
Phomopsichin A–D; Four New Chromone Derivatives from Mangrove Endophytic Fungus Phomopsis sp. 33#


 ,
,
Abstract
:1. Introduction
2. Results
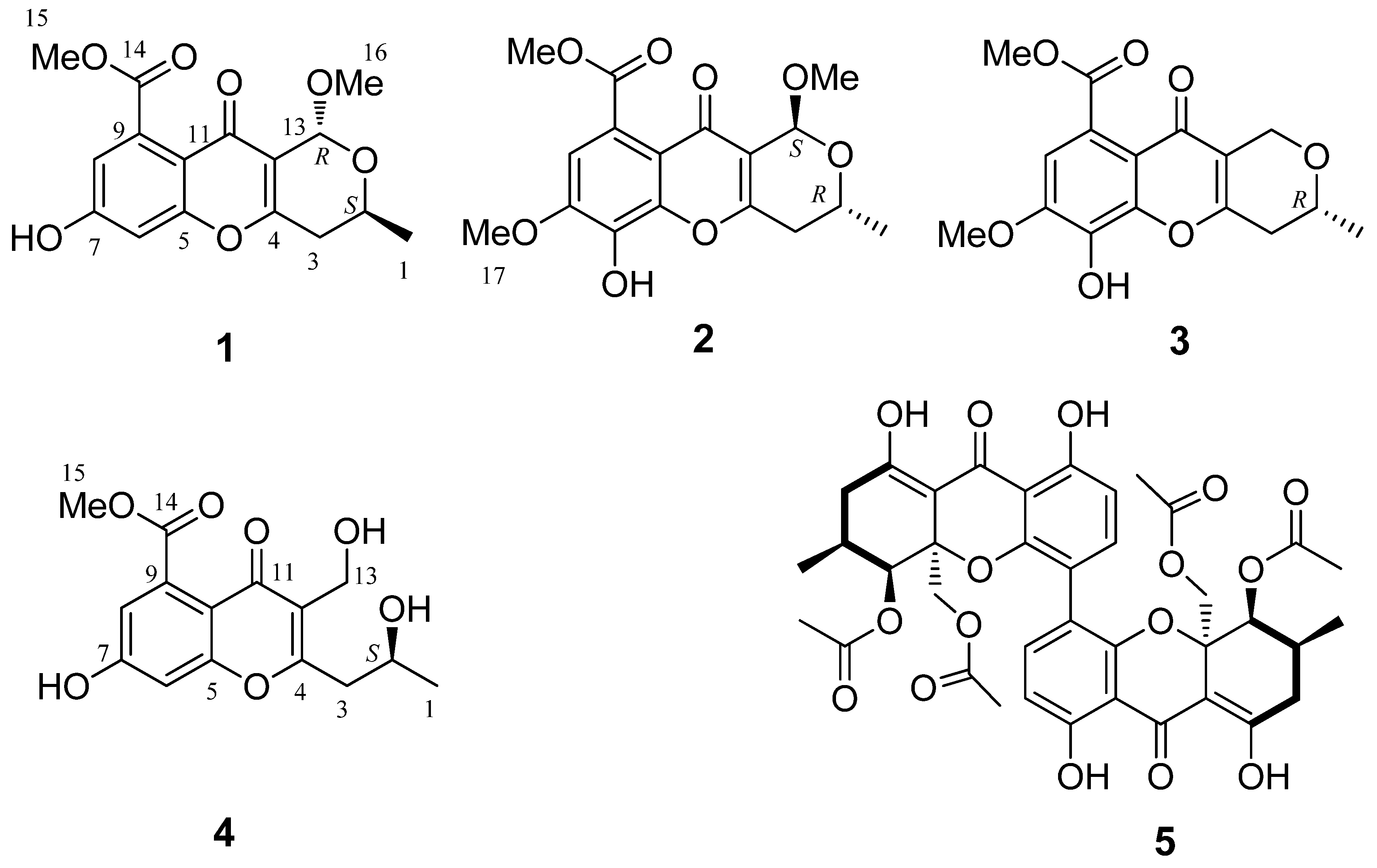
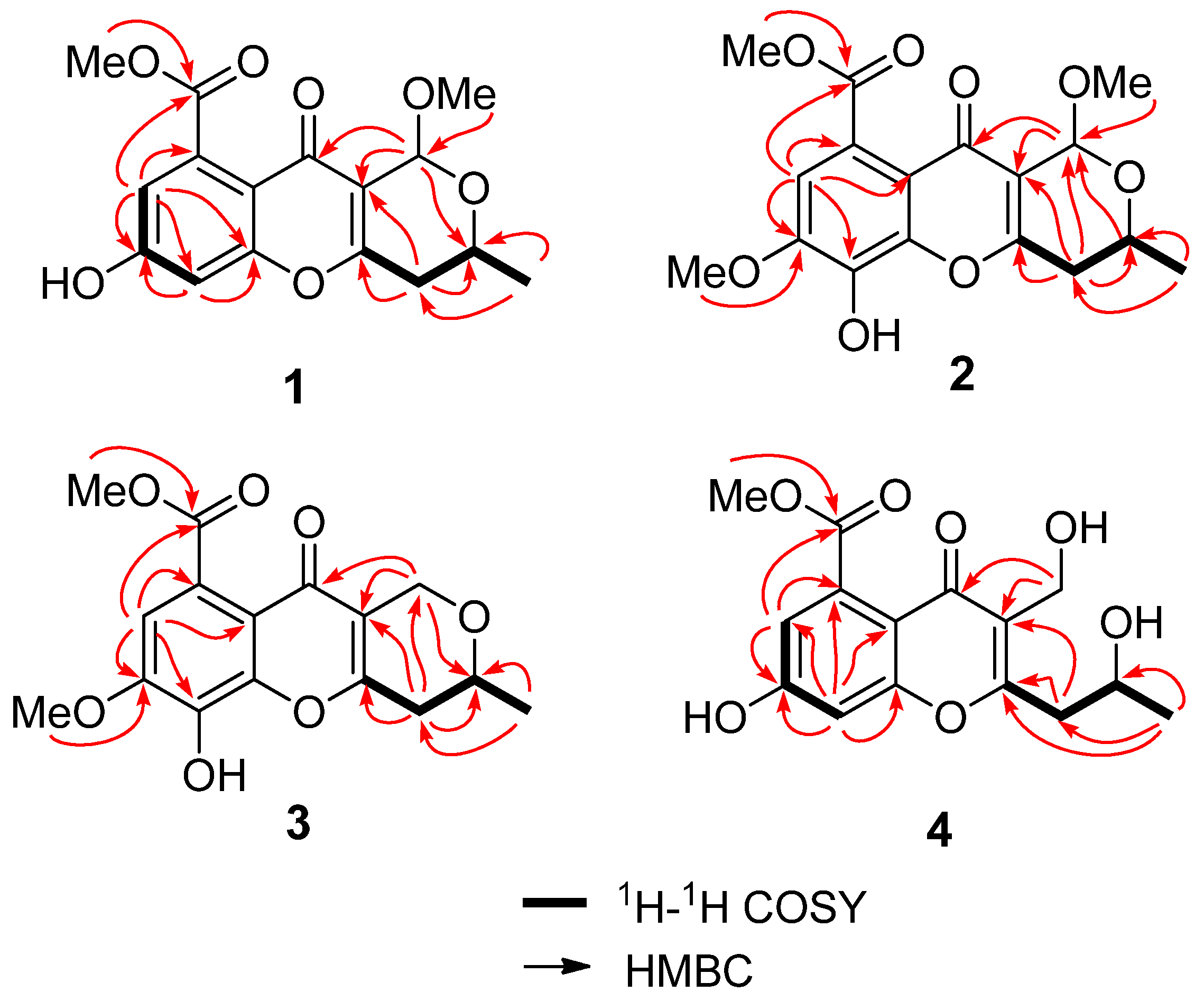
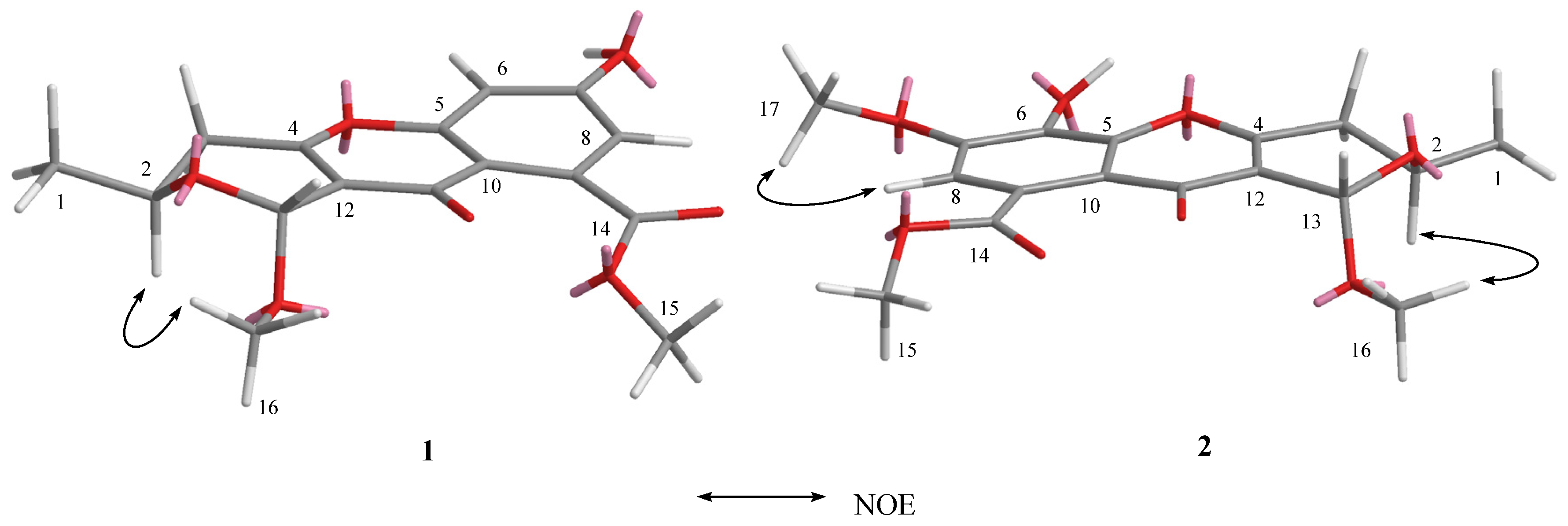
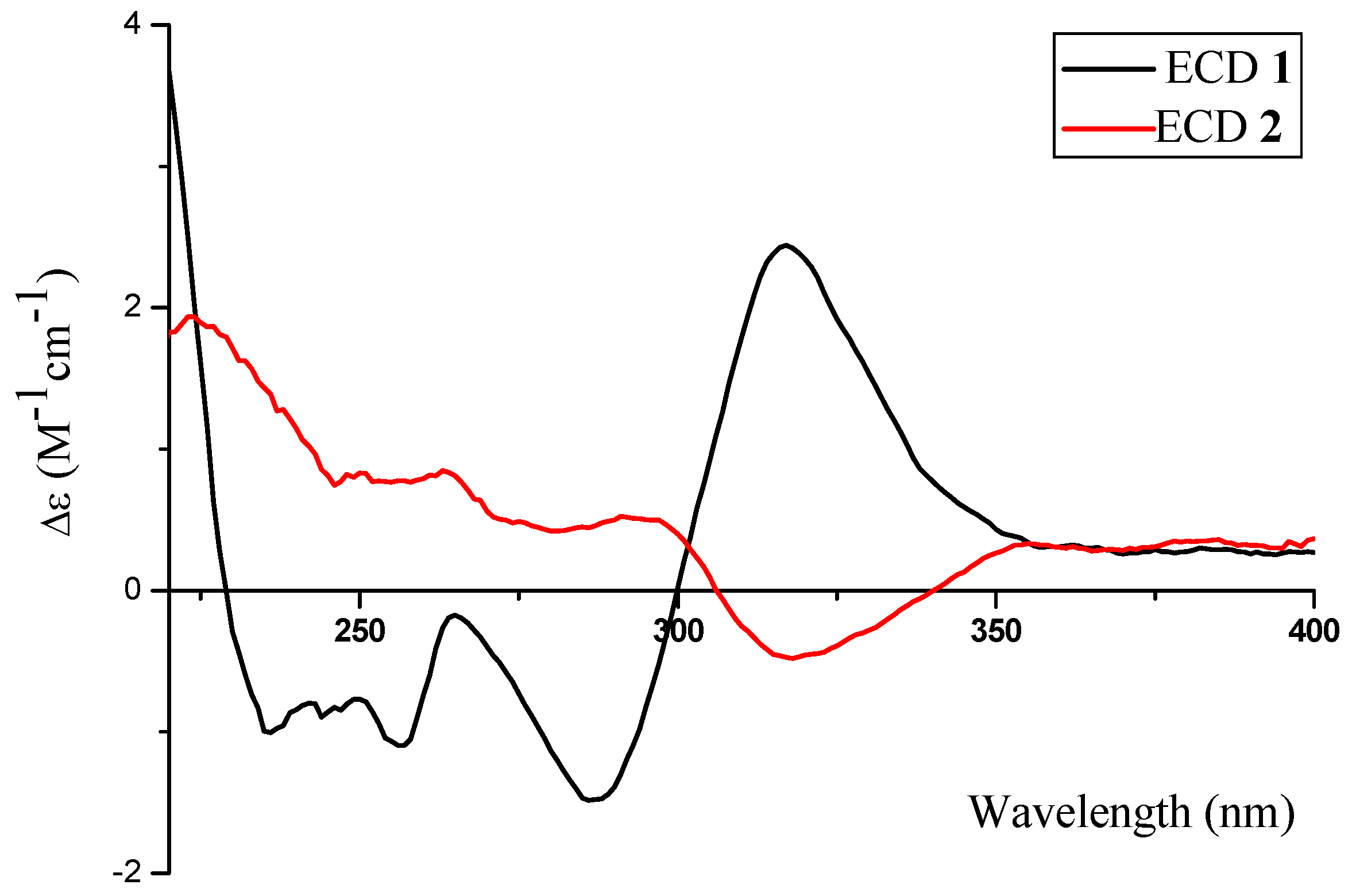
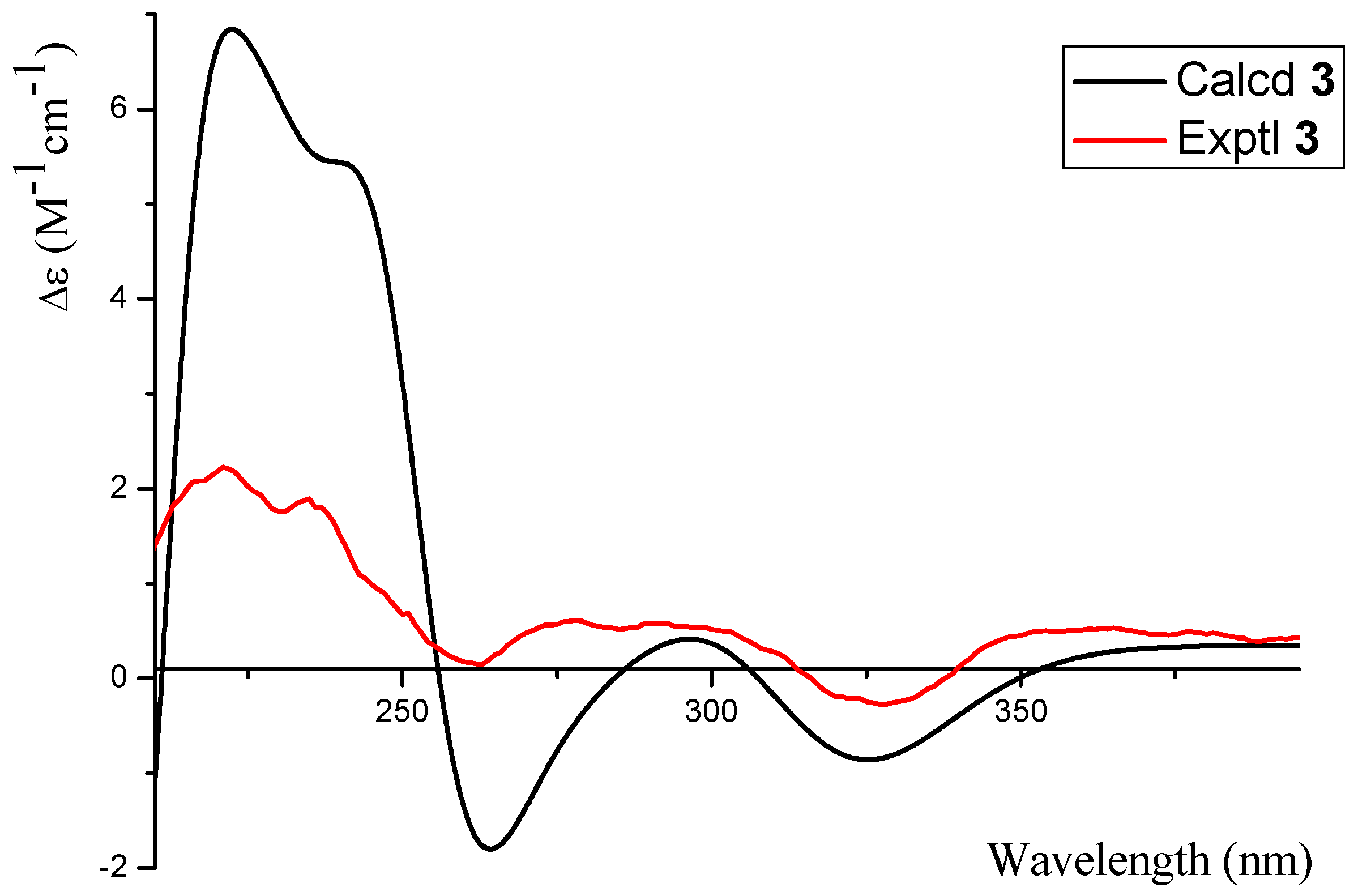
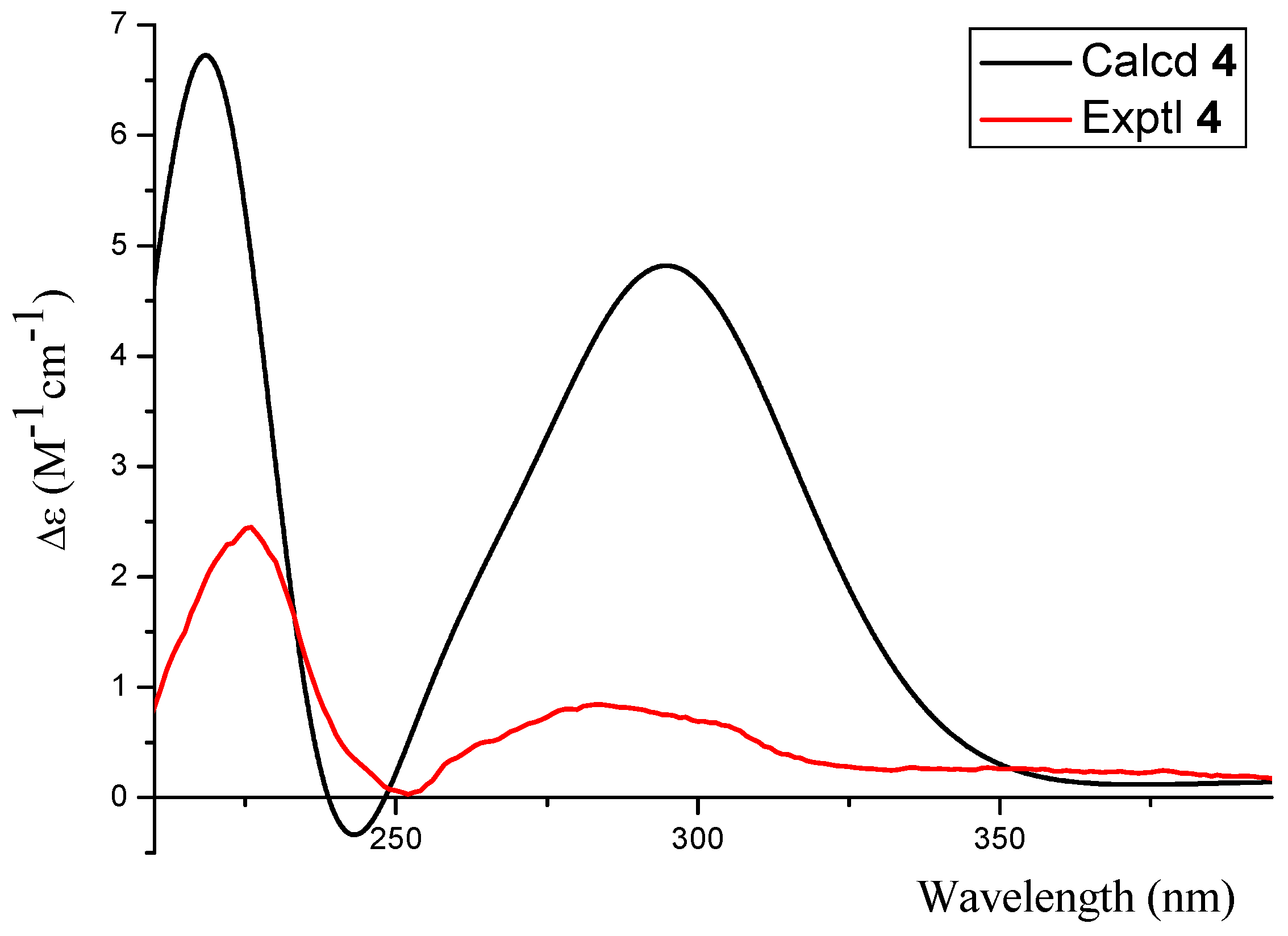
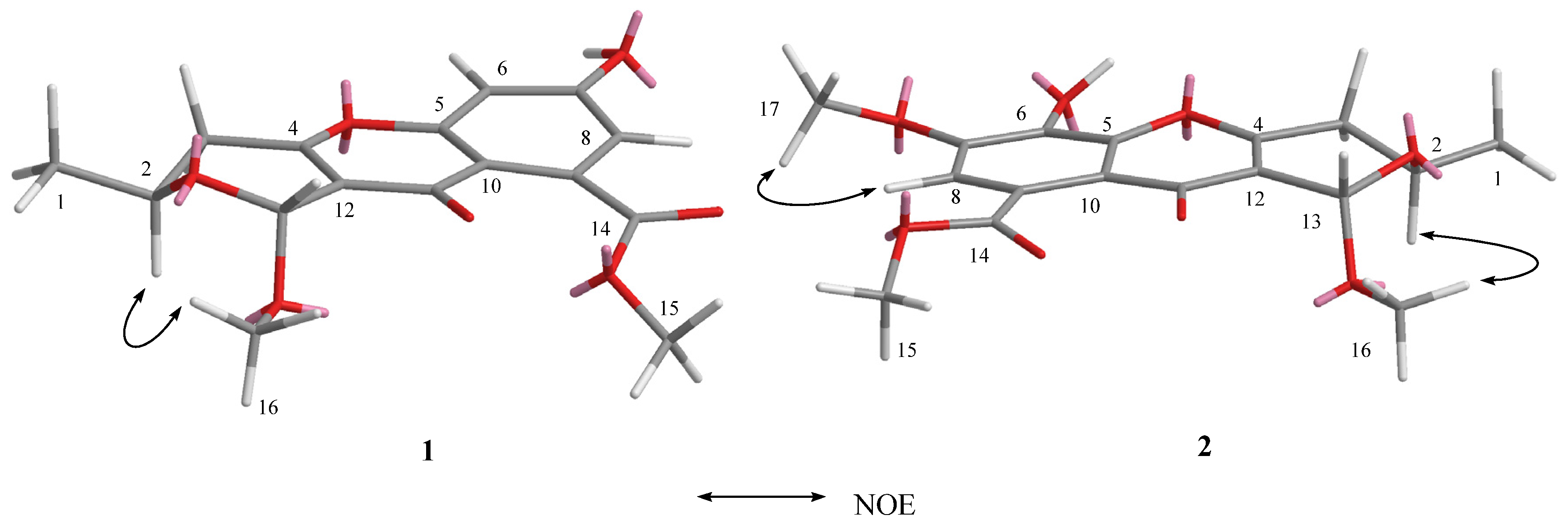
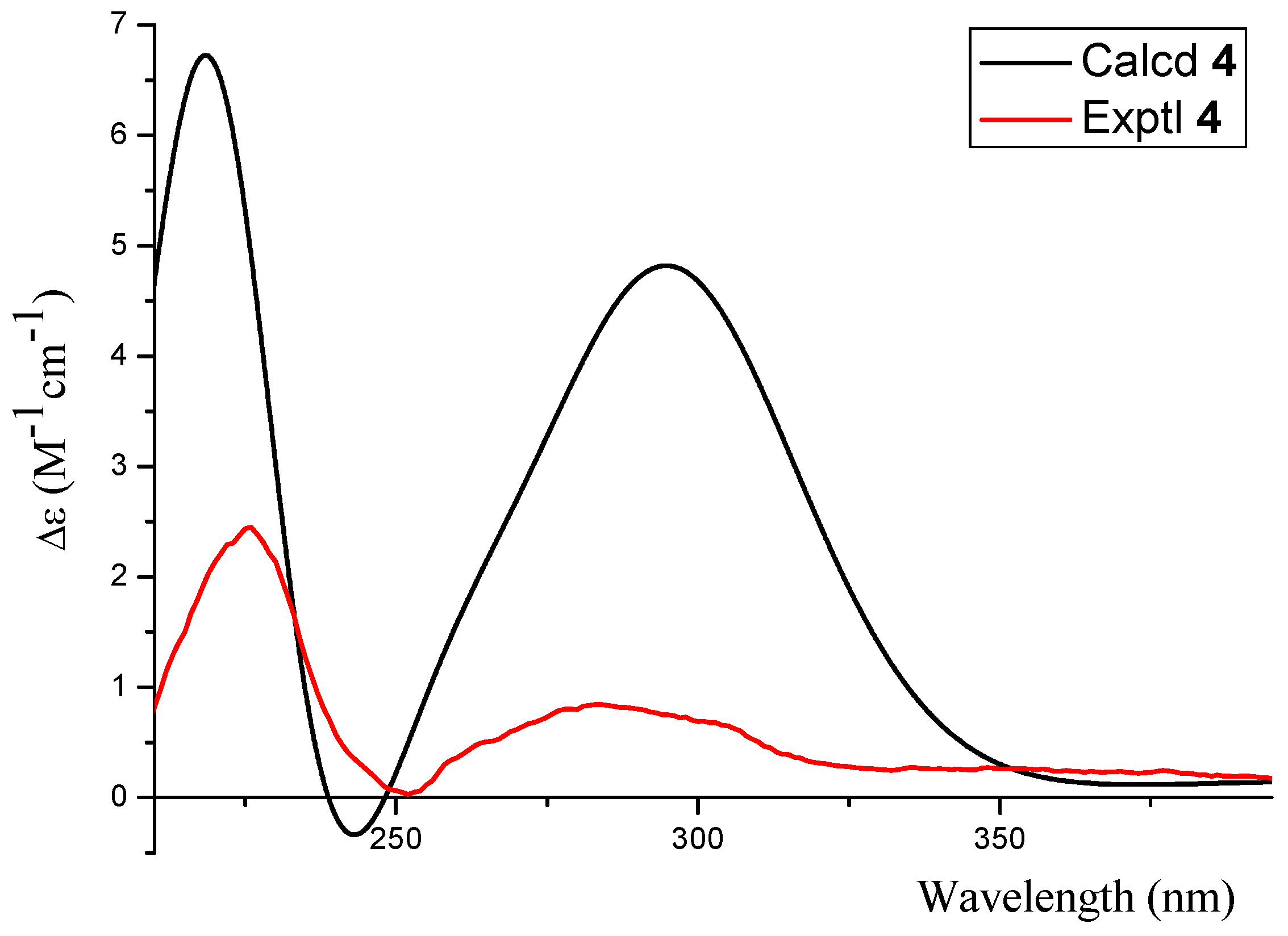
2.1. Structure Elucidation
2.2. Biological Evaluation
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Spectral Data
3.5. Computational Analyses
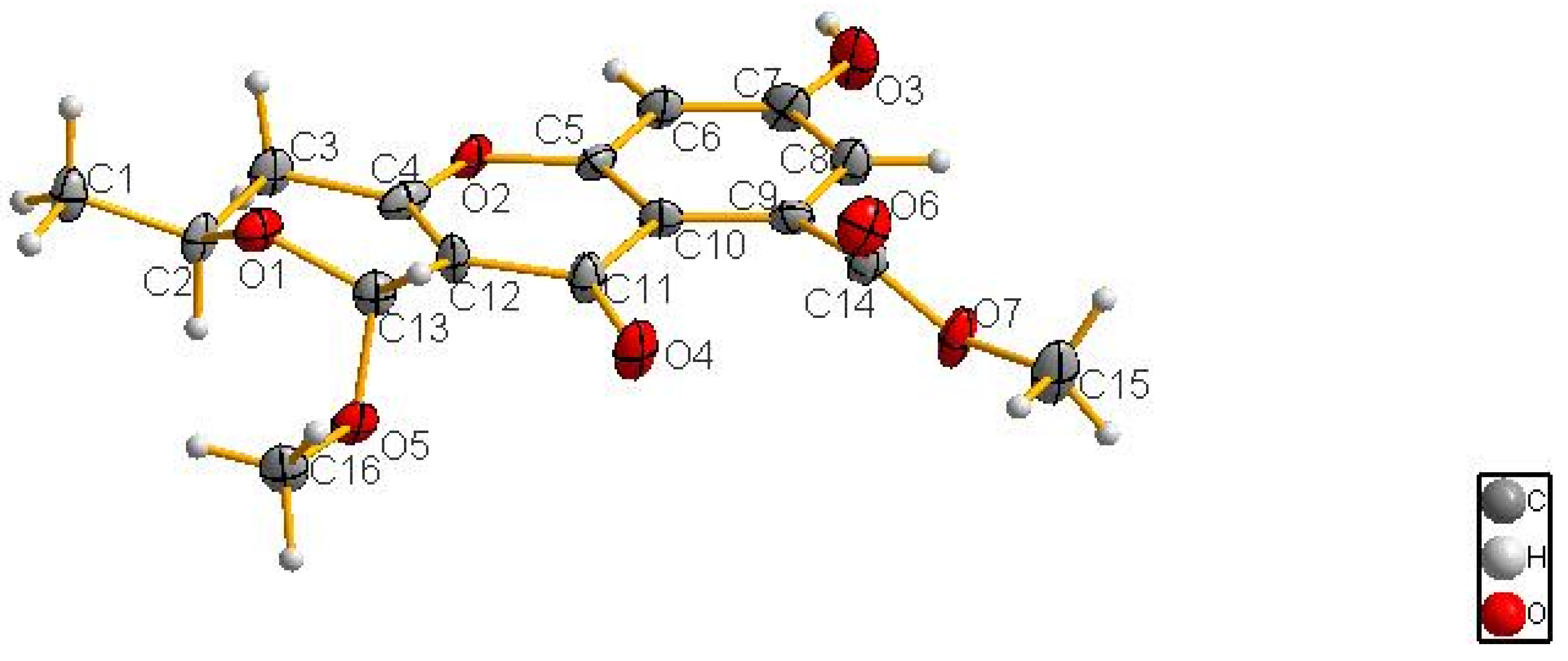
3.6. X-ray Crystallographic Analysis of Compound 1
3.7. AchE Inhibitory Assay
3.8. DPPH Radical Scavenging Assay
3.9. OH-Radical-Scavenging Assay
3.10. α-Glucosidase Inhibitory Assay
3.11. Antibacterial Experiment
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tang, Y.; Ling, J.; Zhang, P.; Zhang, X.; Zhang, N.; Wang, W.; Li, J.; Li, N. Potential therapeutic agents for circulatory diseases from Bauhinia glauca Benth.subsp. pernervosa (Da Ye Guan Men). Bioorg. Med. Chem. Lett. 2015, 25, 3217–3220. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.A.; Salman, M.; Stavinoha, W.B.; Satsangi, N.; Williams, R.F.; Streeper, R.T.; Weintraub, S.T. Antiinflammatory C-glucosyl chromone from Aloe barbadensis. J. Nat. Prod. 1996, 59, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Speranza, G.; Morelli, C.F.; Tubaro, A.; Altinier, G.; Durì, L.; Manitto, P. Aloeresin I. An anti-inflammatory 5-methylchromone from cape aloe. Planta Med. 2005, 71, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Nastasă, C.M.; Duma, M.; Pîrnău, A.; Vlase, L.; Tiperciuc, B.; Oniga, O. Development of new 5-(chromene-3-yl)methylene-2,4-thiazolidinediones as antimicrobial agents. Clujul Med. 2016, 89, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.S.; Xu, S.Q.; Cheng, M.; Chen, Y.; Xia, P.; Qian, K.; Xia, Y.; Yang, Z.Y.; Chen, C.H.; Morris-Natschke, S.L.; et al. Anti-AIDS agents 87. New bio-isosteric dicamphanoyl-dihydropyranochromone (DCP) and dicamphanoyl-khellactone (DCK) analogues with potent anti-HIV activity. Bioorg. Med. Chem. Lett. 2011, 21, 5831–5834. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Shi, Q.; Chen, C.H.; Zhu, H.; Huang, L.; Ho, P.; Lee, K.H. Anti-AIDS agents 79. Design, synthesis, molecular modeling and structure-activity relationships of novel dicamphanoyl-2′,2′-dimethyldihydropyranochromone (DCP) analogs as potent anti-HIV agents. Bioorg. Med. Chem. 2010, 18, 6678–6689. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.D.; Lecerf-Schmidt, F.; Guragossian, N.; Pazinato, J.; Gozzi, G.J.; Winter, E.; Valdameri, G.; Veale, A.; Boumendjel, A.; Di Pietro, A.; et al. New, highly potent and non-toxic, chromone inhibitors of the human breast cancer resistance