
Novel Conopeptides of Largely Unexplored Indo Pacific Conus sp.

 , ,
, ,
Abstract
:1. Introduction
2. Results
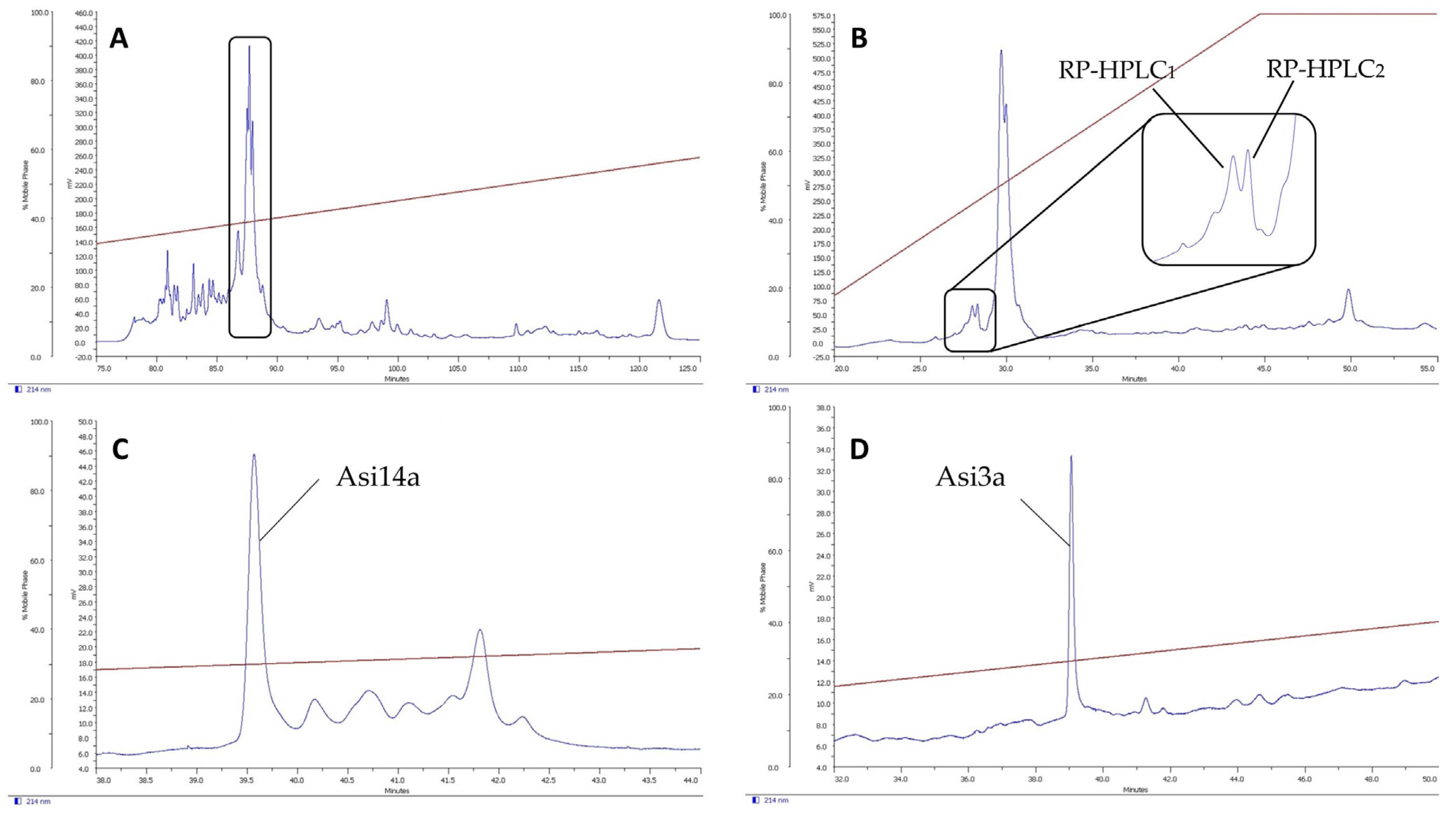
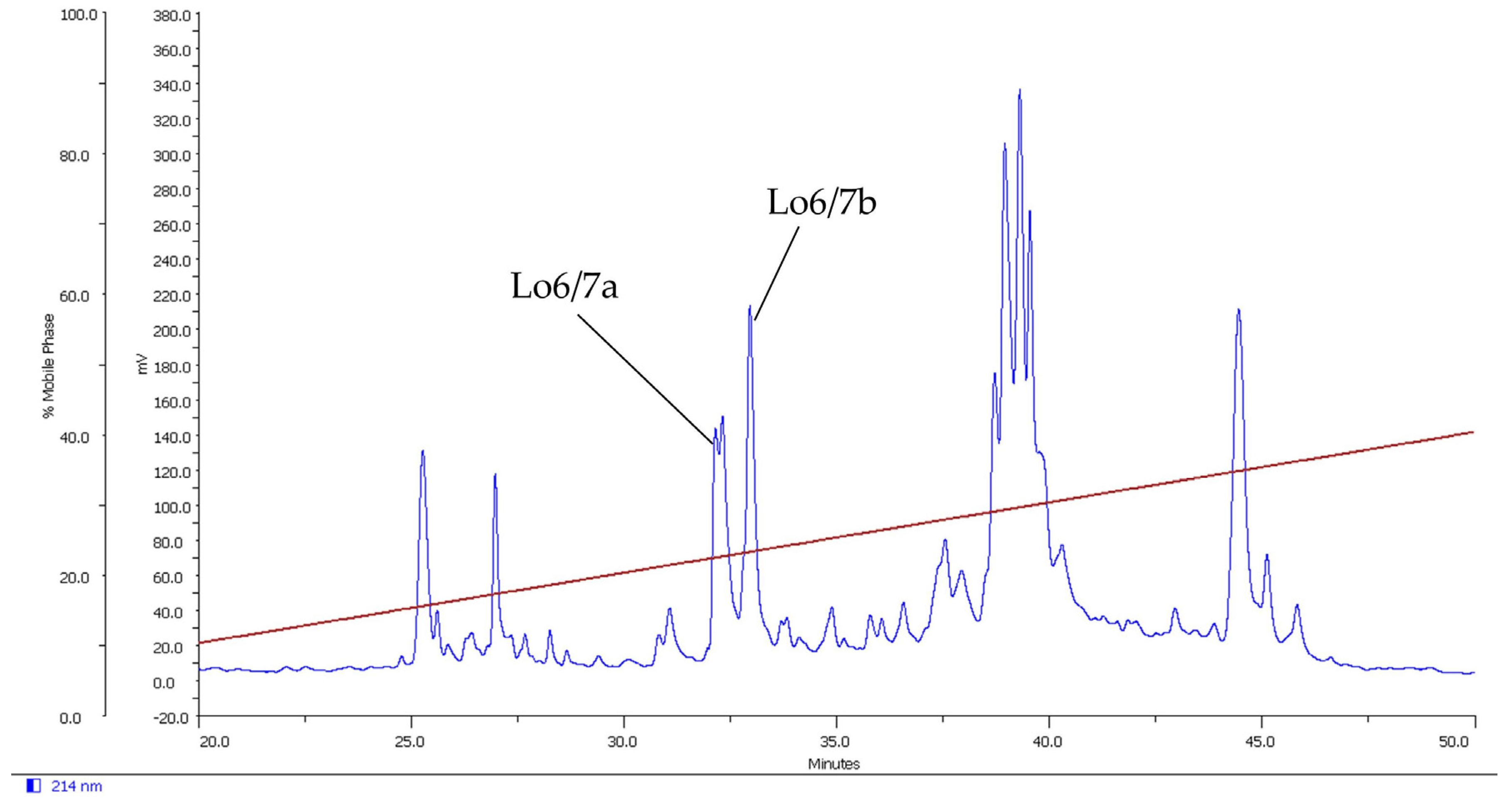
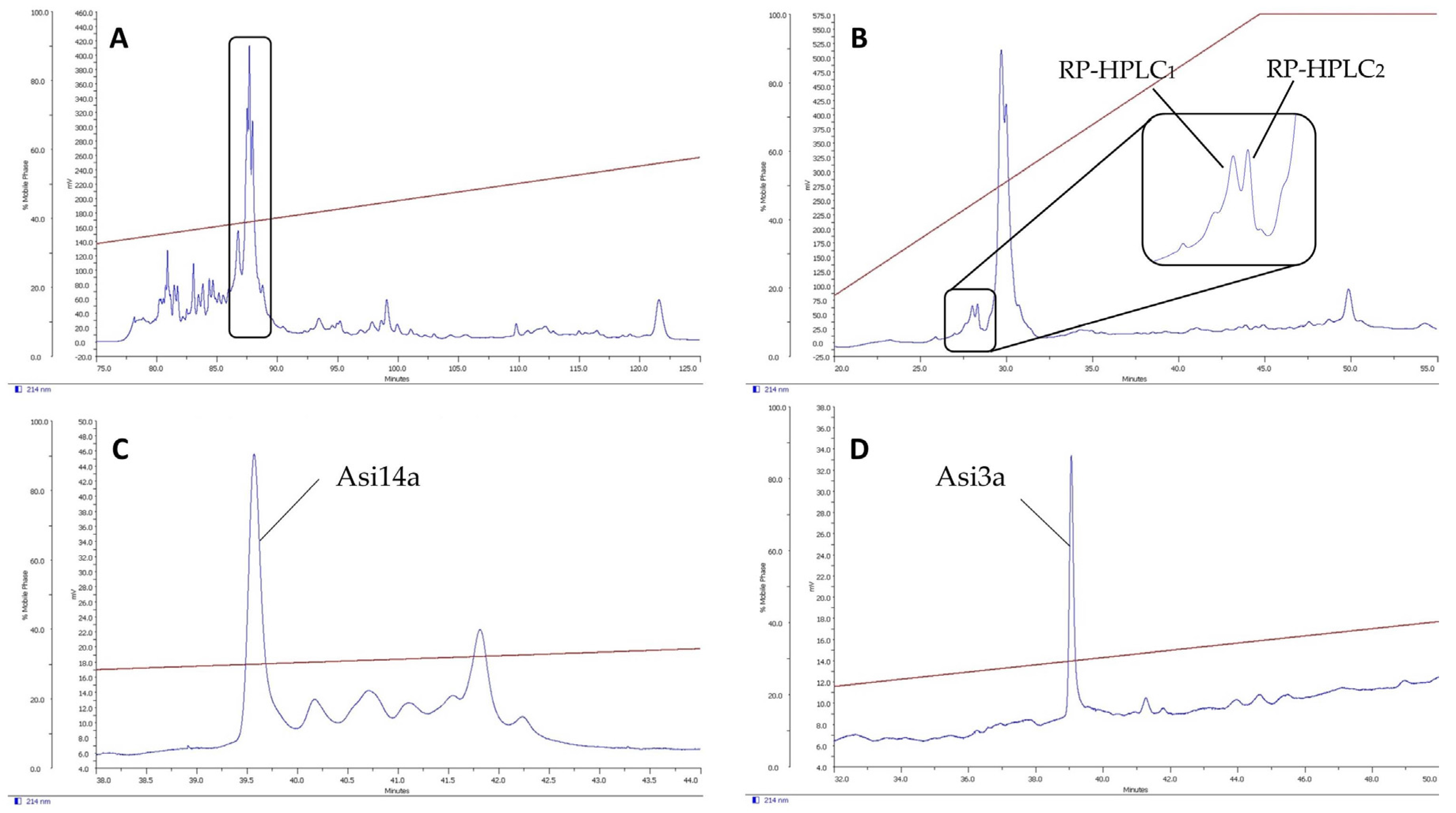
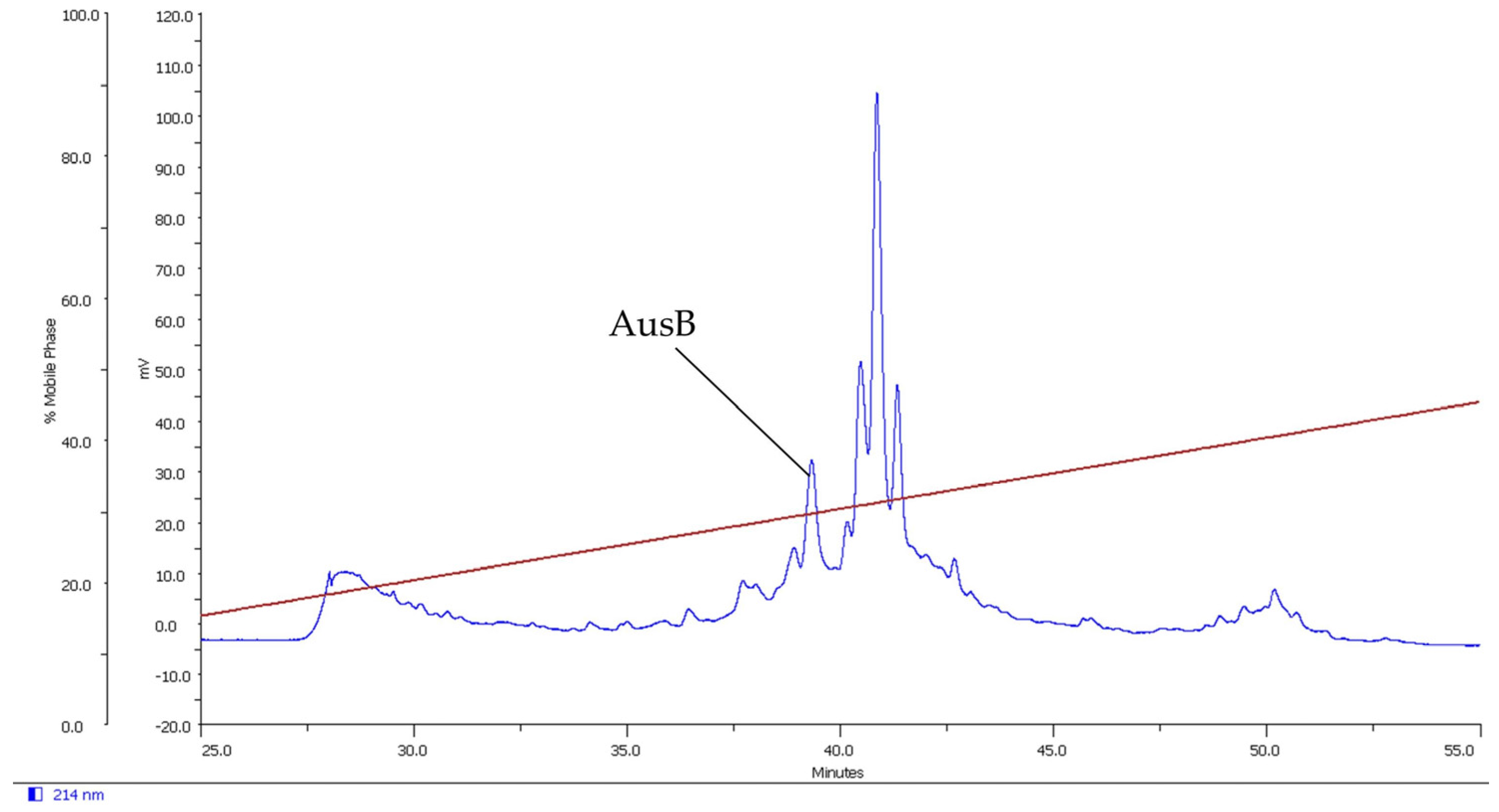
2.1. Isolation of Novel Conotoxins from C. longurionis, C. asiaticus and C. australis
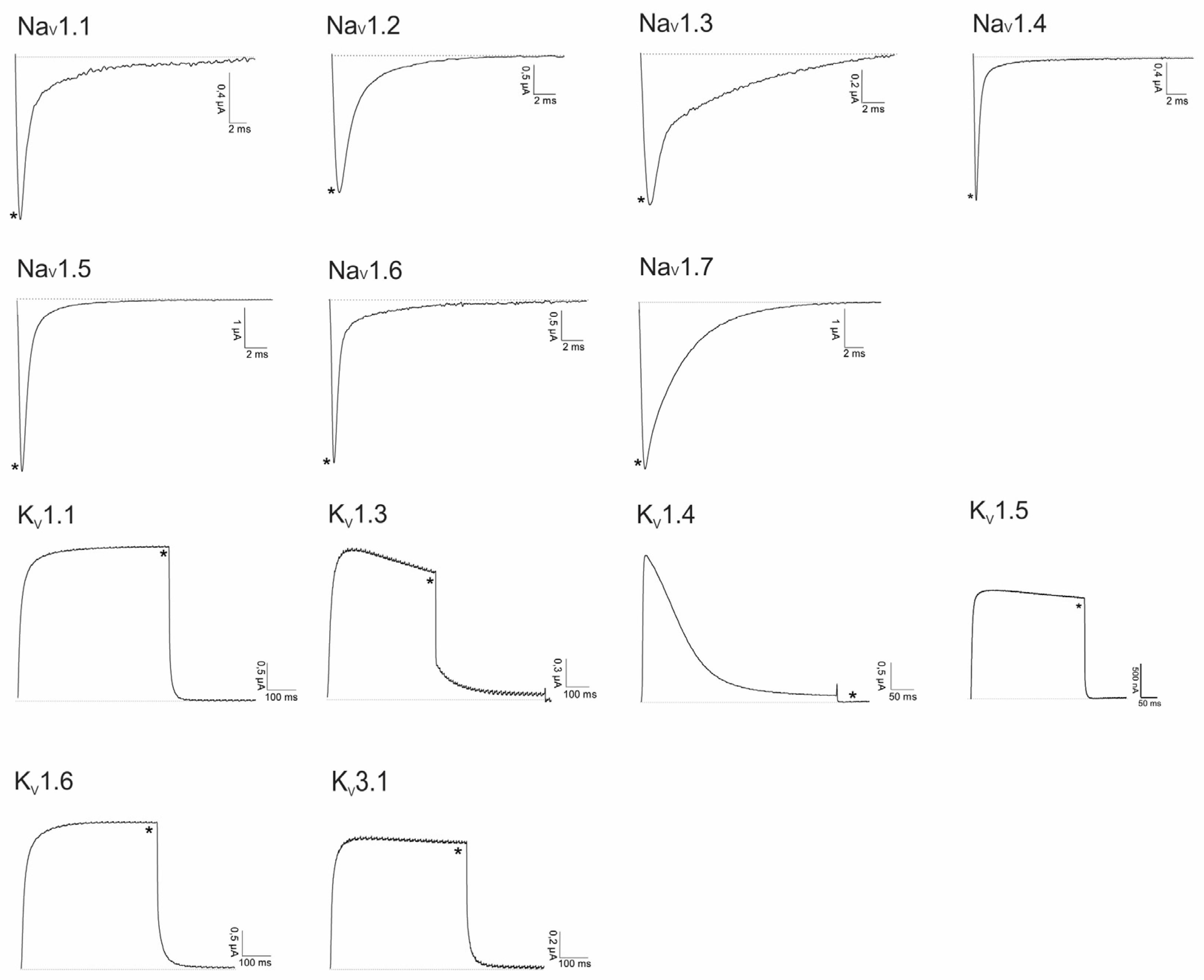
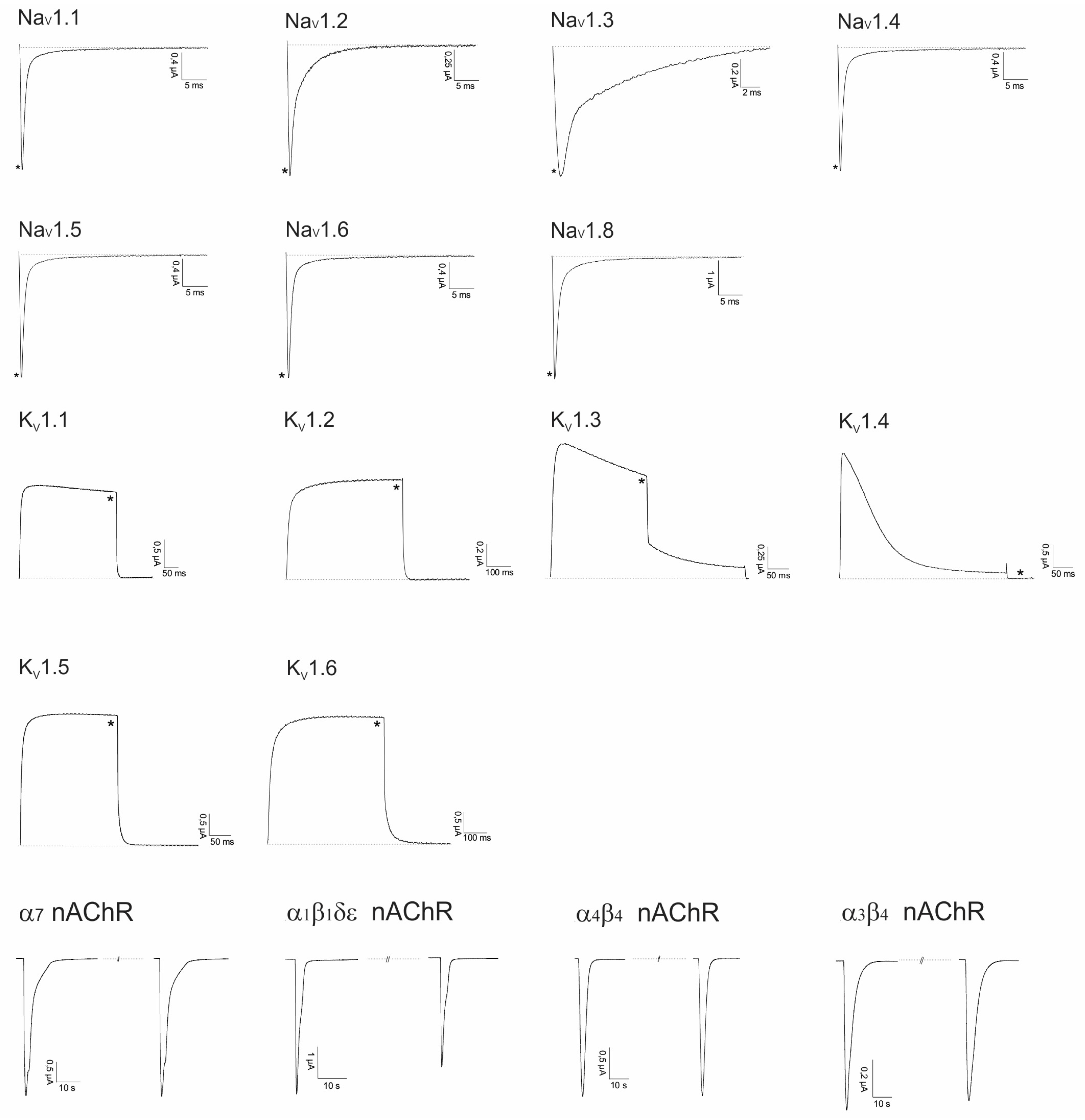
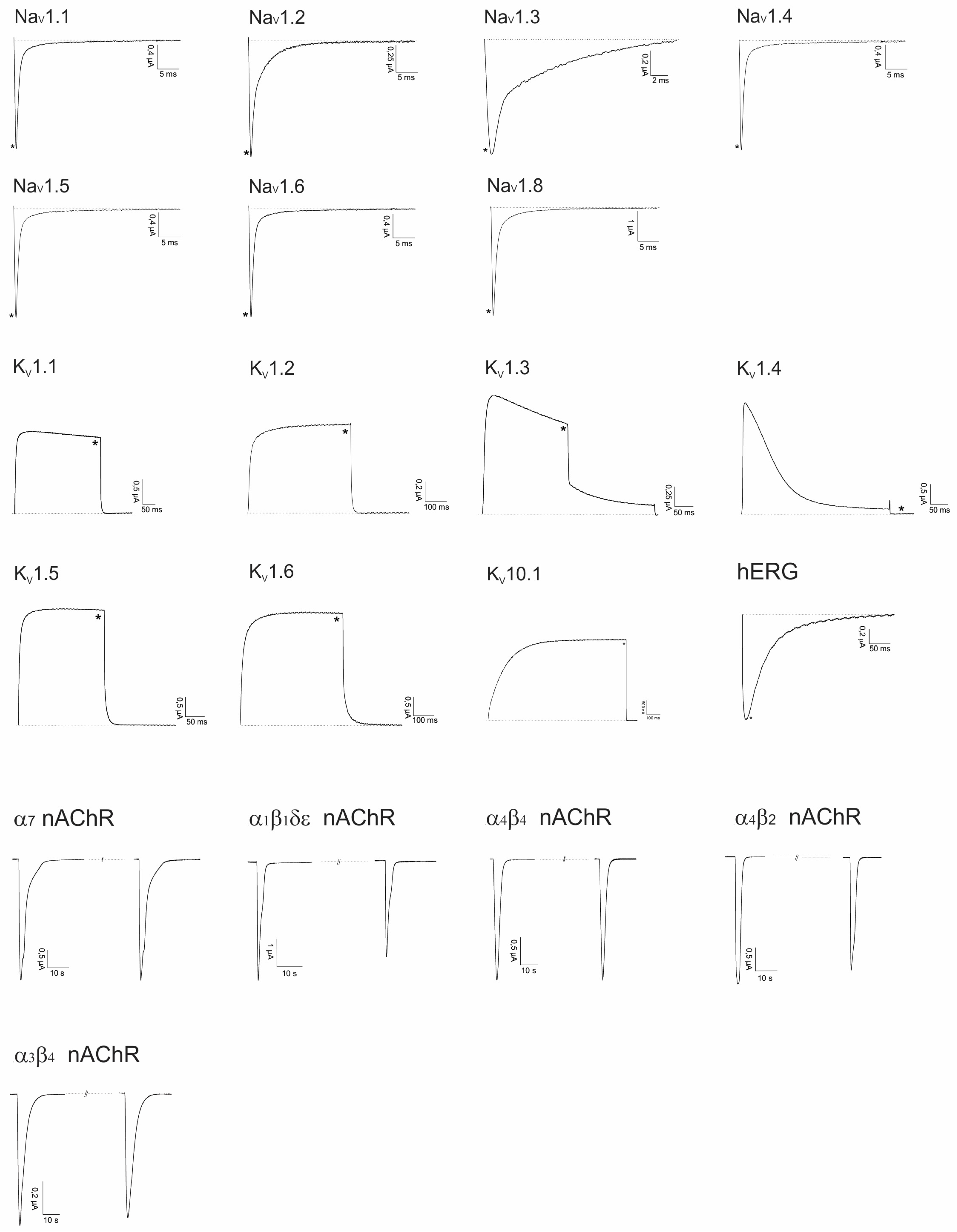
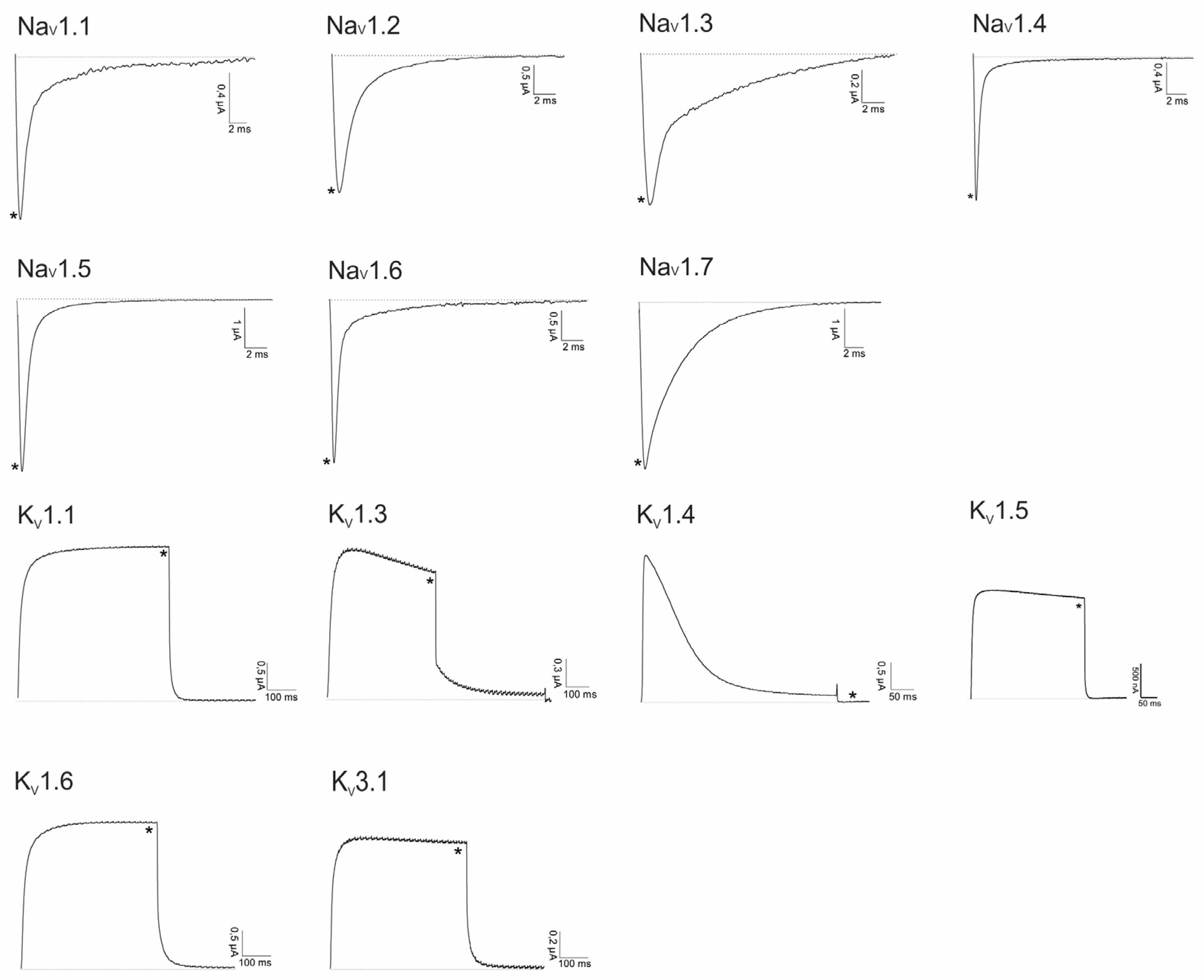
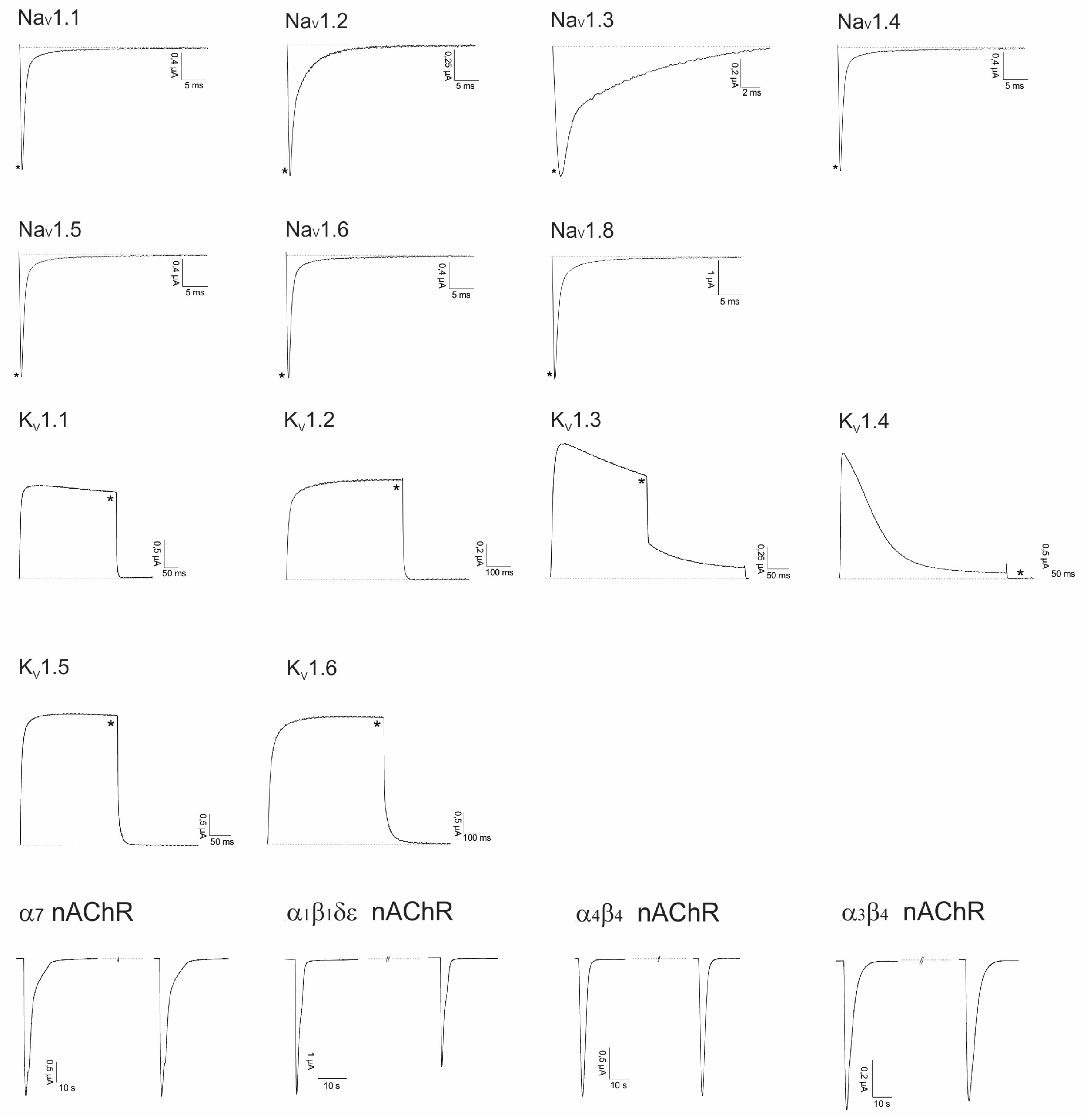
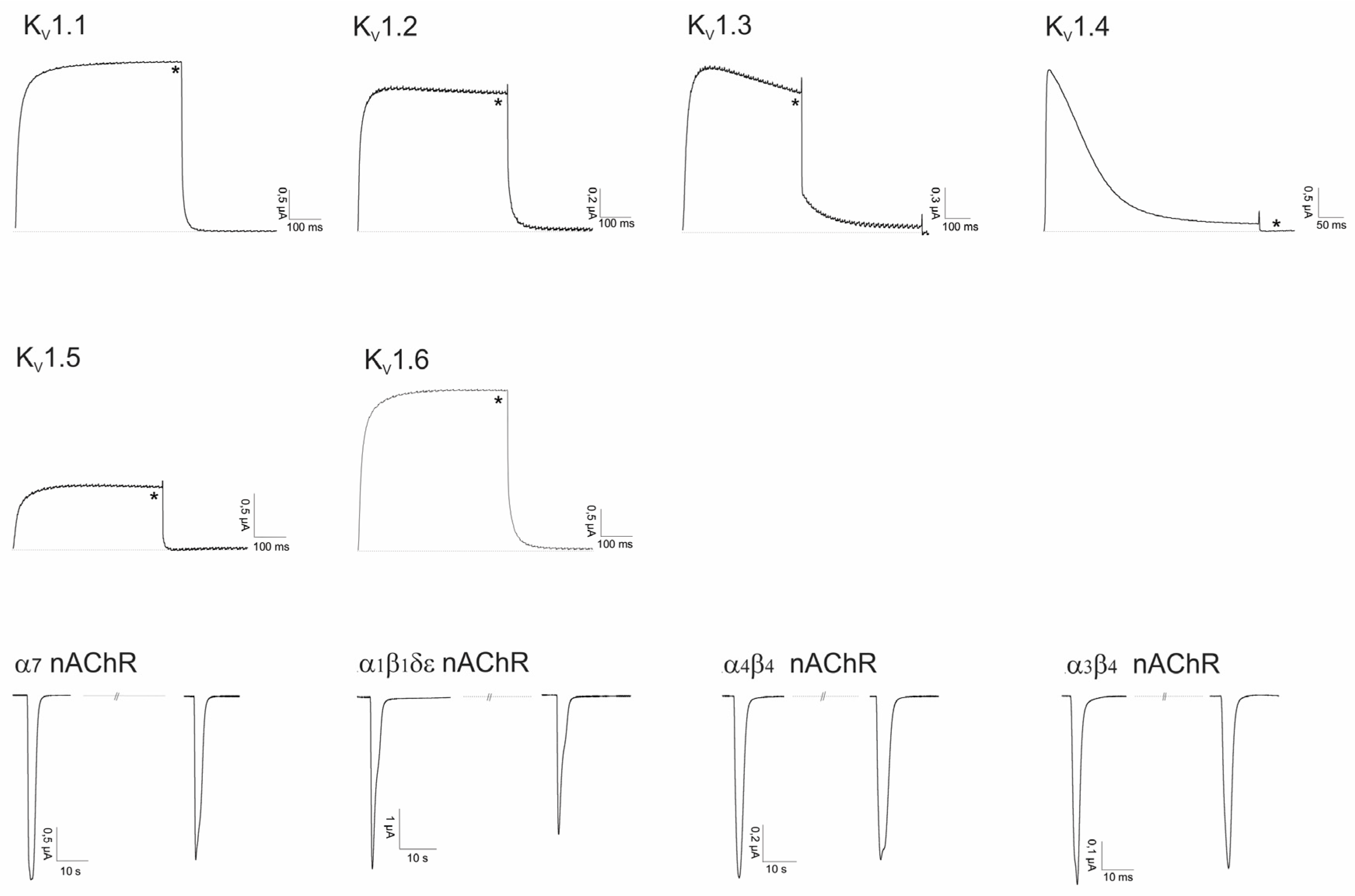
2.2. Electrophysiological Screening against Voltage-Gated and Ligand-Gated Ion Channels
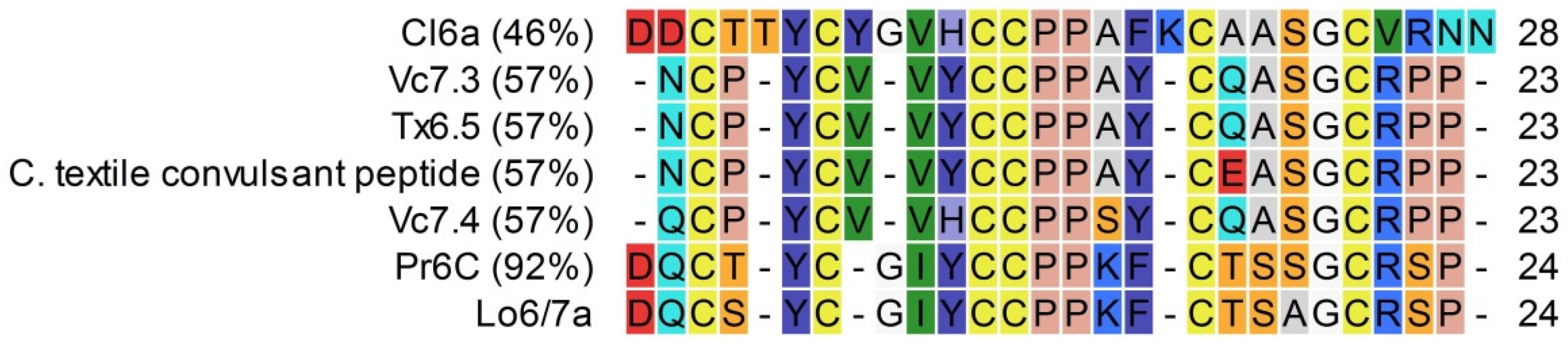
2.2.1. Lo6/7a and Lo6/7b
2.2.2. Asi3a
2.2.3. Asi14a
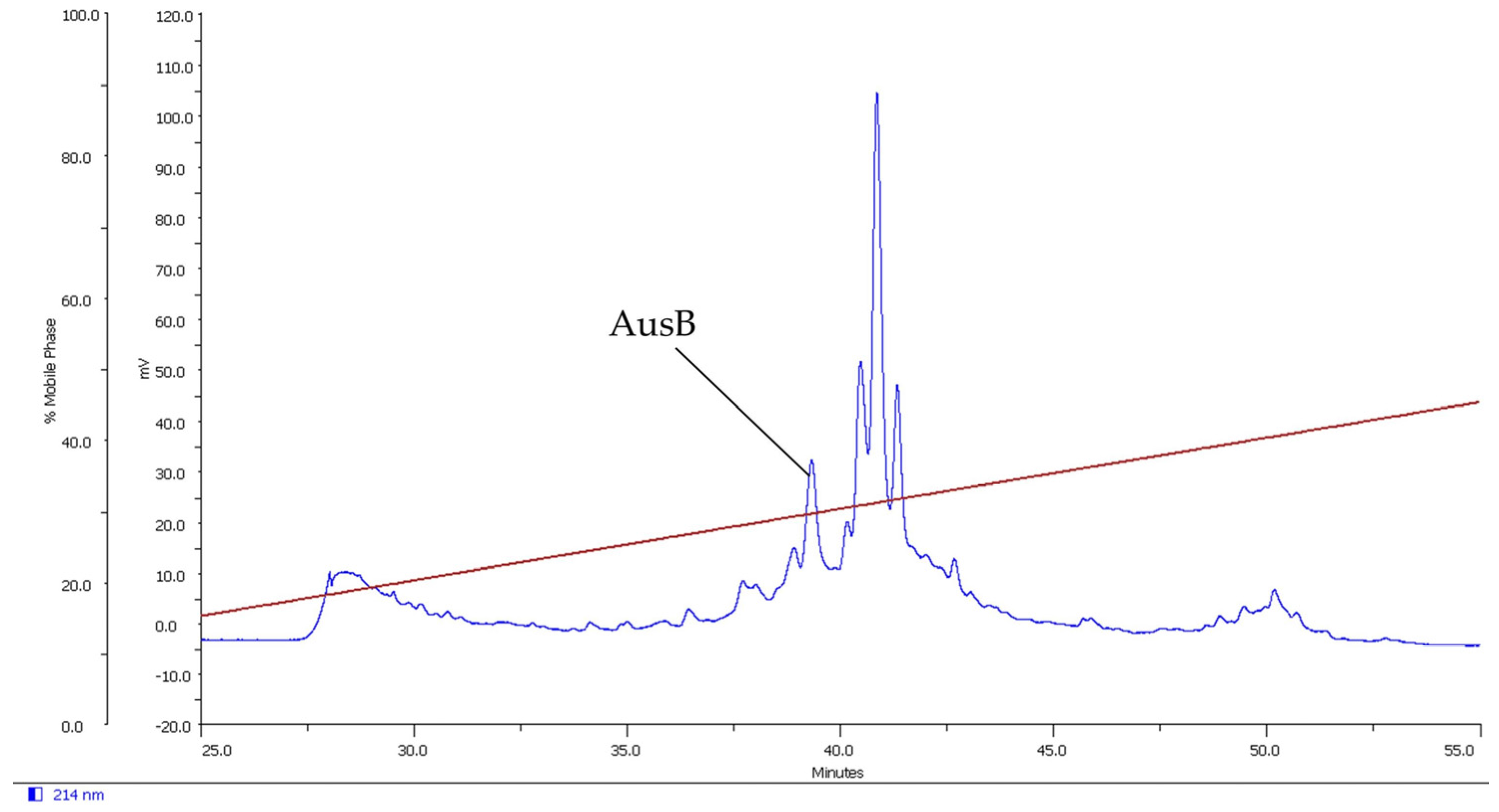
2.2.4. AusB
2.3. Antibacterial Activity
3. Discussion
3.1. Conopeptide Alignment and Classification
3.2. Antagonistic Assays in the Quest of Identifying Novel Pharmacological Targets
4. Materials and Methods
4.1. Cone Snail Specimens and Venom Extraction
4.2. Peptide Fractionation and Purification
4.3. Peptide Sequencing
4.4. Peptide Synthesis and Folding
4.5. Preparation of RNA for Functional Testing in Xenopus Oocytes
4.6. Electrophysiological Recordings
4.7. Growth Media and Micro-Organisms
4.8. Antibacterial Assays
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Olivera, B.M. EE Just lecture, 1996—Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Lewis, R.J. Therapeutic potential of cone snail venom peptides (conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Linley, J.E.; Baker, M.D.; Minett, M.S.; Cregg, R.; Werdehausen, R.; Rugiero, F.; Wood, J.N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135, 2585–2612. