
Three New Cytotoxic Polyhydroxysteroidal Glycosides from Starfish Craspidaster hesperus

Abstract
:1. Introduction
2. Results and Discussion
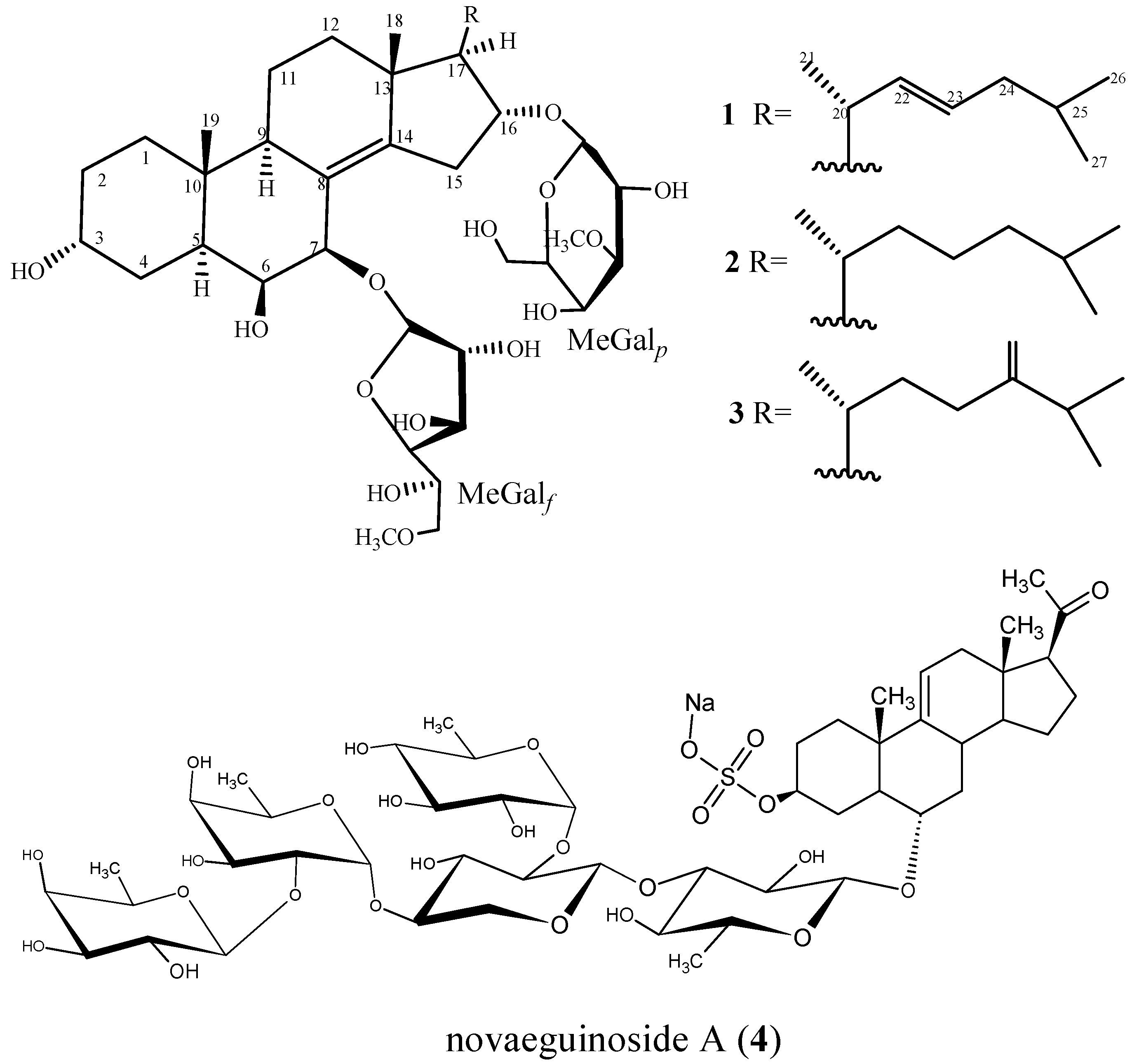
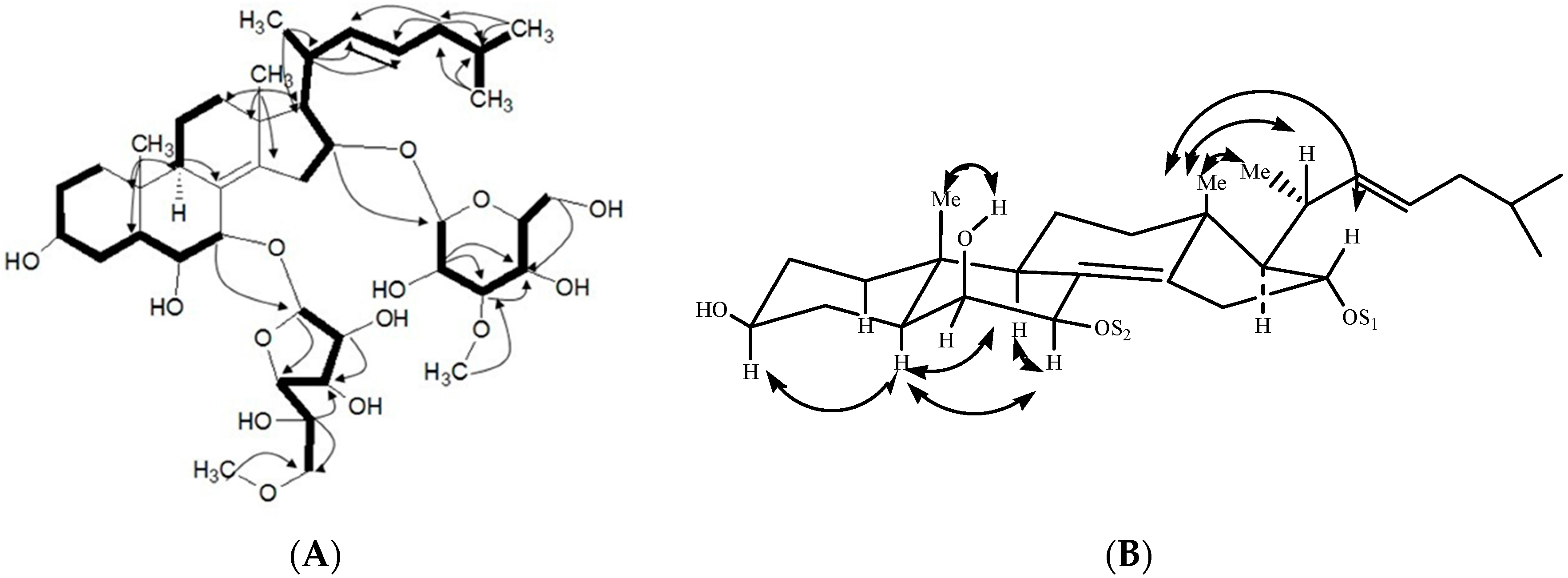
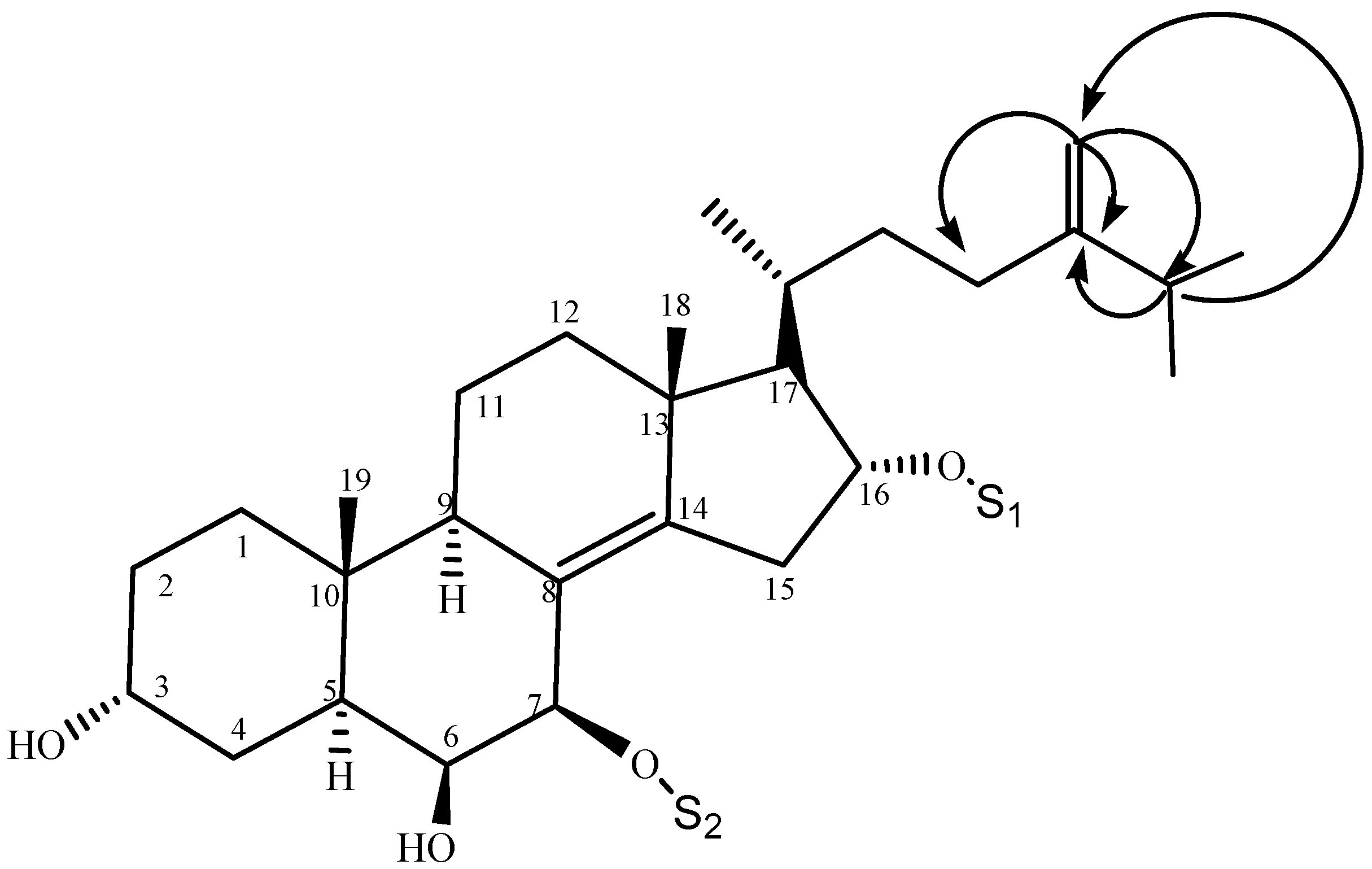
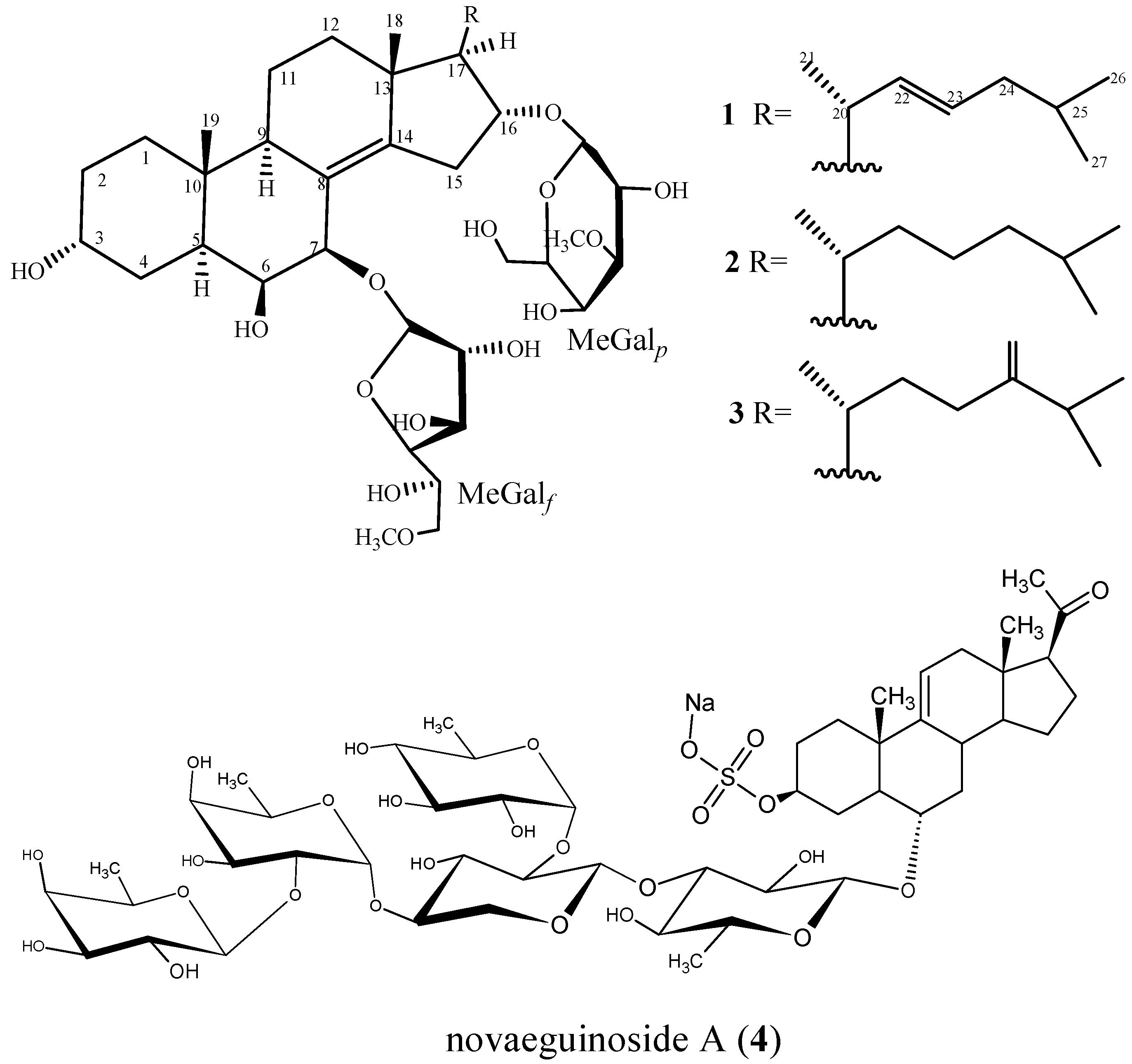
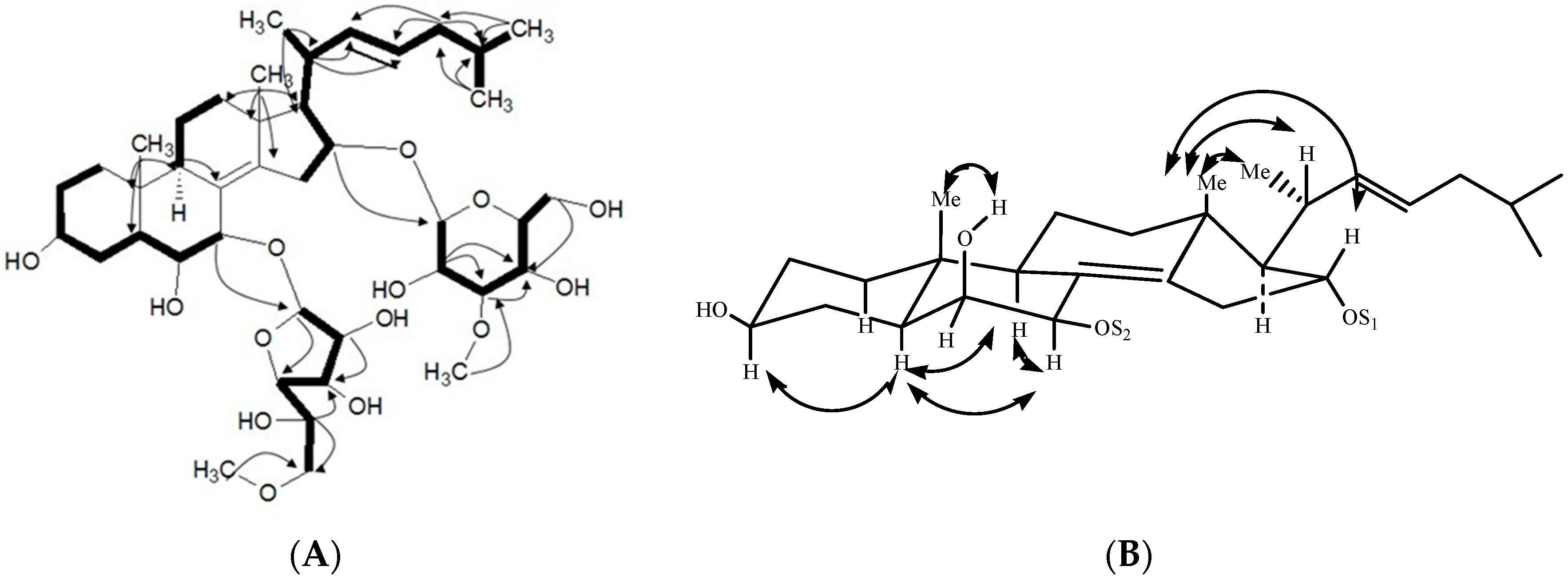
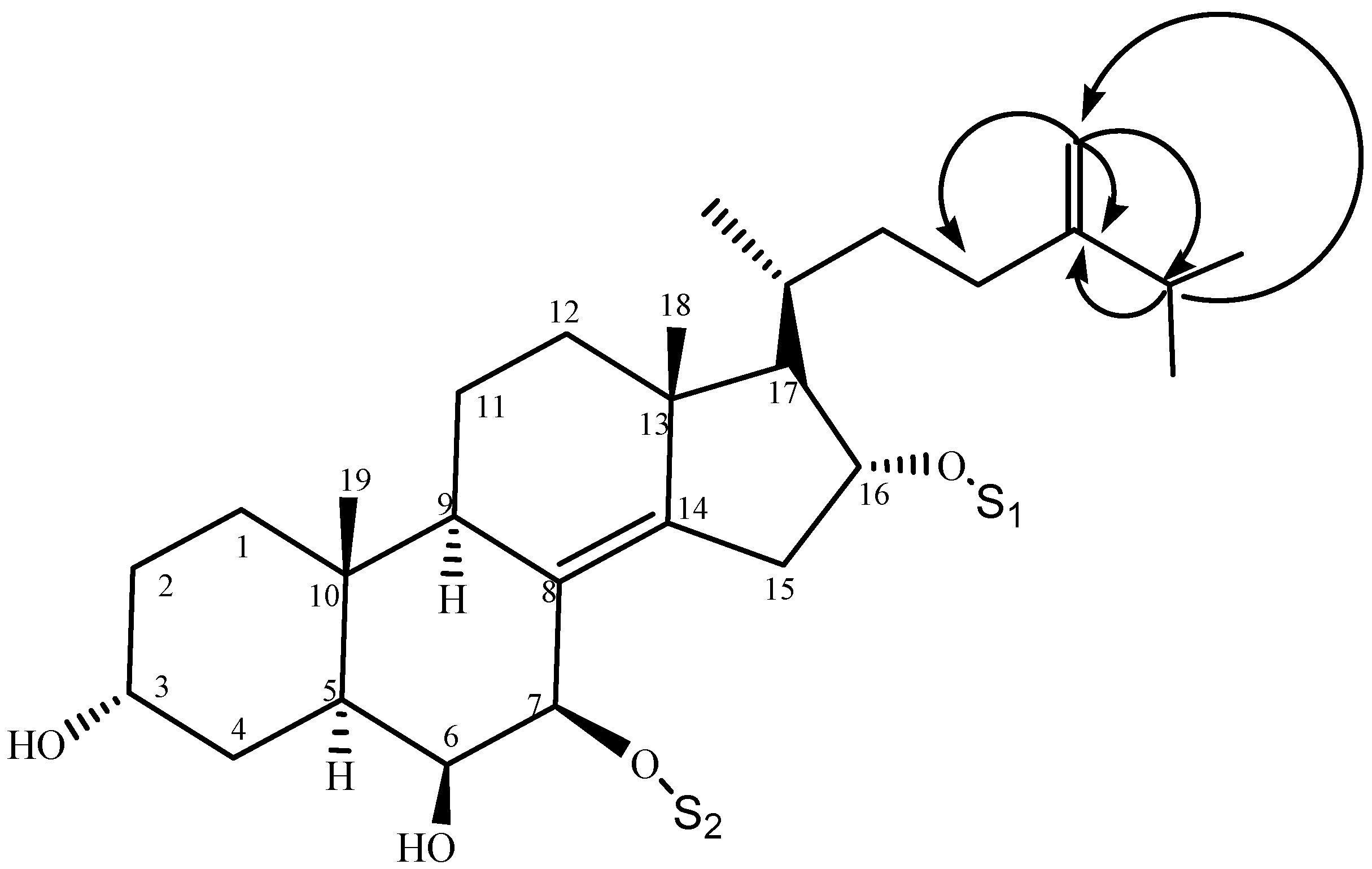
2.1. Characterization of the Compounds
2.2. Cytotoxic Activities
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Methanolysis of 1–3 [15]
3.5. Demethylation and Acid Hydrolysis of 1–3 [15]
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HPLC | High performance liquid chromatography |
| NMR | Nuclear magnetic resonance spectroscopy |
| DEPT | Distortionless enhancement by polarization transfer |
| COSY | Two dimensional 1H correlation |
| HMQC | Heteronuclear multiple-quantum correlation |
| HMBC | 1H-detected heteronuclear multiple-bond correlation |
| NOESY | Nuclear overhauser effect spectroscopy |
| GC–MS | Gas chromatography-mass spectrometer |
| TLC | Thin layer chromatography |
| SRB | Sulforhodamine B |
References