protein ABCG2. Eur. J. Med. Chem. 2016, 122, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Guru, S.K.; Jain, S.K.; Joshi, P.; Gandhi, S.G.; Bharate, S.B.; Bhushan, S.; Bharate, S.S.; Vishwakarma, R.A. A chromatography-free isolation of rohitukine from leaves of Dysoxylum binectariferum: Evaluation for in vitro cytotoxicity, CDK inhibition and physicochemical properties. Bioorg. Med. Chem. Lett. 2016, 26, 3457–3463. [Google Scholar] [CrossRef] [PubMed]
- Bolós, J.; Anglada, L.; Gubert, S.; Planas, J.M.; Agut, J.; Príncep, M.; De la Fuente, N.; Sacristán, A.; Ortiz, J.A. 7-[3-(1-piperidinyl)propoxy]chromenones as potential atypical antipsychotics. 2. Pharmacological profile of 7-[3-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)-piperidin-1-yl]propoxy]-3-(hydroxymeth yl)chromen-4-one (abaperidone. FI-8602). J. Med. Chem. 1998, 41, 5402–5409. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Yang, J.T.; Lu, C.C.; Chang, S.F.; Chen, C.N.; Su, Y.P.; Lee, K.C. Fulvic acid attenuates resistin-induced adhesion of HCT-116 colorectal cancer cells to endothelial cells. Int. J. Mol. Sci. 2015, 16, 29370–29382. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Singh, A.; Mishra, A. The effect of fulvic acid on pre- and postaggregation state of Aβ(17-42): Molecular dynamics simulation studies. Biochim. Biophys. Acta 2013, 