
Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid

Abstract
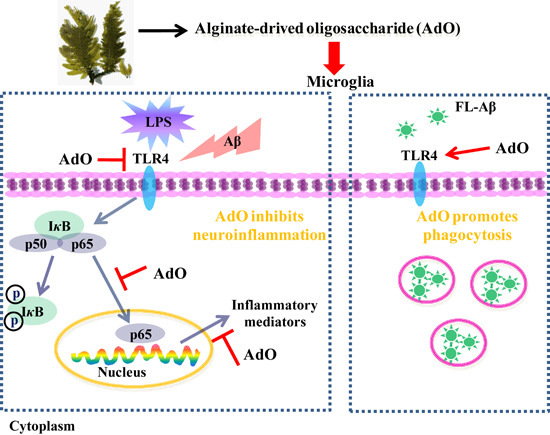
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
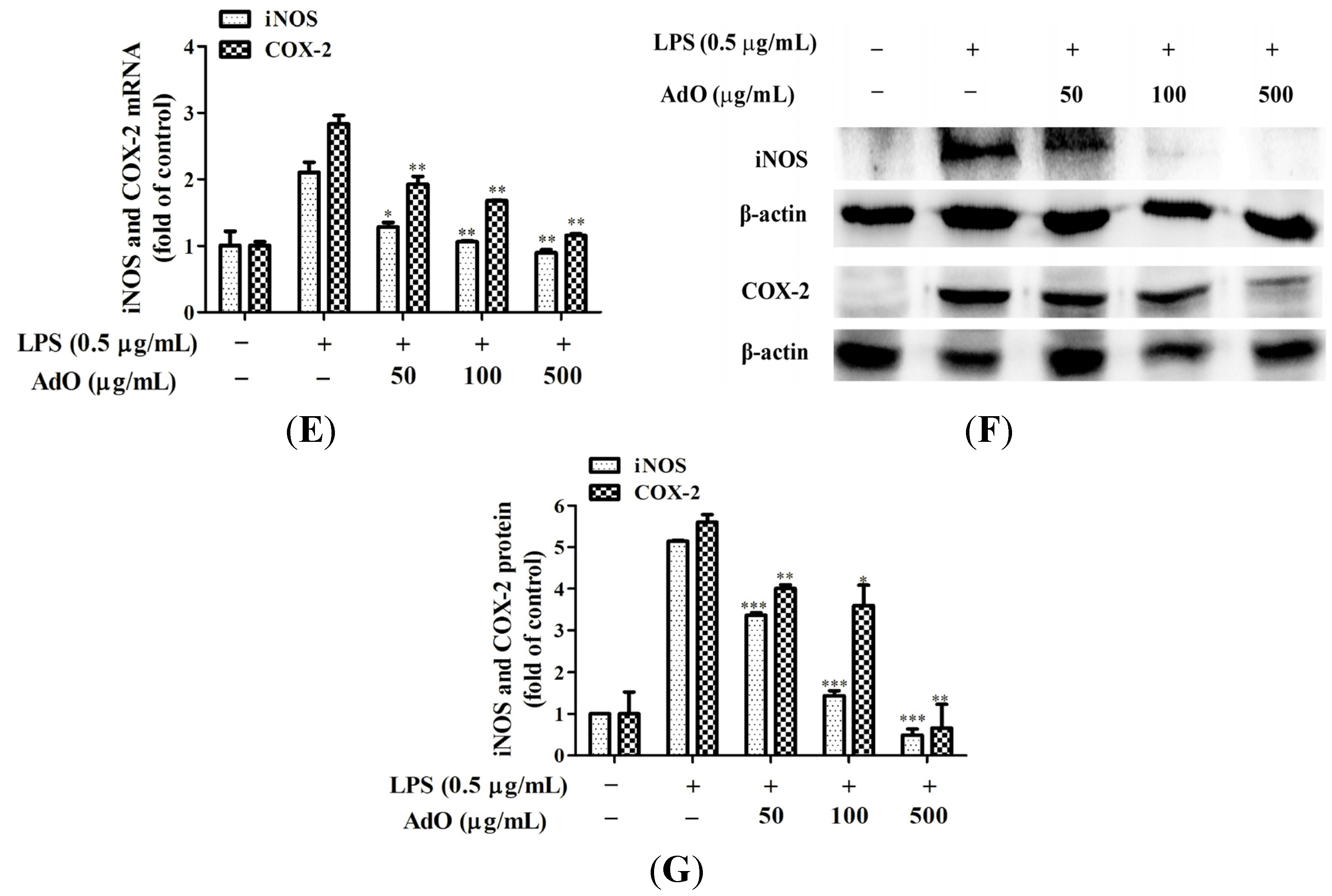
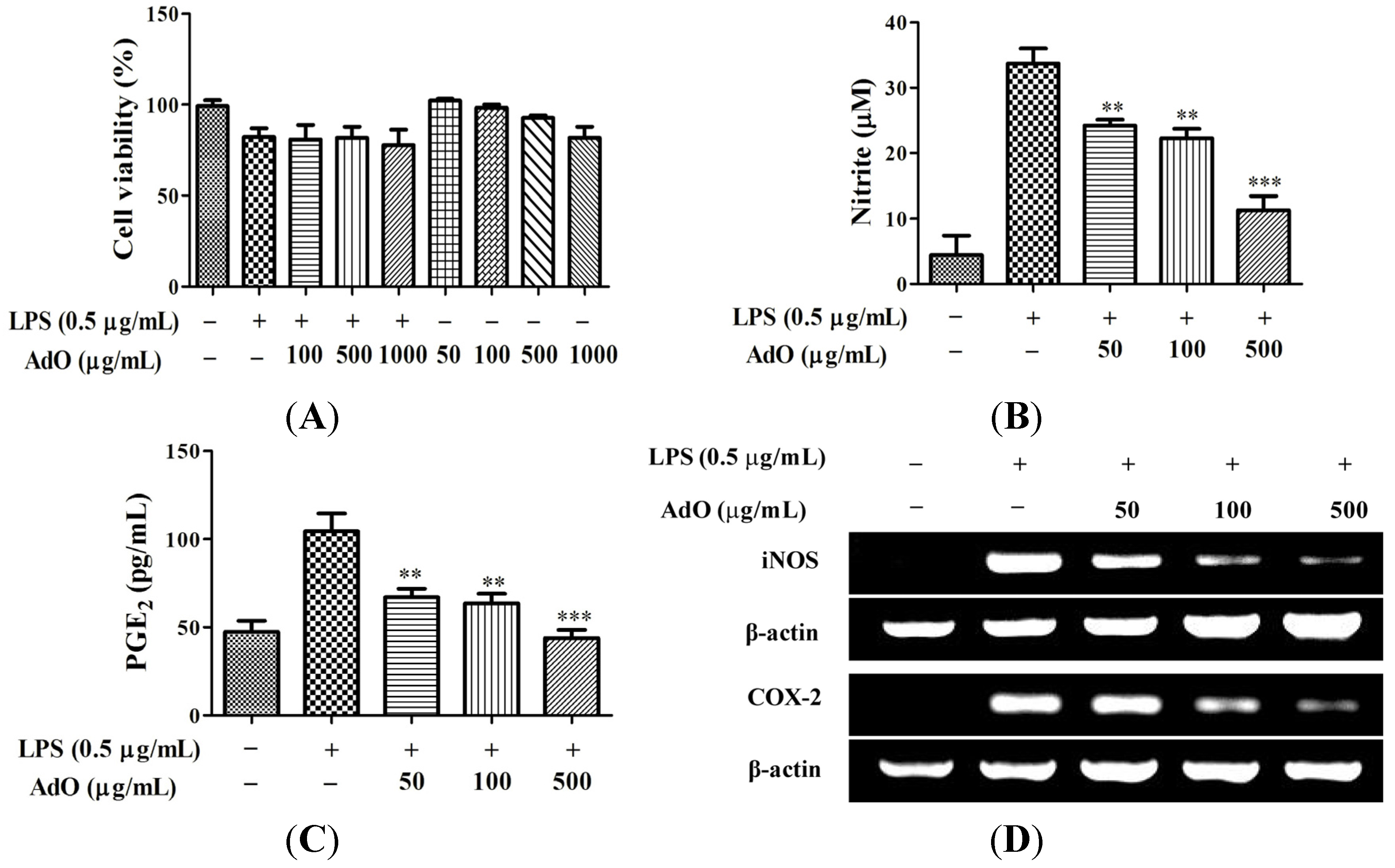
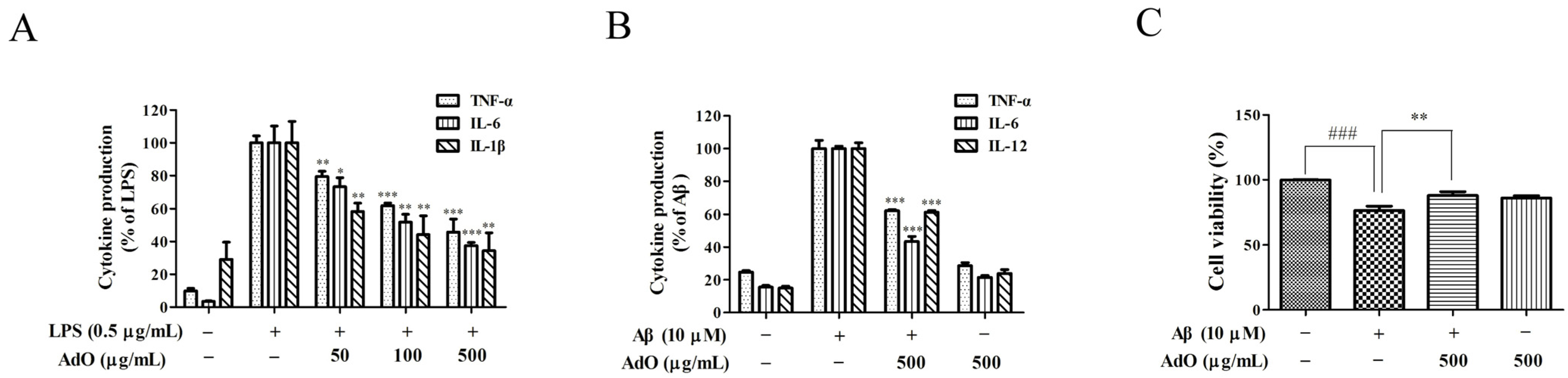
2.1. AdO Suppresses LPS-Induced Production of Inflammatory Mediators in BV2 Cells
2.2. AdO Inhibits LPS/Aβ-Activated Secretion of Inflammatory Cytokines in BV2 Cells
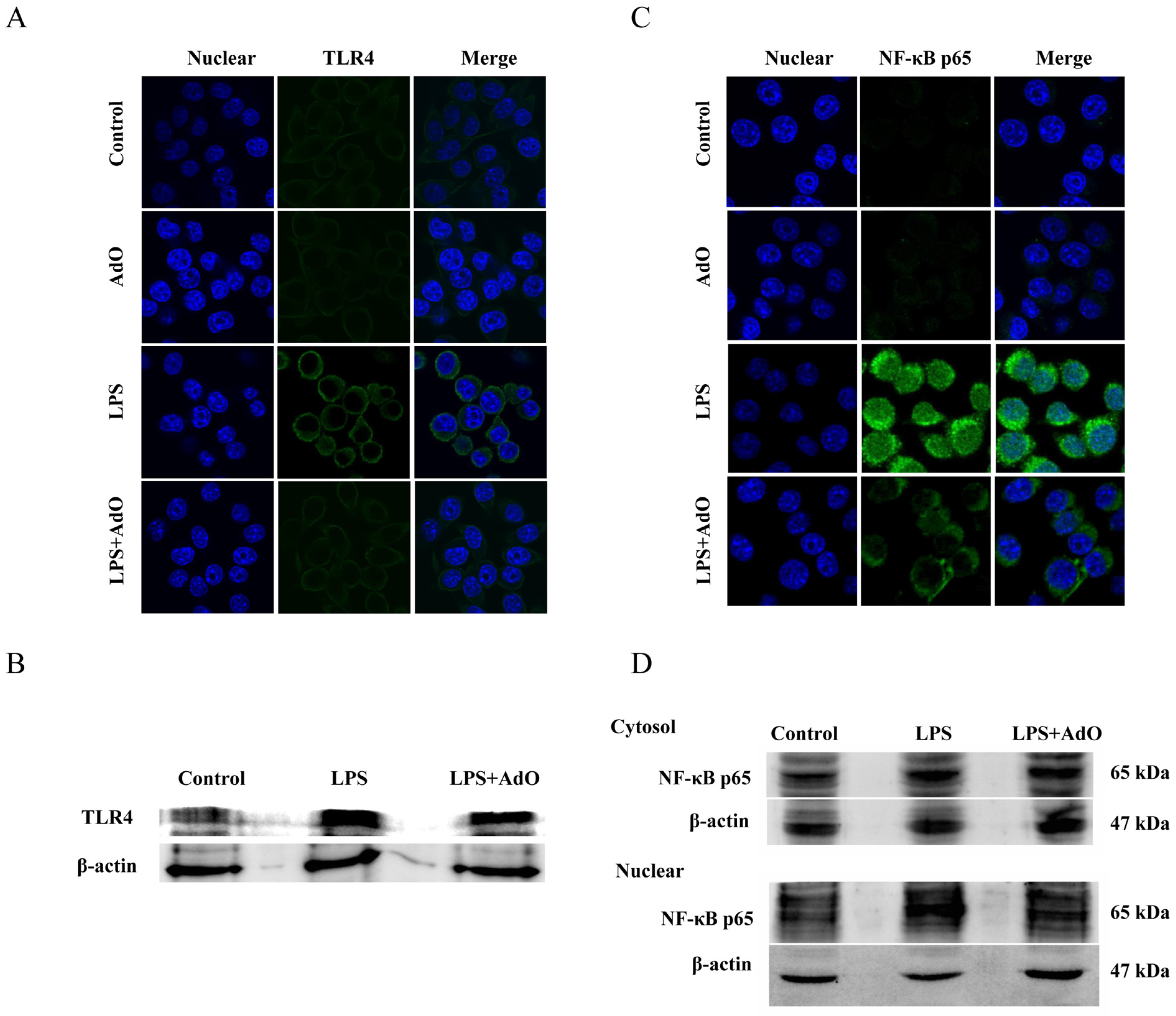
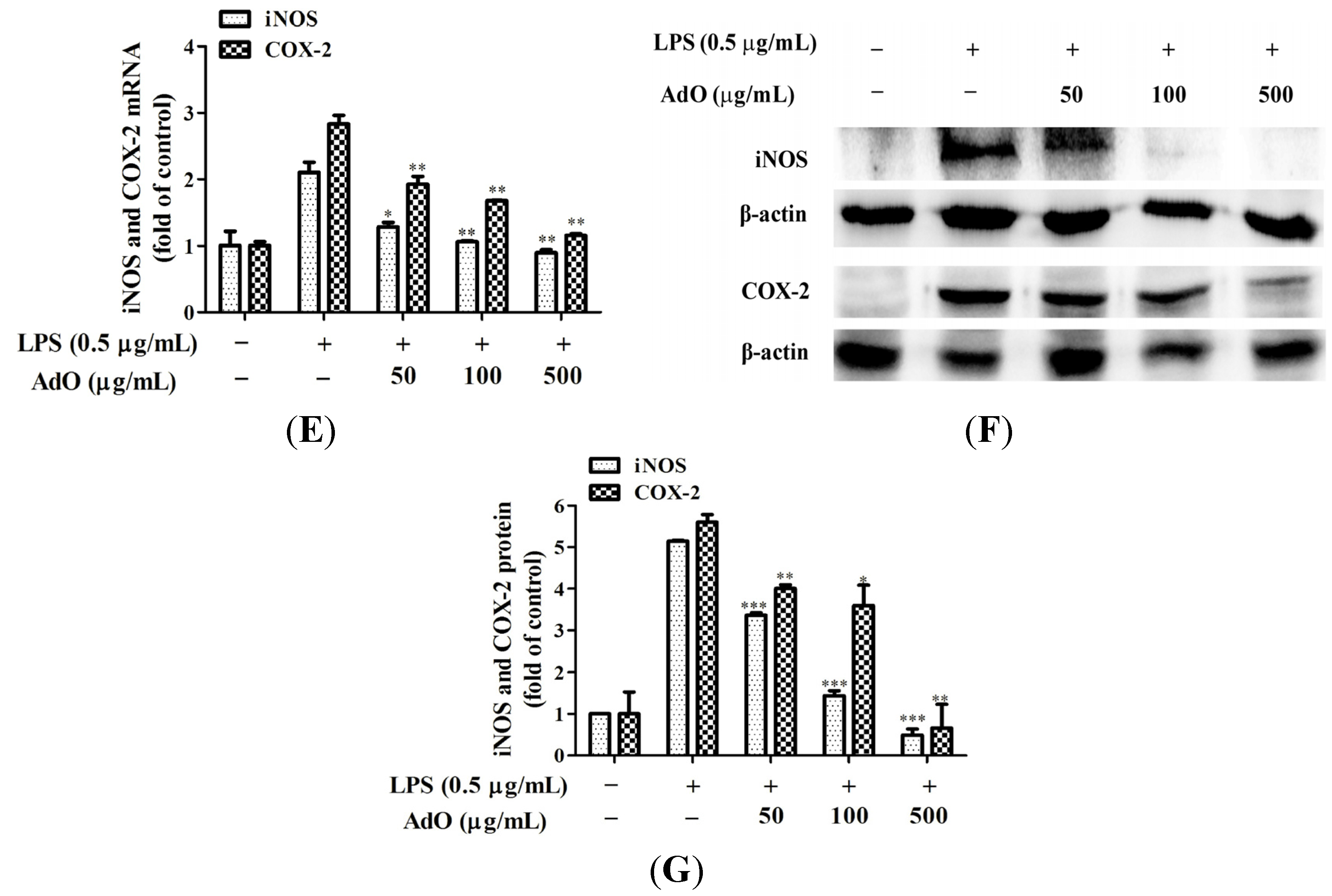
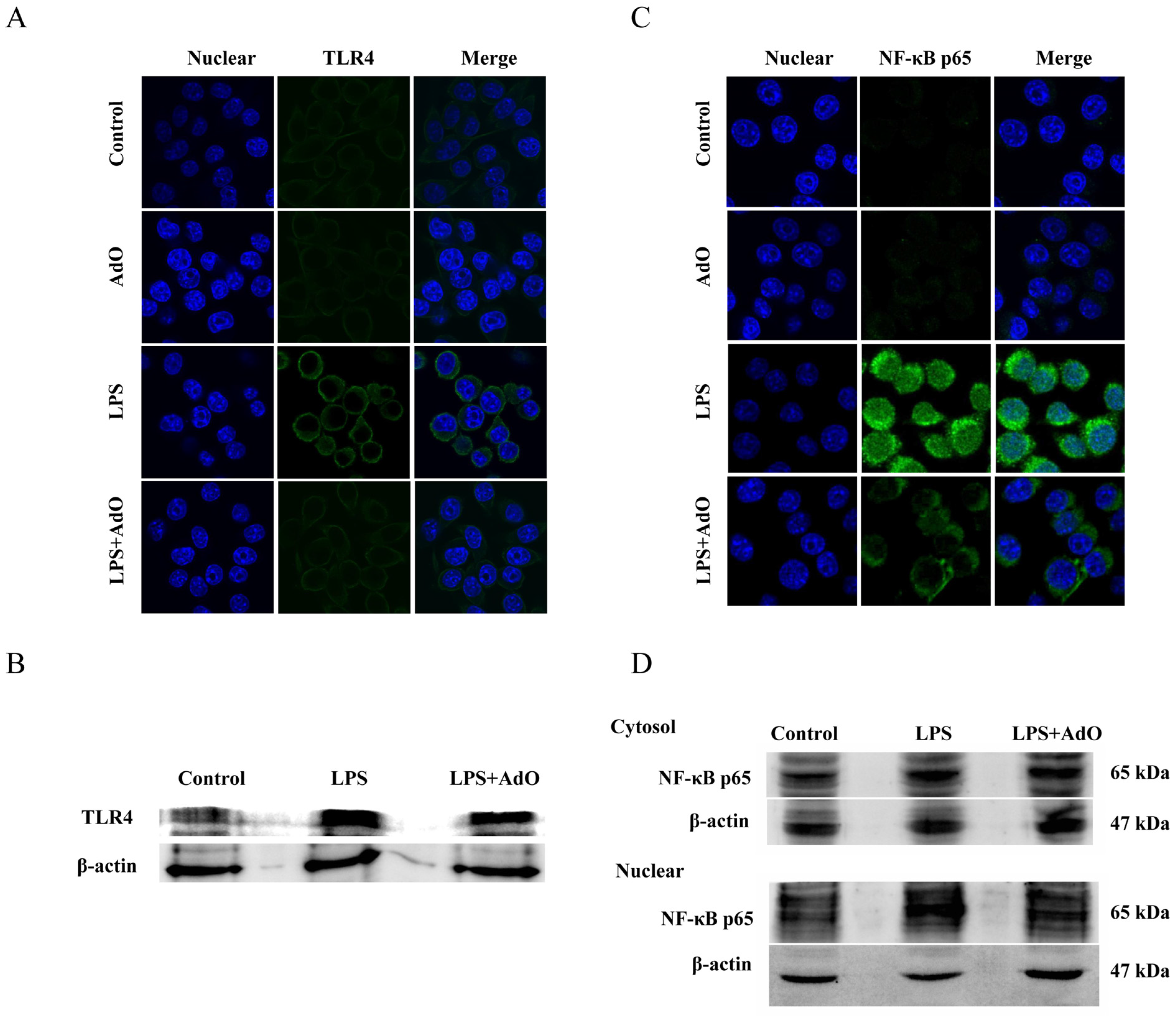
2.3. AdO Inhibits LPS-Activated Signaling Pathway in BV2 Cells
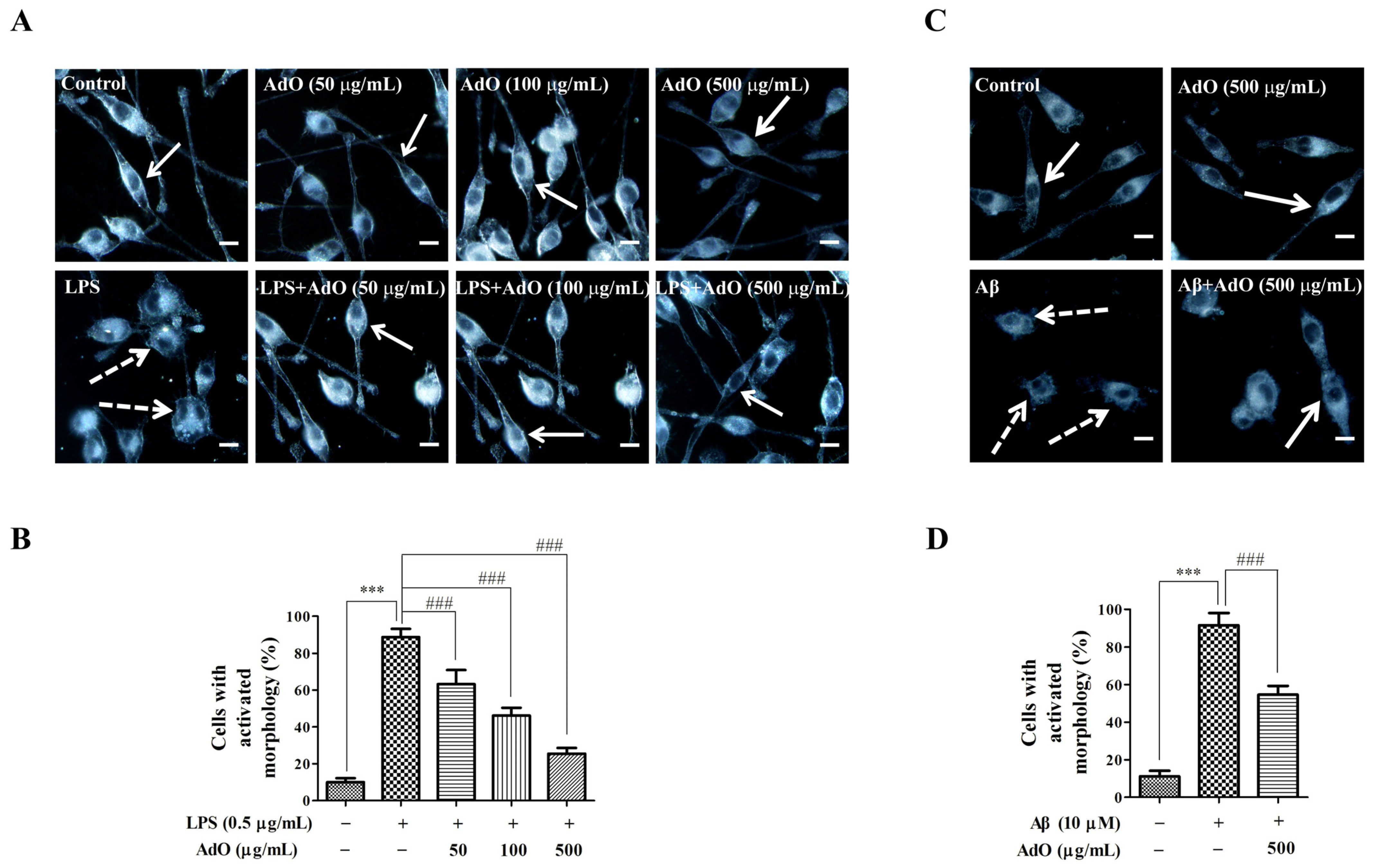
2.4. Effect of AdO on LPS/Aβ-Activated Morphological Changes of the BV2 Cells