- Ivanchina, N.V.; Kicha, A.A.; Stonik, V.A. Steroid glycosides from marine organisms. Steroids 2011, 76, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Shubina, L.K.; Fedorov, S.N.; Levina, E.V.; Andriyaschenko, P.V.; Kalinovsky, A.I.; Stonik, V.A.; Smirnov, I.S. Comparative study on polyhydroxylated steroids from echinoderms. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 119, 505–511. [Google Scholar] [CrossRef]
- Iorizzi, M.; Minale, L.; Riccio, R.; Higa, T.; Tnanka, J. Starfish saponins, part 46. Steroidal glycosides and polyhydroxysteroids from the starfish Culcita novaeguineae. J. Nat. Prod. 1991, 54, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Herz, W.; Kirby, G.W.; Moore, R.E.; Steglich, W.; Tamm, C. Progress in the Chemistry of Organic Natural Products; Springer: New York, NY, USA, 1993; Volume 62, pp. 75–308. [Google Scholar]
- D’Auria, M.V.; Minale, L.; Riccio, R. Polyoxygenated steroids of marine origin. Chem. Rev. 1993, 93, 1839–1895. [Google Scholar] [CrossRef]
- Minale, L.; Riccio, R.; Zollo, F. Structure and Chemistry (Part C). In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier Science B.V.: Amsterdam, NY, USA, 1995; Volume 15, pp. 43–110. [Google Scholar]
- Stonik, V.A.; Ivanchina, N.V.; Kicha, A.A. New polar steroids from starfish. Nat. Prod. Commun. 2008, 3, 1587–1592. [Google Scholar]
- Iorizzi, M.; De Marino, S.; Zollo, F. Steroidal Oligoglycosides from the Asteroidea. Curr. Org. Chem. 2001, 5, 951–973. [Google Scholar] [CrossRef]