1834, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Hueso-Rodríguez, J.A.; Boyd, H.; Concha, N.O.; Janson, C.A.; Gilpin, M.; Bateson, J.H.; Cheever, C.; Niconovich, N.L.; Pearson, S.; et al. Identification of a series of tricyclic natural products as potent broad-spectrum inhibitors of metallo-beta-lactamases. Antimicrob. Agents Chemother. 2002, 46, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Zhang, Y.; Li, L.; Wang, X.; Ding, G. Chaetochromones A and B, two new polyketides from the fungus Chaetomium indicum (CBS.860.68). Molecules 2013, 18, 10944–10952. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wang, H.Y.; Wu, C.S.; Jiao, Y.; Li, M.; Wang, Y.Y.; Wang, S.Q.; Zhao, Z.T.; Lou, H.X. Austdiol. Fulvic acid and citromycetin derivatives from an endolichenic fungus, Myxotrichum sp. Phytochem. Lett. 2013, 6, 662–666. [Google Scholar]
- Lösgen, S.; Schlörke, O.; Meindl, K.; Herbst-Irmer, R.; Zeeck, A. Structure and biosynthesis of chaetocyclinones, new polyketides produced by an endosymbiotic Fungus. Eur. J. Org. Chem. 2007, 2191–2196. [Google Scholar] [CrossRef]
- Elsässer, B.; Krohn, K.; Flörke, U.; Root, N.; Aust, H.-J.; Draeger, S.; Schulz, B.; Antus, S.; Kurtán, T. X-ray structure determination, absolute configuration and biological activity of phomoxanthone A. Eur. J. Org. Chem. 2005, 21, 4563–4570. [Google Scholar] [CrossRef]