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Sudarslal, S.; Singaravadivelan, G.; Ramasamy, P.; Ananda, K.; Sarma, S.P.; Sikdar, S.K.; Krishnan, K.S.; Balaram, P. A novel 13 residue acyclic peptide from the marine snail, Conus monile, targets potassium channels. Biochem. Biophys. Res. Commun. 2004, 317, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, S.; Andreotti, N.; Jouirou, B.; Sabatier, J.M. Animal toxins acting on voltage-gated potassium channels. Curr. Pharm. Des. 2008, 14, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Orts, D.J.; Peigneur, S.; Madio, B.; Cassoli, J.S.; Montandon, G.G.; Pimenta, A.M.; Bicudo, J.E.; Freitas, J.C.; Zaharenko, A.J.; Tytgat, J. Biochemical and electrophysiological characterization of two sea anemone type 1 potassium toxins from a geographically distant population of Bunodosoma caissarum. Mar. Drugs 2013, 11, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Peigneur, S.; Wijesekara, I.; Tytgat, J. Conotoxins targeting nicotinic acetylcholine receptors: An overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Peigneur, S.; Maiti, M.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Ulens, C.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Structure-function elucidation of a new α-conotoxin, Lo1a, from Conus longurionis. J. Biol. Chem. 2014, 289, 9573–9583. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Peigneur, S.; Maiti, M.; Mille, B.G.