- Yang, S.-W.; Chan, T.-M.; Buevich, A.; Priestley, T.; Crona, J.; Wright, A.E.; Patel, M.; Gullo, V.; Chen, G.-D.; Pramanik, B.; et al. Novel steroidal saponins, Sch 725737 and Sch 725739, from a marine starfish, Novodinia antillensis. Biorgan. Med. Chem. Lett. 2007, 17, 5543–5547. [Google Scholar] [CrossRef] [PubMed]
- Kicha, A.A.; Kalinovsky, A.I.; Ivanchina, N.V.; Malyarenko, T.V.; Dmitrenok, P.S.; Ermakova, S.P.; Stonik, V.A. Four new asterosaponins, Hippasteriosides A–D, from the Far eastern starfish Hippasteria kurilensis. Chem. Biodivers. 2011, 8, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, Y.-H.; Xu, Q.-Z.; La, M.-P.; Zhang, H.-W. Two new cytotoxic triterpene glycosides from the sea cucumber Holothuria scabra. Planta Med. 2009, 75, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, Q.-Z.; Yi, Y.-H.; Gong, W.; Jiao, B.-H. Two new cytotoxic disulfated holostane glycosides from the sea cucumber Pentacta quadrangularis. Chem. Biodivers. 2010, 7, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Han, H.; Chen, X.F.; Yi, Y.H.; Sun, H.X. Cytotoxic and apoptosis-inducing activity of triterpene glycosides from Holothuria scabra and Cucumaria frondosa against HepG2 cells. Mar. Drugs 2014, 12, 4274–4290. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Zou, Z.R.; Yi, Y.H.; Han, H.; Li, L.; Pan, M.X. Variegatusides: New non-sulphated triterpene glycosides from the sea cucumber Stichopus variegates semper. Mar. Drugs 2014, 12, 2004–2018. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Tang, H.-F.; Qu, F.; Lin, H.-W.; Tian, X.-R.; Yao, M.-N. Polyhydroxysteroidal glycosides from the Starfish Anthenea chinensis. J. Nat. Prod. 2010, 73, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Shashkov, A.S.; Chizhov, O.S. C13 NMR spectroscopy in chemistry of carbohydrates and related compounds. Bioorg. Khim. 1976, 2, 437–497. [Google Scholar]
- Wang, H.; Sun, L.-H.; Glazebnik, S.; Zhao, K. Peralkylation of saccharides under aqueous conditions. Tetrahedron Lett. 1995, 36, 2953–2956. [Google Scholar] [CrossRef]
- Araki, S.; Abe, S.; Odani, S.; Ando, S.; Fujii, N.; Satake, M. Structure of a triphosphonopentaosylceramide containing 4-O-Methyl-N-acetylglucosamine from the skin of the sea hare Aplysia kurodai. J. Biol. Chem. 1987, 262, 14141–14145. [Google Scholar] [PubMed]
- Kicha, A.A.; Ivanchin, N.V.; Kalinovsky, A.I.; Dmitrenok, P.S.; Stonik, V.A. Asterosaponin P2 from the Far-Eastern starfish patiria (asterina) pectinifera. Russ. Chem. Bull. 2000, 49, 1794–1795. [Google Scholar] [CrossRef]