- Rönsberg, D.; Debbab, A.; Mándi, A.; Vasylyeva, V.; Böhler, P.; Stork, B.; Engelke, L.; Hamacher, A.; Sawadogo, R.; Diederich, M.; et al. Pro-apoptotic and immunostimulatory tetrahydroxanthone dimmers from the endophytic fungus Phomopsislongicolla. J. Org. Chem. 2013, 78, 12409–12425. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Qiu, Z.; You, J.; Tan, H.; Zhou, S. Isolation and characterization of endophytic streptomycete antagonists of fusarium wilt pathogen from surface-sterilized banana roots. FEMS Microbiol. Lett. 2005, 247, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision a.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Van Zandt, M.C.; Jones, M.L.; Gunn, D.E.; Geraci, L.S.; Jones, J.H.; Sawicki, D.R.; Sredy, J.; Jacot, J.L.; Dicioccio, A.T.; Petrova, T.; et al. Discovery of 3-[(4,5,7-trifluorobenzothiazol-2-yl)methyl]indole-N-acetic acid (lidorestat) and congeners as highly potent and selective inhibitors of aldose reductase for treatment of chronic diabetic complications. J. Med. Chem. 2005, 48, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, J.V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–92. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Wu, W.H.; Wang, J.; Lan, M.B. Antioxidant properties of polysaccharide from the brown seaweed Sargassum graminifolium (Turn.), and its effects on calcium oxalate crystallization. Mar. Drugs 2012, 10, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, L.; Fang, Z.; Hu, Y.; Chen, S.; Sugawara, T.; Ye, X. The Effect of the Molecular Architecture on the Antioxidant Properties