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Discovery of a new subclass of α-conotoxins in the venom of Conus australis. Toxicon 2014, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Ramilo, C.A.; Corpuz, G.P.; Olivera, B.M. Conus peptides: Phylogenetic range of biological activity. Biol. Bull. 1992, 183, 159–164. [Google Scholar] [CrossRef]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Olivera, B.M. Divergent M- and O-superfamily peptides from venom of fish-hunting Conus parius. Peptides 2010, 31, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Collodoro, M.; Gilles, N.; Turtoi, A.; De Pauw, E.; Quinton, L. Secretion and maturation of conotoxins in the venom ducts of Conus textile. Toxicon 2012, 60, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Kil, Y.J.; Ueberheide, B.; Chait, B.T.; Tayo, L.; Cruz, L.; Lu, B.; Yates, J.R.; Bern, M. Constrained de novo sequencing of conotoxins. J. Proteome Res. 2012, 11, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Biggs, J.S.; Watkins, M.; Puillandre, N.; Ownby, J.P.; Lopez-Vera, E.; Christensen, S.; Moreno, K.J.; Bernaldez, J.; Licea-Navarro, A.; Corneli, P.S.; et al. Evolution of Conus peptide toxins: Analysis of Conus californicus Reeve, 1844. Mol. Phylogenet. Evol. 2010, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pi, C.; Liu, J.; Chen, S.; Peng, C.; Sun, D.; Zhou, M.; Xiang, H.; Ren, Z.; Xu, A. Identification and characterization of a novel O-superfamily conotoxin from Conus litteratus. J. Pept. Sci. 2008, 14, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Remigio, E.A. Variation and evolution of toxin gene expression patterns of six closely related venomous marine snails. Mol. Ecol. 2008, 17, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Melaun, C.; Mebs, D. Direct cDNA cloning of novel conopeptide precursors of the O-superfamily. Peptides 2005, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genom. 