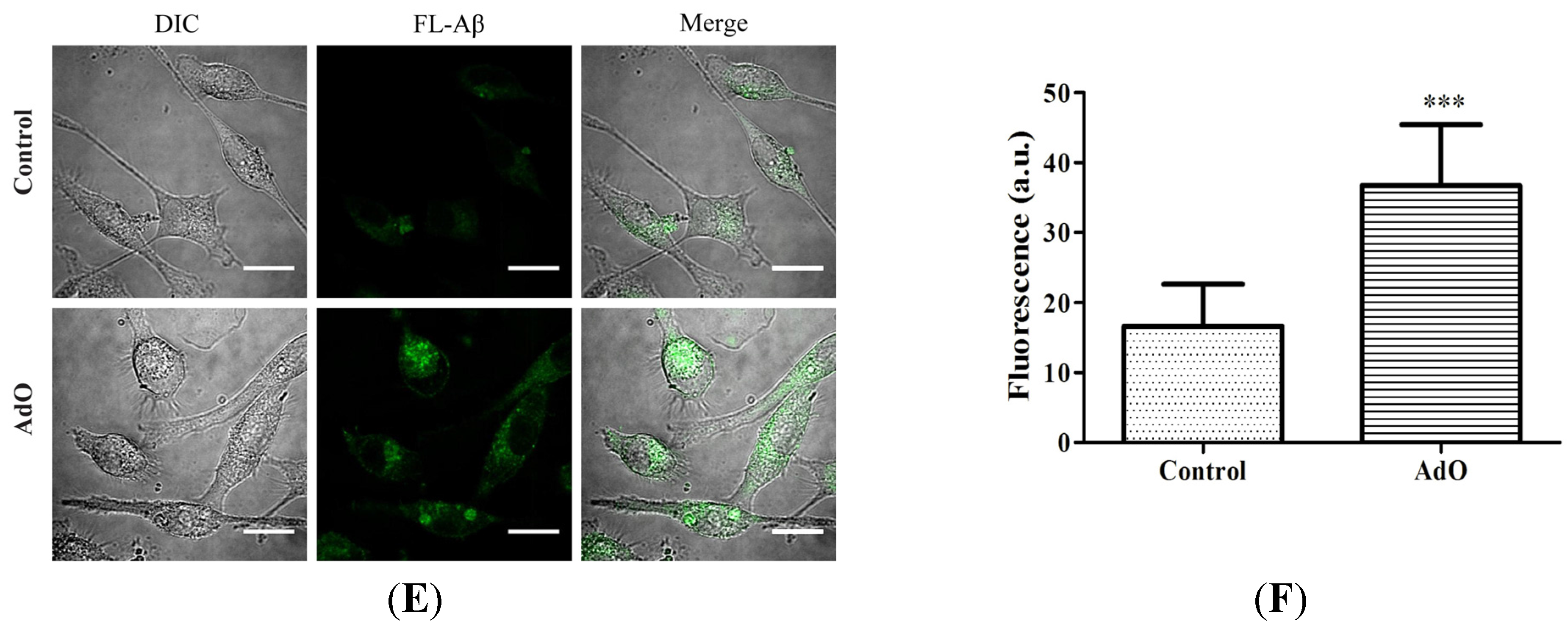
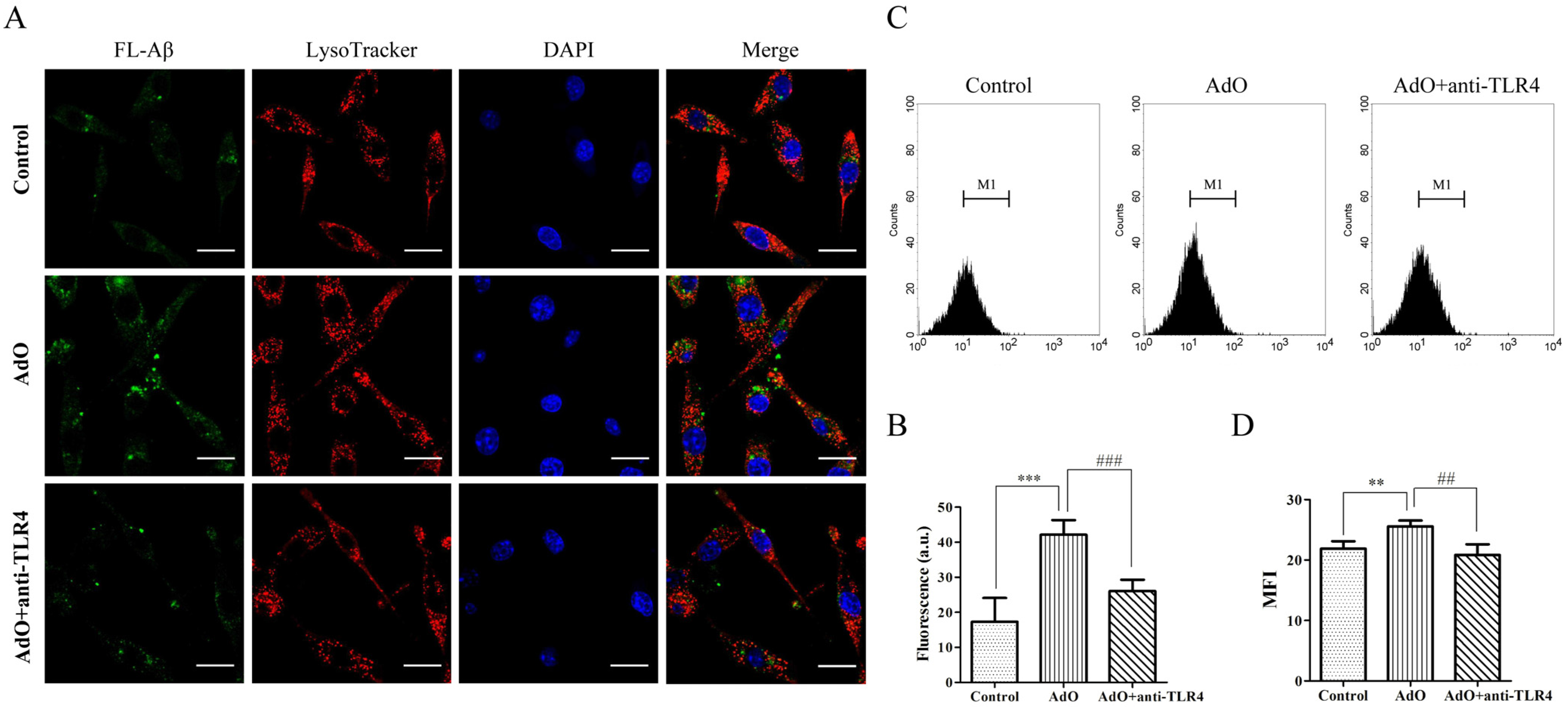
2.5. AdO Promotes the Phagocytosis of Aβ in BV2 Cells
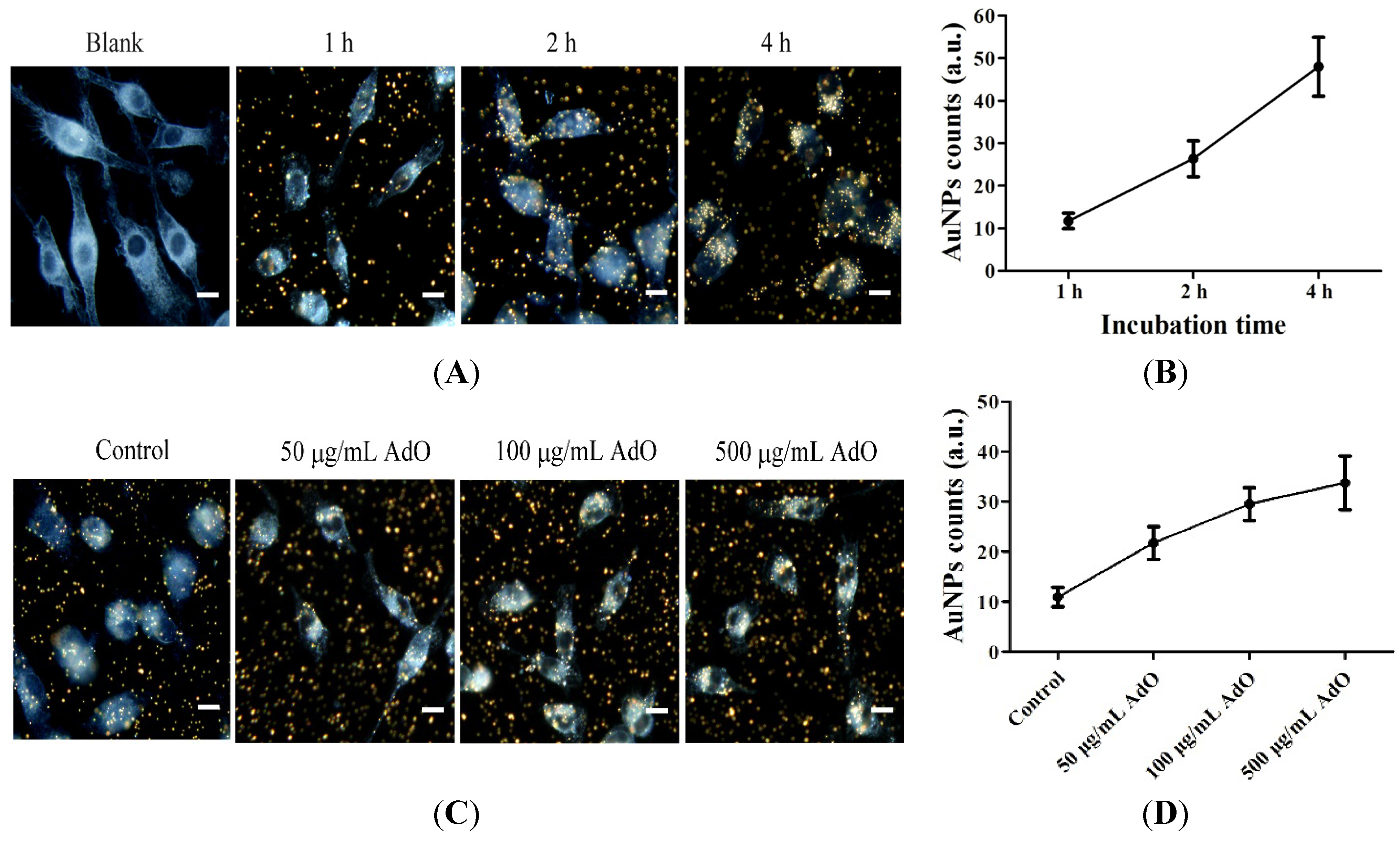
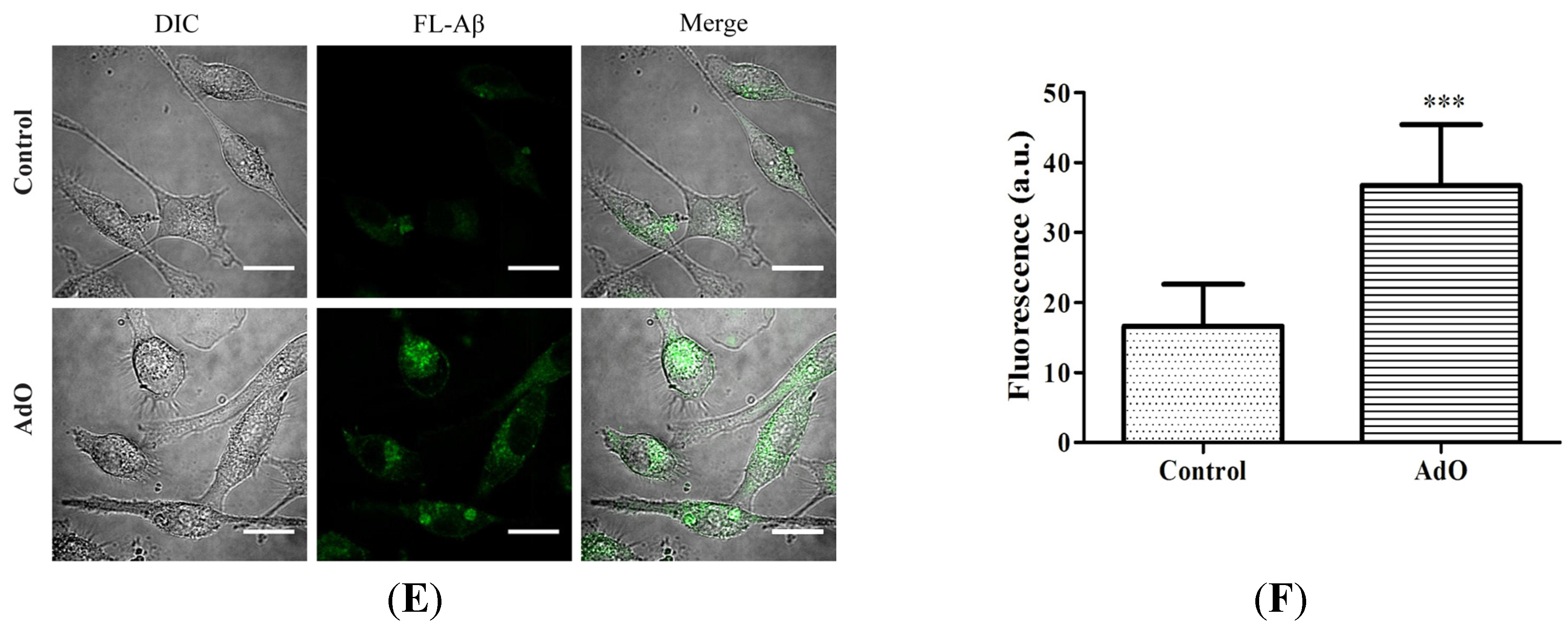
2.6. TLR4 Is Involved in the AdO-Promoted Microglial Phagocytosis
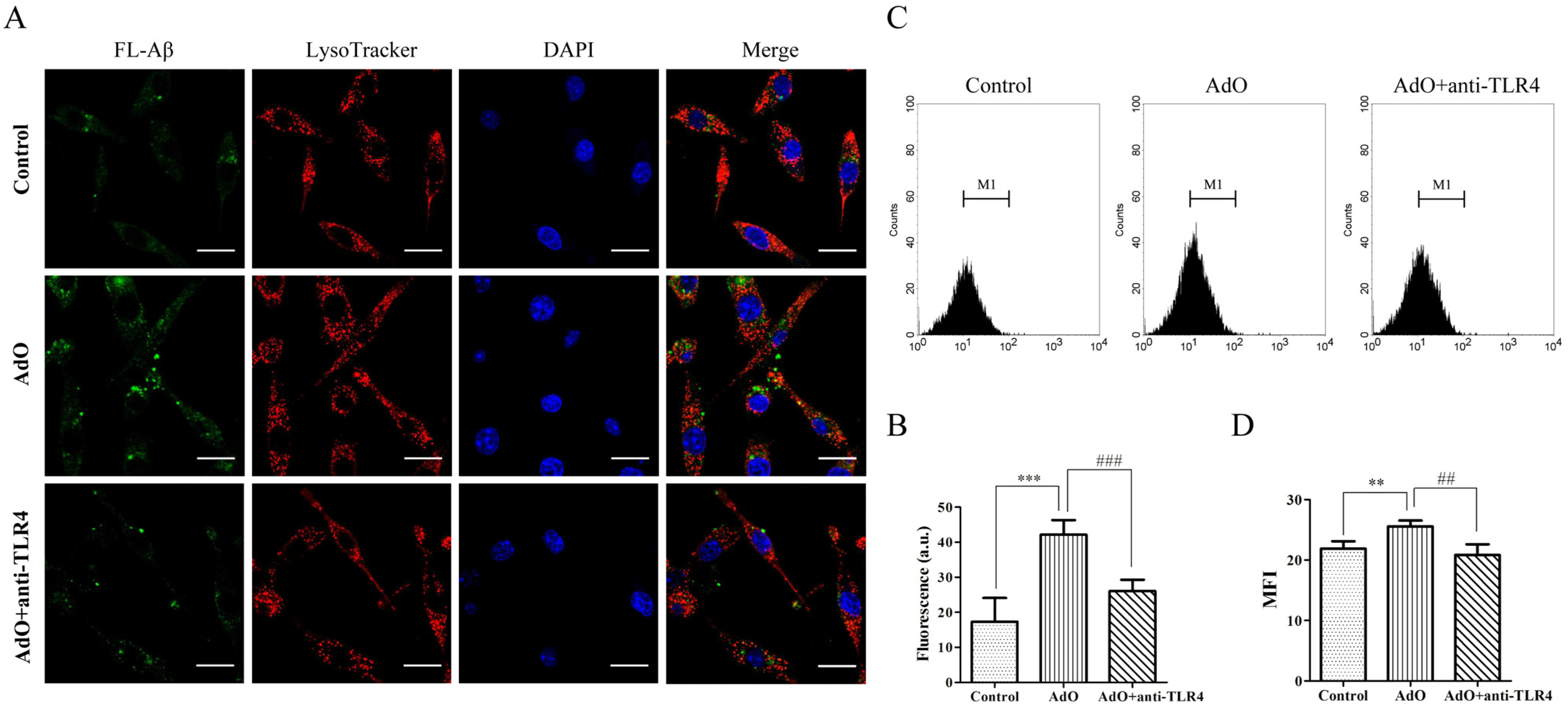
3. Discussion
4. Materials and Methods
4.1. Materials
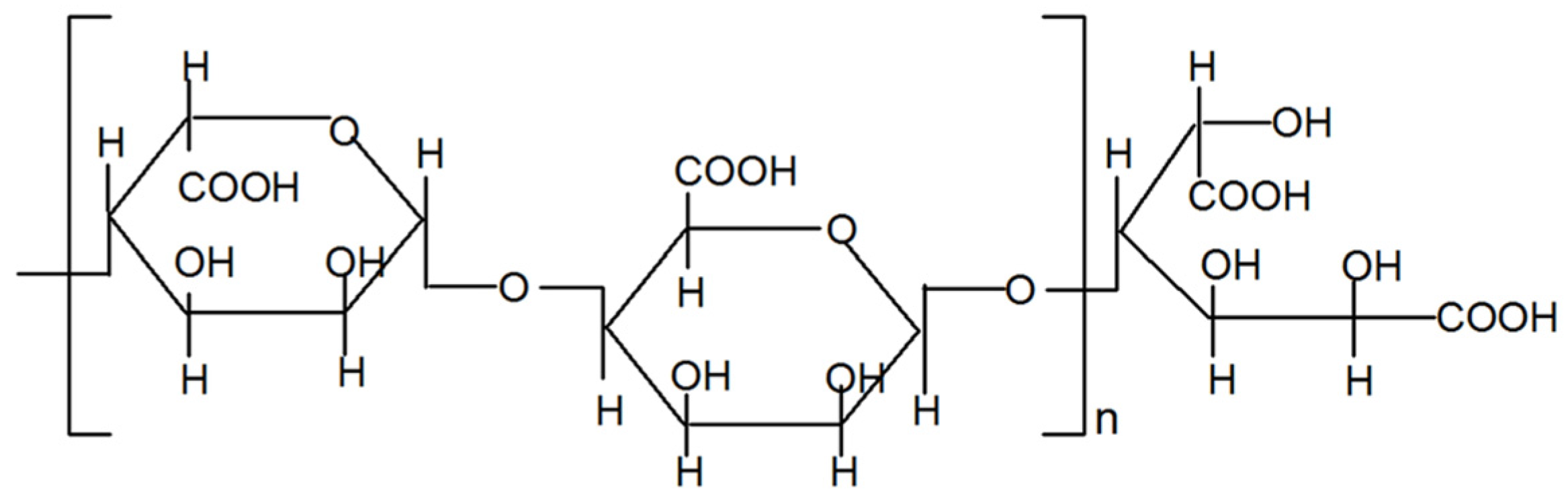
4.2. Preparation of AdO
4.3. Cell Culture
4.4. Cytotoxicity Assay
4.5. Measurement of NO and PGE2
4.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.7. Western Blot Analysis
4.8. Measurement of Cytokines
4.9. Immunofluorescence Analysis
4.10. Cell Morphology
4.11. Phagocytosis Assay