- Tang, H.-F.; Cheng, G.; Wu, J.; Chen, X.-L.; Zhang, S.-Y.; Wen, A.-D.; Lin, H.-W. Cytotoxic asterosaponins capable of promoting polymerization of tubulin from the Starfish Culcita novaeguineae. J. Nat. Prod. 2009, 72, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.-F.; Yi, Y.-H.; Li, L.; Sun, P.; Zhang, S.-Q.; Zhao, Y.-P. Bioactive asterosaponins from the Starfish Culcita novaeguineae. J. Nat. Prod. 2005, 68, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- DeLean, A.D.; Munson, P.J.; Rodbard, D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978, 235, E97. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 |
|---|---|---|---|
| 1 | 34.6, CH2 | 34.4, CH2 | 33.8, CH2 |
| 2 | 30.5, CH2 | 30.5, CH2 | 29.6, CH2 |
| 3 | 66.7, CH | 66.7, CH | 65.7, CH |
| 4 | 34.3, CH2 | 34.3, CH2 | 33.7, CH2 |
| 5 | 38.5, CH | 38.4, CH | 37.5, CH |
| 6 | 74.5, CH | 74.6, CH | 78.0, CH |
| 7 | 78.5, CH | 78.8, CH | 76.9, CH |
| 8 | 127.4, qC | 127.2, qC | 126.3, qC |
| 9 | 46.0, CH | 45.7, CH | 44.9, CH |
| 10 | 39.2, qC | 39.2, qC | 38.6, qC |
| 11 | 19.5, CH2 | 19.7, CH2 | 18.6, CH2 |
| 12 | 36.7, CH2 | 37.3, CH2 | 36.7, CH2 |
| 13 | 45.0, qC | 45.0, qC | 44.5, qC |
| 14 | 146.0, qC | 147.1, qC | 146.5, qC |
| 15 | 34.5, CH2 | 34.7, CH2 | 34.5, CH2 |
| 16 | 78.9, CH | 79.6, CH | 77.8, CH |
| 17 | 62.1, CH | 62.2, CH | 61.2, CH |
| 18 | 20.7, CH3 | 20.3, CH3 | 19.3, CH3 |