2011, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Lluisma, A.O.; Milash, B.A.; Moore, B.; Olivera, B.M.; Bandyopadhyay, P.K. Novel venom peptides from the cone snail Conus pulicarius discovered through next-generation sequencing of its venom duct transcriptome. Mar. Genom. 2012, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Gilad, Y.; Avidan, N.; Ben-Asher, E.; Levy, Z.; Fainzilber, M. Mechanisms for evolving hypervariability: The case of conopeptides. Mol. Biol. Evol. 2001, 18, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.C.; Zhou, M.; Peng, C.; Shao, X.X.; Guo, Z.Y.; Chi, C.W. Novel conopeptides in a form of disulfide-crosslinked dimer. Peptides 2010, 31, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Fan, C.X.; Hu, K.P.; Wei, K.H.; Zhong, M.N. Studies on conotoxins of Conus betulinus. J. Nat. Toxins 1999, 8, 341–349. [Google Scholar] [PubMed]
- Biggs, J.S.; Rosenfeld, Y.; Shai, Y.; Olivera, B.M. Conolysin-Mt: A conus peptide that disrupts cellular membranes. Biochemistry 2007, 46, 12586–12593. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, Y.; Li, L.; Weng, L.; Wang, Q.; Zhang, S.; Jia, B.; Hu, H.; He, Y.; Jacob, Y.; et al. Inhibition of influenza virus replication by constrained peptides targeting nucleoprotein. Antivir. Chem. Chemother. 