4.12. Flow Cytometric Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maccioni, R.B.; Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farías, G.A. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 1–9. [Google Scholar]
- Tansey, M.G.; Frank-Cannon, T.C.; McCoy, M.K.; Lee, J.K.; Martinez, T.N.; McAlpine, F.E.; Ruhn, K.A.; Tran, T.A. Neuroinflammation in Parkinson’s disease: Is there sufficient evidence for mechanism-based interventional therapy? Front. Biosci. 2008, 13, 709–717. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Carret-Rebillat, A.-S.; Pace, C.; Gourmaud, S.; Ravasi, L.; Montagne-Stora, S.; Longueville, S.; Tible, M.; Sudol, E.; Chang, R.C.-C.; Paquet, C.; Mouton-Liger, F.; Hugon, J. Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; Dolga, A.; Koepke, J.; Mengel, D.; Culmsee, C.; Dodel, R.; Koczulla, A.; Bach, J.-P. α-antitrypsin modulates microglial-mediated neuroinflammation and protects microglial cells from amyloid-β-induced toxicity. J. Neuroinflammation 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, C.M.; Wang, J.M.; Zhao, S.Q.; Zhang, K.; Wang, J.; Zhang, W.; Wu, C.; Yang, J. Pseudoginsenoside-F11 (PF11) exerts anti-neuroinflammatory effects on LPS-activated microglial cells by inhibiting TLR4-mediated TAK1/IKK/NF-κB, MAPKs and Akt signaling pathways. Neuropharmacology 2014, 79, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhou, A.; Xu, L.; Zhang, X. The role of TLR4-mediated PTEN/PI3K/AKT/NF-κB signaling pathway in neuroinflammation in hippocampal neurons. Neurosci. 