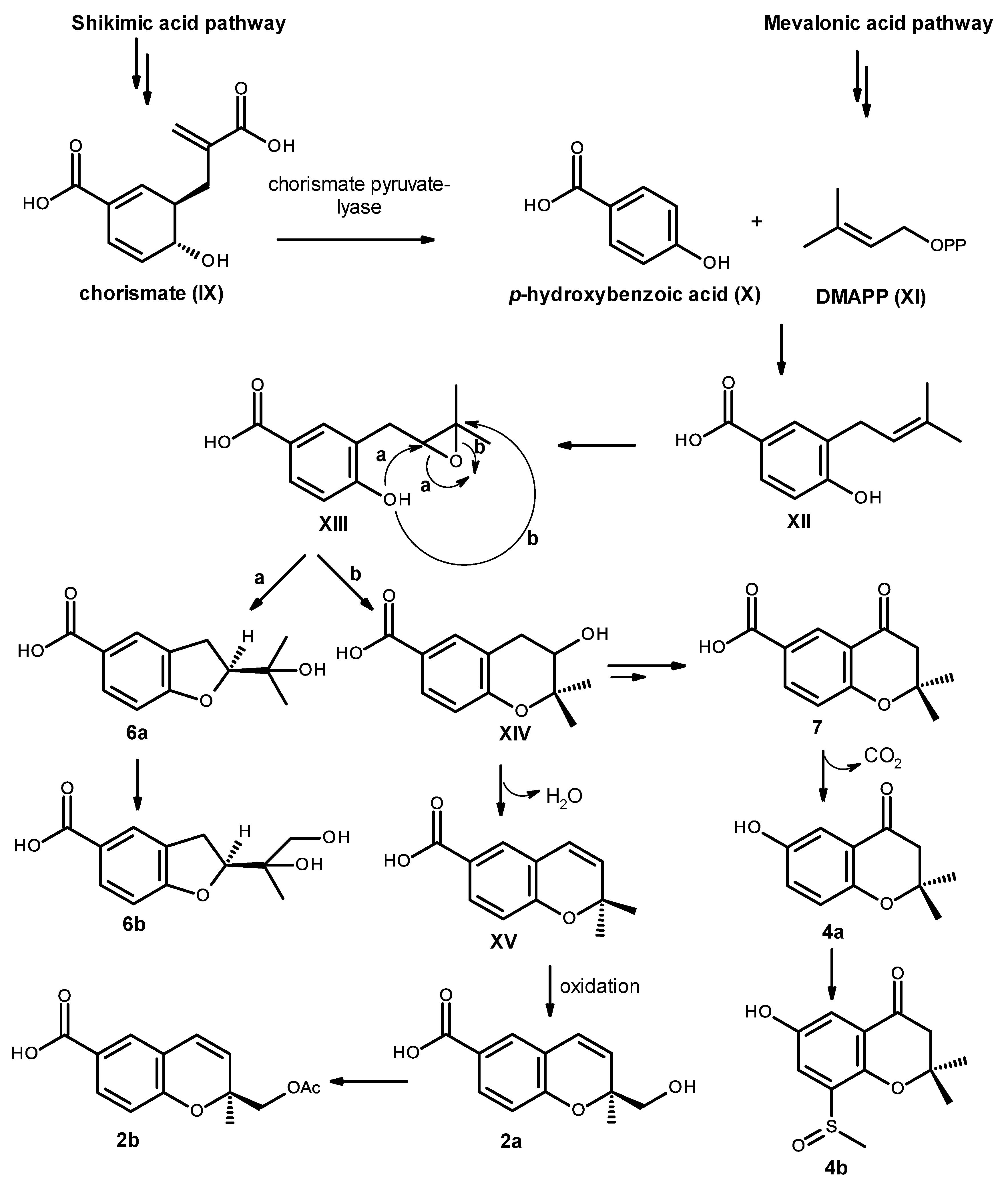
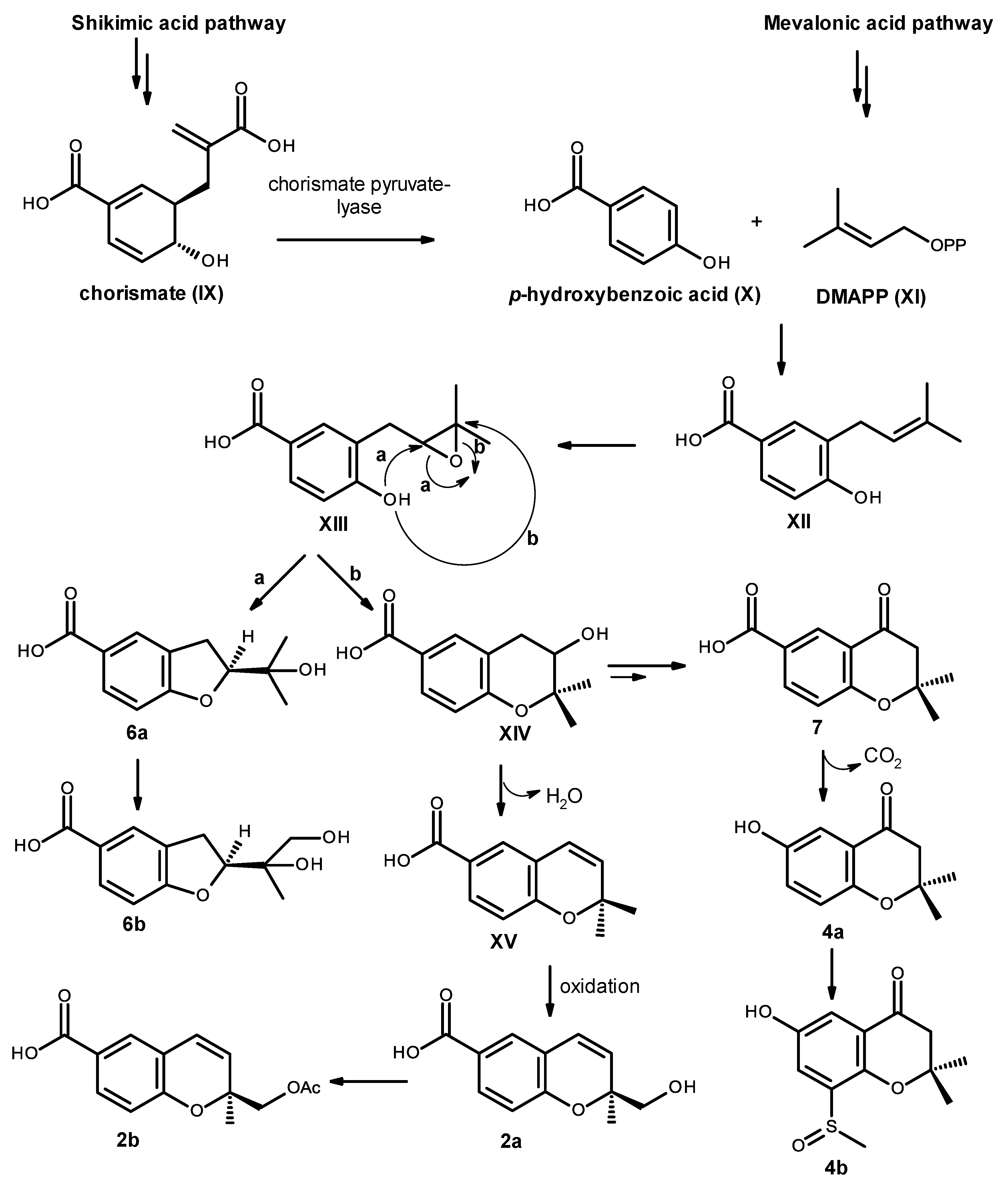
Biosynthetically, compounds
2a,
2b,
4a,
4b,
6a,
6b and
7 are of mixed origin, i.e., shikimic acid and mevalonic acid pathways, similar to that proposed for fomannoxin [
19], as depicted in
Figure 19. Elimination of pyruvate from chorismate (IX) by chorismate pyruvate lyase leads to the formation of
p-hydroxybenzoic acid (X), which after prenylation by DMAPP (XI), originates the intermediate XII. Epoxidation and cyclization of XII, via Route
a, leads to a formation of the furan ring in compound
6a and after oxidation of one of the methyl groups leads to compound
6b. On the other hand, cyclization via Route
b leads to the formation of the pyran ring in XIV. Dehydration and oxidation of one of the methyl groups originates compound
2a, which after acetylation of the primary alcohol function of the side chain will originate compound
2b. Alternatively, the intermediate XIV can also undergo dehydration, reduction and oxidation to give the ketone function in compound
7. Oxidative decarboxylation of compound
7 leads to the formation of compound
4a, which after sulfinylation of the benzene ring originates compound
4b. However, it is possible that the introduction of the methyl sulfoxide group to the aromatic ring could happen before cyclization.