of Chitosan Gallate. Mar. Drugs 2016, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Kelman, D.; Posner, E.K.; McDermid, K.J.; Tabandera, N.K.; Wright, P.R.; Wright, A.D. Antioxidant activity of Hawaiian marine algae. Mar. Drugs 2012, 10, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Harasit, K.M.; Tapas, M.; Ambikesh, M. Kinetics of the basic hydrolysis of tris (1,10-phenenthroline) Fe (II): Influence of polymer-surfactant interactions. Colloid Surf. A-Physicochem. Eng. Asp. 2011, 380, 300–307. [Google Scholar]
- Moradi-Afrapoli, F.; Asghari, B.; Saeidnia, S.; Ajani, Y.; Mirjani, M.; Malmir, M.; Dolatabadi Bazaz, R.; Hadjiakhoondi, A.; Salehi, P.; Hamburger, M.; et al. In vitro α-glucosidase inhibitory activity of phenolic constituents from aerial parts of Polygonum hyrcanicum. Daru 2012, 20, 37. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.G.; Uppuluri, P.; Tristan, A.R.; Wormley, F.L., Jr.; Mowat, E.; Ramage, G.; Lopez-Ribot, J.L. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat. Protoc. 2008, 3, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Debbab, A.; Aly, A.H.; Proksch, P. Endophytes and associated marine derived fungi-ecological and chemical perspectives. Fungal Divers. 2012, 57, 45–83. [Google Scholar] [CrossRef]