2014, 269, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.H.; O’Banion, M.K. Neuroinflammation and anti-inflammatory therapy for Alzheimer’s disease. Adv. Drug. Deliv. Rev. 2002, 54, 1627–1656. [Google Scholar] [CrossRef]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; Wyss-Coray, T. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Gibbons, H.M.; Dragunow, M. Valproic acid enhances microglial phagocytosis of amyloid-β1-42. Neuroscience 2010, 169, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Tan, H.P. Alginate-Based Biomaterials for Regenerative Medicine Applications. Materials. 2013, 6, 1285–1309. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, Y.; Xu, X.; Tamura, T.; Oda, T.; Muramatsu, T. Enzymatically depolymerized alginate oligomers that cause cytotoxic cytokine production in human mononuclear cells. Biosci. Biotechnol. Biochem. 2003, 67, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Tusi, S.K.; Khalaj, L.; Ashabi, G.; Kiaei, M.; Khodagholi, F. Alginate oligosaccharide protects against endoplasmic reticulum- and mitochondrial-mediated apoptotic cell death and oxidative stress. Biomaterials 2011, 32, 5438–5458. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Khodagholi, F.; Abdi, A.; Maghsoudi, N. Alginate protects NT2 neurons against H2O2-induced neurotoxicity. Carbohydr. Polym. 2010, 79, 1063–1072. [Google Scholar] [CrossRef]