2011, 22, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Hamada, T.; Hirota, H.; Nakao, Y.; Matsunaga, S.; van Soest, R.W.; Fusetani, N. Asteropine A, a sialidase-inhibiting conotoxin-like peptide from the marine sponge Asteropus simplex. Chem. Biol. 2006, 13, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Aponte, C.A.; Silva-Sanchez, J.; Quintero-Hernandez, V.; Rodriguez-Romero, A.; Balderas, C.; Possani, L.D.; Gurrola, G.B. Vejovine, a new antibiotic from the scorpion venom of Vaejovis mexicanus. Toxicon 2011, 57, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, Z.; Zhang, R.; Wu, Y.; Li, W.; Cao, Z. StCT2, a new antibacterial peptide characterized from the venom of the scorpion Scorpiops tibetanus. Peptides 2012, 36, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, U.; Mujaddad-Ur-Rehman, M.; Khalid, N.; Fawad, S.A.; Fatima, A. Antibacterial activity of the venom of Heterometrus xanthopus. Indian J. Pharmacol. 2012, 44, 509–511. [Google Scholar] [PubMed]
- Conde, R.; Zamudio, F.Z.; Rodriguez, M.H.; Possani, L.D. Scorpine, an anti-malaria and anti-bacterial agent purified from scorpion venom. FEBS Lett. 2000, 471, 165–168. [Google Scholar] [CrossRef]
- Corzo, G.; Escoubas, P.; Villegas, E.; Barnham, K.J.; He, W.; Norton, R.S.; Nakajima, T. Characterization of unique amphipathic antimicrobial peptides from venom of the scorpion Pandinus imperator. Biochem. J. 2001, 359, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Corzo, G.; Naoki, H.; Andriantsiferana, M.; Nakajima, T. Purification, structure-function analysis, and molecular characterization of novel linear peptides from scorpion Opisthacanthus madagascariensis. Biochem. Biophys. Res. Commun. 2002, 293, 1514–1522. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, Y.; Dai, C.; Zhao, R.; Li, S.; Wu, Y.; Cao, Z.; Li, W. Imcroporin, a new cationic antimicrobial peptide from the venom of the scorpion Isometrus maculates. Antimicrob. Agents. Chemother. 2009, 53, 3472–3477. [Google Scholar] [CrossRef] [PubMed]