- Imhoff, J.F. Natural Products from Marine Fungi-Still an Underrepresented Resource. Mar. Drugs 2016, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Cai, X.; Xu, F.; She, Z.; Chan, W.L.; Vrijmoed, L.L.; Jones, E.B.; Lin, Y. Three metabolites from the mangrove endophytic fungus Sporothrix sp. (#4335) from the South China Sea. J. Org. Chem. 2009, 74, 1093–1098. [Google Scholar] [PubMed]
- Ma, Y.H.; Li, J.; Huang, M.X.; Liu, L.; Wang, J.; Lin, Y.C. Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#. Mar. Drugs 2015, 13, 6306–6318. [Google Scholar] [PubMed]
- Li, J.; Xue, Y.Y.; Yuan, J.; Lu, Y.J.; Zhu, X.; Lin, Y.C.; Liu, L. Lasiodiplodins from Mangrove Endophytic Fungus Lasiodiplodia sp. 318#. Nat. Prod. Res. 2015, 30, 1–6. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 (in C3D6O) | 2 (in CDCl3) | 3 (in CDCl3) | 4 (in CD3OD) | |||||
|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 21.0 q | 1.34 d 6.0 | 21.0 q | 1.39 d 6.4 | 21.3 q | 1.38 d 6.0 | 23.8 q | 1.31 d 6.0 |
| 2 | 62.8 d | 4.34 m | 62.1 d | 4.41 m | 70.0 d | 3.83 m | 66.7 t | 4.23 m |
| 3 | 34.6 t | 2.67 dd 18.0, 4.0 | 34.4 t | 2.63 m | 34.6 t | 2.64 m | 42.1 t | 2.94 m |
| 2.58 dd 18.0,10.8 | ||||||||
| 4 | 164.1 s | 163.6 s | 160.7 s | 167.7 s | ||||
| 5 | 158.4 s | 144.4 s | 144.8 s | 159.3 s | ||||
| 6 | 104.3 d | 6.93 d 2.4 | 134.7 s | 134.9 s | 104.4 d | 6.87 d 2.4 | ||
| 7 | 162.6 s | 149.2 s | 149.0 s | 164.2 s | ||||
| 8 | 113.8 d | 6.85 d 2.4 | 108.0 d | 6.89 s | 107.9 d | 6.93 s | 114.8 d | 6.77 d 2.4 |
| 9 | 114.6 s | 124.5 s | 124.0 s | 113.7 s | ||||