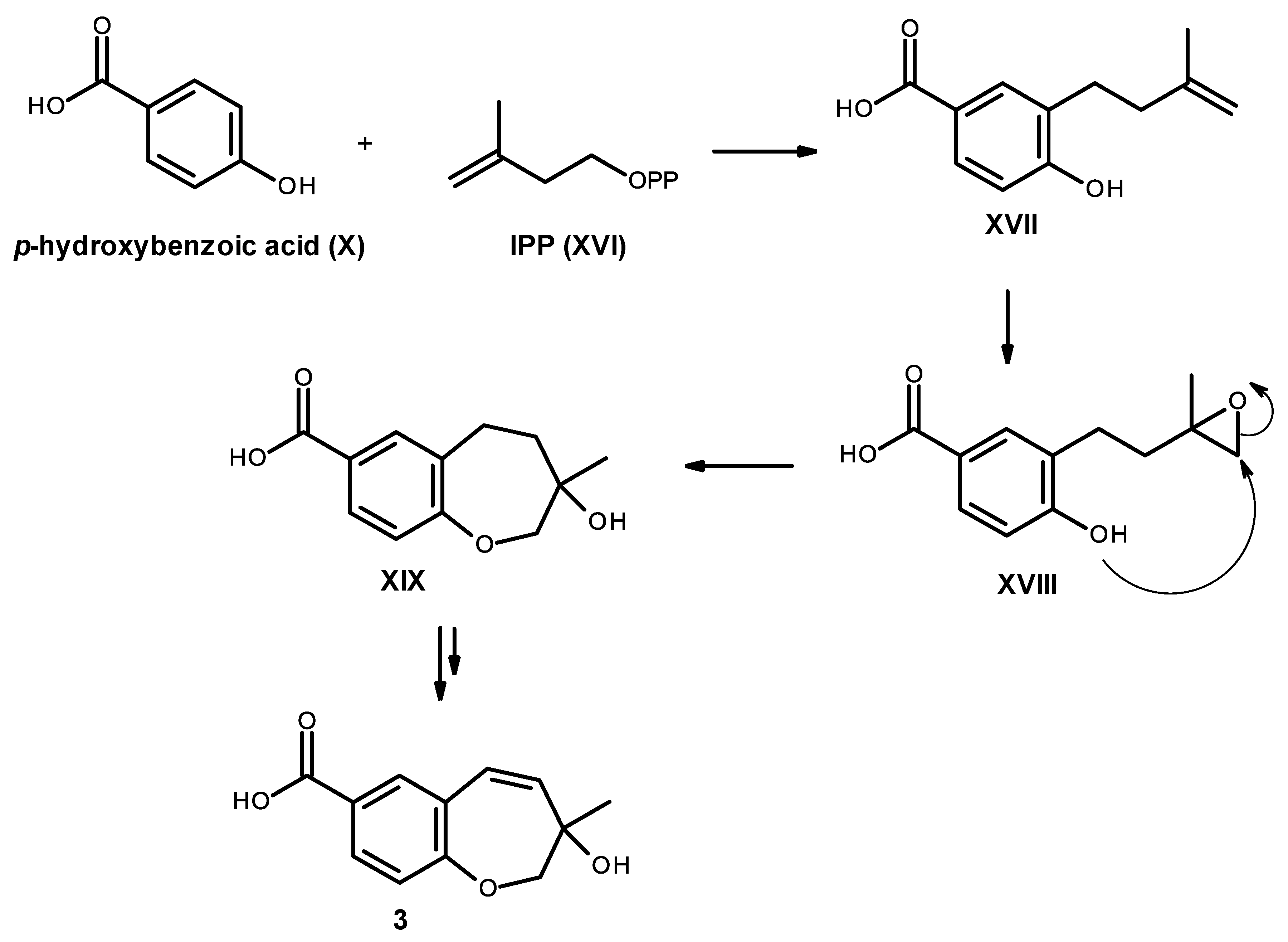
Compound
3 is also derived from a prenylation of
p-hydroxybenzoic acid (X); however, it can occur with IPP (XVI) instead of DMAPP (XI). Epoxidation of the double bond of the side chain of XVII, followed by cyclization of XVIII leads to the formation of an oxepin ring in XIX. Oxidation and dehydration of the oxepin ring will lead to the formation of compound
3, as depicted in
Figure 20.
Compounds
1–
7 were evaluated for their antibacterial activity against Gram-positive and Gram-negative bacteria, as well as multidrug-resistant isolates from the environment, according to the previously described protocol [
6], as well as for their antifungal activity against yeast (
Candida albicans ATCC 10231), filamentous fungus (
Aspergillus fumigatus ATCC 46645) and dermatophyte (
Trichophyton rubrum FF5) in the antifungal assay [
20]. The results showed that none of the tested compounds exhibited significant antibacterial activity (MIC > 256 μg/mL) or antifungal activity (MIC > 512 μg/mL). These compounds were also evaluated for their in vitro growth inhibitory activity against the MCF-7 (breast adenocarcinoma), NCI-H460 (non-small cell lung cancer) and A375-C5 (melanoma) cell lines by the protein binding dye SRB method [
21], and they did not show any activity in this assay (GI
50 > 150 mM).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}