| 19 | 16.2, CH3 | 16.2, CH3 | 15.4, CH3 |
| 20 | 36.2, CH | 33.6, CH | 33.2, CH |
| 21 | 24.7, CH3 | 21.2, CH3 | 21.9, CH3 |
| 22 | 138.0, CH | 35.4, CH2 | 25.0, CH2 |
| 23 | 128.6, CH | 25.9, CH2 | 33.7, CH2 |
| 24 | 42.9, CH2 | 40.3, CH2 | 157.1, qC |
| 25 | 29.5, CH | 28.9, CH | 35.8, CH |
| 26 | 23.2, CH3 | 23.2, CH3 | 23.9, CH3 |
| 27 | 23.2, CH3 | 23.2, CH3 | 23.9, CH3 |
| 28 | 107.5, CH2 | ||
| MeGalf | |||
| 1′ | 108.5, CH | 108.5, CH | 108.3, CH |
| 2′ | 83.4, CH | 83.5, CH | 82.5, CH |
| 3′ | 78.7, CH | 78.6, CH | 78.7, CH |
| 4′ | 85.5, CH | 85.3, CH | 84.7, CH |
| 5′ | 71.0, CH | 71.0, CH | 71.0, CH |
| 6′ | 76.1, CH2 | 76.1, CH2 | 74.6, CH2 |
| 6′OCH3 | 59.4, CH3 | 59.5, CH3 | 58.8, CH3 |
| MeGalp | |||
| 1′′ | 103.4, CH | 103.2, CH | 102.9, CH |
| 2′′ | 75.5, CH | 75.4, CH | 75.5, CH |
| 3′′ | 88.7, CH | 88.6, CH | 87.7, CH |
| 4′′ | 71.8, CH | 71.9, CH | 71.8, CH |
| 5′′ | 78.5, CH | 78.5, CH | 78.5, CH |
| 6′′ | 63.8, CH2 | 63.8, CH2 | 62.6, CH2 |
| 3′′-OCH3 | 61.5, CH3 | 61.4, CH3 | 60.4, CH3 |
| Position | 1 | 2 | 3 |
|---|---|---|---|
| 1 | 2.06 m; 1.50 m | 2.06 m; 1.50 m | 1.52 m; 1.32 m |
| 2 | 1.85 m; 2.02 m | 1.88 m; 2.02 m | 1.80 m; 1.21 m |
| 3 | 4.52 m | 4.50 m | 4.31 m |
| 4 | 1.92 m; 2.48 t (14.6, 16.1) | 1.92 m; 2.48 t (16.3, 16.3) | 1.90 m; 2.31 m |