- Leandro, L.F.; Mendes, C.A.; Casemiro, L.A.; Vinholis, A.H.; Cunha, W.R.; de Almeida, R.; Martins, C.H. Antimicrobial activity of apitoxin, melittin and phospholipase A2 of honey bee (Apis mellifera) venom against oral pathogens. An. Acad. Bras. Cienc. 2015, 87, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Samy, R.P.; Sethi, G.; Lim, L.H. A brief update on potential molecular mechanisms underlying antimicrobial and wound-healing potency of snake venom molecules. Biochem. Pharmacol. 2016, 115, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.M.; Woodhams, D.; Howard, C.; LaFonte, B.E.; Gregory, J.R.; Johnson, P.T. Role of antimicrobial peptides in amphibian defense against trematode infection. EcoHealth 2016, 13, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.L.; Abdel-Rahman, M.A.; Miller, K.; Strong, P.N. Antimicrobial peptides from scorpion venoms. Toxicon 2014, 88, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.K.; Tenorio, M.J. Systematic Classification of Recent and Fossil Conoidean Gastropods: With Keys to the Genera of Cone Shells; ConchBooks: Harxheim, Germany, 2009. [Google Scholar]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 25 October 2016).
- Green, B.R.; Bulaj, G. Oxidative folding of conotoxins in immobilized systems. Protein Pept. Lett. 2006, 13, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Amino Acid Sequence | Cysteine Arrangement | Cysteine Framework | Source |