| 10 | 136.4 s | 116.0 s | 115.6 s | 136.2 s | ||||
| 11 | 173.2 s | 173.5 s | 174.1 s | 177.2 s | ||||
| 12 | 117.4 s | 116.6 s | 116.5 s | 122.1 s | ||||
| 13 | 95.2 d | 5.40 s | 94.5 d | 5.57 s | 62.5 t | 4.82 d 15.2 | 55.0 t | 4.55 s |
| 4.48 d 15.2 | ||||||||
| 14 | 169.5 s | 169.8 s | 170.0 s | 171.6 s | ||||
| 15 | 52.8 q | 3.85 s | 53.2 q | 3.95 s | 53.2 q | 3.96 s | 53.3 q | 3.91 s |
| 16 | 55.6 q | 3.42 s | 55.9 q | 3.55 s | ||||
| 17 | 56.8 q | 3.98 s | 56.9 q | 3.99 s | ||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Li, J.; Liu, L.; Yin, S.; Wang, J.; Lin, Y. Phomopsichin A–D; Four New Chromone Derivatives from Mangrove Endophytic Fungus Phomopsis sp. 33#. Mar. Drugs 2016, 14, 215. https://doi.org/10.3390/md14110215
Huang M, Li J, Liu L, Yin S, Wang J, Lin Y. Phomopsichin A–D; Four New Chromone Derivatives from Mangrove Endophytic Fungus Phomopsis sp. 33#. Marine Drugs. 2016; 14(11):215. https://doi.org/10.3390/md14110215
Chicago/Turabian StyleHuang, Meixiang, Jing Li, Lan Liu, Sheng Yin, Jun Wang, and Yongcheng Lin. 2016. "Phomopsichin A–D; Four New Chromone Derivatives from Mangrove Endophytic Fungus Phomopsis sp. 33#" Marine Drugs 14, no. 11: 215. https://doi.org/10.3390/md14110215
APA StyleHuang, M., Li, J., Liu, L., Yin, S., Wang, J., & Lin, Y. (2016). Phomopsichin A–D; Four New Chromone Derivatives from Mangrove Endophytic Fungus Phomopsis sp. 33#. Marine Drugs, 14(11), 215. https://doi.org/10.3390/md14110215