| 5 | 2.91 brd (15.0) | 2.94 brd (16.2) | 2.79 brd (13.0) |
| 6 | 4.34 m | 4.33 m | 4.66 m |
| 7 | 4.96 d (2.2) | 4.96 m | 3.71 d (2.4) |
| 8 | |||
| 9 | 2.80 brt (8.9) | 2.80 brt (8.6) | 2.64 m |
| 10 | |||
| 11 | 1.75 m; 1.74 m | 1.76 m; 1.74 m | 1.52 m; 1.62 m |
| 12 | 1.38 m; 1.86 m | 1.48 m; 1.96 m | 1.32 m; 1.79 m |
| 13 | |||
| 14 | |||
| 15 | 3.10 m; 3.21 m | 3.06 m; 3.27 m | 2.90 m; 3.10 m |
| 16 | 4.79 m | 4.78 m | 4.79 m |
| 17 | 1.72 d (9.9) | 1.72 d (9.9) | 1.60 dd (9.6, 2.5) |
| 18 | 1.06 s | 1.06 s | 0.91 s |
| 19 | 1.32 s | 1.32 s | 1.23 s |
| 20 | 2.57 m | 1.76 m | 1.71 m |
| 21 | 1.24 d (7.9) | 1.09 d (7.2) | 0.78 d (7.0) |
| 22 | 6.15 m | 1.86 m; 1.60 m | 1.38 m |
| 23 | 5.45 m | 1.50 m; 1.26 m | 1.90 m; 2.31 m |
| 24 | 2.09 m | 1.27 m | |
| 25 | 1.68 m | 1.58 m | 2.42 m |
| 26 | 0.96 d (5.1) | 0.92 d (7.6) | 1.11 d (7.6) |
| 27 | 0.98 d (5.1) | 0.92 d (7.6) | 1.11 d (7.6) |
| 28 | 4.75 s; 4.77 s | ||
| MeGalf | |||
| 1′ | 5.80 d (3.2) | 5.81 d (1.1) | 5.61 d (3.2) |
| 2′ | 4.76 m | 4.73 m | 4.56 m |
| 3′ | 4.87 m | 4.86 m | 4.87 m |
| 4′ | 4.70 m | 4.69 m | 4.54 m |
| 5′ | 4.48 m | 4.46 m | 4.48 m |
| 6′ | 4.03 m | 4.00 m | 3.80 m |
| 6′-OCH3 | 3.45 s | 3.44 s | 3.29 s |
| MeGalp | |||
| 1′′ | 4.80 d (7.7) | 4.81 d (7.6) | 4.70 d (7.7) |
| 2′′ | 3.94 m | 3.93 m | 3.94 m |