|---|---|---|---|---|
| Lo6/7a | DQCSYCGIYCCPPKFCTSAGCRSP * | C–C–CC–C–C | VI/VII | C. longurionis |
| Lo6/7b | SCLSSGALCGIDSNCCNGCNVPRNQCY * | C–C–CC–C–C | VI/VII | C. longurionis |
| Asi3a | CCQWPCSHGCIPCCY * | CC–C–C–CC | III | C. asiaticus |
| Asi14a | SCGYPCSHCGIPGCYPG * | C–C–C–C | XIV | C. asiaticus |
| AusB | GAYFDGFDVPCVPRRDDC | C–C | N.A. | C. australis |
| Gram-Negative Bacteria | Gram-Positive Bacteria | ||
|---|---|---|---|
| 1 | Aeromonas hydrophila ATCC7966 | 1 | Bacillus megaterium ATCC13632 |
| 2 | Agrobacterium tumefaciens A208 | 2 | Bacillus subtilis LMG 7135 |
| 3 | Azospirillum brasilense Sp7 | 3 | Brevibacterium linens ATCC9172 |
| 4 | Bordetella avium 197N | 4 | Corynebacterium glutamicum DSM20300 |
| 5 | Brevundimonas diminuta LMG 2088 | 5 | Lactobacillus plantarum LMG-P21295 |
| 6 | Burkholderia cepacia LMG 1222 | 6 | Lactobacillus rhamnosus GG LMG 6400 |
| 7 | Burkholderia gladioli LMG 2216 | 7 | Mycobacterium smegmatis DSM43756 |
| 8 | Burkholderia vietnamensis LMG 10927 | 8 | Rhodococcus erythropolis N11 |
| 9 | Chromobacterium violaceum CV026 | 9 | Staphylococcus aureus ATCC6358 |
| 10 | Citrobacter freundii ATCC8090 | 10 | Streptomyces lividans TK24 |
| 11 | Enterobacter aerogenes ATCC13048 | ||
| 12 | Erwinia amylovora CFBP1430 | Yeast | |
| 13 | Erwinia carotovora LMG 2458 | 1 | Candida albicans CAI4 |
| 14 | Proteus vulgaris LMM2011 | 2 | Saccharomyces cerevisiae W303-1A |
| 15 | Pseudomonas aeruginosa PA14 | ||
| 16 | Pseudomonas entomophila L48 | ||
| 17 | Pseudomonas fluorescens Pf0-1 | ||
| 18 | Pseudomonas putida KT2440 | ||