| 3′′ | 3.76 t | 3.74 t | 3.58 t |
| 4′′ | 4.15 m | 4.09 m | 4.15 m |
| 5′′ | 3.89 m | 3.89 m | 3.89 m |
| 6′′ | 4.37 m; 4.53 m | 4.34 m; 4.53 m | 4.32 m; 4.73 m |
| 3′′-OCH3 | 3.91 s | 3.90 s | 3.72 s |
| Cell Line | 1 | 2 | 3 | HCP a |
|---|---|---|---|---|
| A-549 | 3.62 ± 1.08 * | 1.84 ± 0.65 * | 2.40 ± 0.73 * | 0.84 ± 0.05 |
| MOLT-4 | 2.59 ± 0.94 * | 0.68 ± 0.12 | 2.12 ± 0.81 * | 0.16 ± 0.03 |
| BEL-7402 | 5.26 ± 0.36 * | 2.67 ± 0.54 * | 5.72 ± 0.82 * | 0.03 ± 0.05 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.-X.; Kang, Y.-F.; Han, H. Three New Cytotoxic Polyhydroxysteroidal Glycosides from Starfish Craspidaster hesperus. Mar. Drugs 2016, 14, 189. https://doi.org/10.3390/md14100189
Kang J-X, Kang Y-F, Han H. Three New Cytotoxic Polyhydroxysteroidal Glycosides from Starfish Craspidaster hesperus. Marine Drugs. 2016; 14(10):189. https://doi.org/10.3390/md14100189
Chicago/Turabian StyleKang, Jun-Xia, Yong-Feng Kang, and Hua Han. 2016. "Three New Cytotoxic Polyhydroxysteroidal Glycosides from Starfish Craspidaster hesperus" Marine Drugs 14, no. 10: 189. https://doi.org/10.3390/md14100189
APA StyleKang, J.-X., Kang, Y.-F., & Han, H. (2016). Three New Cytotoxic Polyhydroxysteroidal Glycosides from Starfish Craspidaster hesperus. Marine Drugs, 14(10), 189. https://doi.org/10.3390/md14100189