| 19 | P. syringae pv. tabaci LMG 5192 | ||
| 20 | Rhizobium etli CNPAF512 | ||
| 21 | Salmonella enteritidis ATCC13076 | ||
| 22 | Serratia entomophila DSM12358 | ||
| 23 | Shigella flexneri LMG 10472 | ||
| 24 | Sphingomonas wittichii RW1 | ||
| 25 | Variovorax paradoxus LMG 1797 | ||
| 26 | Vibrio harveyi BB120 | ||
| 27 | Xanthomonas axonopodis pv. manihotis LMG 784 | ||
| 28 | Xanthomonas alfalfae pv. alfalfae LMG 497 | ||
| 29 | Yersinia enterocolitica LMG 7899 | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebbe, E.K.M.; Ghequire, M.G.K.; Peigneur, S.; Mille, B.G.; Devi, P.; Ravichandran, S.; Waelkens, E.; D’Souza, L.; De Mot, R.; Tytgat, J. Novel Conopeptides of Largely Unexplored Indo Pacific Conus sp. Mar. Drugs 2016, 14, 199. https://doi.org/10.3390/md14110199
Lebbe EKM, Ghequire MGK, Peigneur S, Mille BG, Devi P, Ravichandran S, Waelkens E, D’Souza L, De Mot R, Tytgat J. Novel Conopeptides of Largely Unexplored Indo Pacific Conus sp. Marine Drugs. 2016; 14(11):199. https://doi.org/10.3390/md14110199
Chicago/Turabian StyleLebbe, Eline K. M., Maarten G. K. Ghequire, Steve Peigneur, Bea G. Mille, Prabha Devi, Samuthirapandian Ravichandran, Etienne Waelkens, Lisette D’Souza, René De Mot, and Jan Tytgat. 2016. "Novel Conopeptides of Largely Unexplored Indo Pacific Conus sp." Marine Drugs 14, no. 11: 199. https://doi.org/10.3390/md14110199
APA StyleLebbe, E. K. M., Ghequire, M. G. K., Peigneur, S., Mille, B. G., Devi, P., Ravichandran, S., Waelkens, E., D’Souza, L., De Mot, R., & Tytgat, J. (2016). Novel Conopeptides of Largely Unexplored Indo Pacific Conus sp. Marine Drugs, 14(11), 199. https://doi.org/10.3390/md14110199