- Xu, X.; Wu, X.T.; Wang, Q.Q.; Cai, N.; Zhang, H.; Jiang, Z.; Wan, M.; Oda, T. Immunomodulatory effects of alginate oligosaccharides on murine macrophage RAW264.7 cells and their structure-activity relationships. J. Agri. Food Chem. 2014, 62, 3168–3176. [Google Scholar]
- Xu, X.; Bi, D.-C.; Li, C.; Fang, W.-S.; Zhou, R.; Li, S.-M.; Chi, L.-L.; Wan, M.; Shen, L.-M. Morphological and proteomic analyses reveal that unsaturated guluronate oligosaccharide modulates multiple functional pathways in murine macrophage RAW264.7 cells. Mar. Drugs 2015, 13, 1798–1818. [Google Scholar] [CrossRef] [PubMed]
- Mirshafiey, A.; Khodadadi, A.; Rehm, B.H.; Khorramizadeh, M.R.; Eslami, M.B.; Razavi, A.; Saadat, F. Sodium alginate as a novel therapeutic option in experimental colitis. Scand. J. Immunol. 2005, 61, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.-J.; Son, E.-W.; Rhee, D.-K.; Pyo, S. Modulation of tnf-α-induced icam-1 expression, no and h202 production by alginate, allicin and ascorbic acid in human endothelial cells. Arch. Pharm. Res. 2003, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, J.; Guan, H. Preparation and characterization of oligomannuronates from alginate degraded by hydrogen peroxide. Carbohydr. Polym. 2004, 58, 115–121. [Google Scholar] [CrossRef]
- Zhou, R.; Shi, X.Y.; Gao, Y.; Cai, N.; Jiang, Z.D.; Xu, X. Anti-inflammatory activity of guluronate oligosaccharides obtained by oxidative degradation from alginate in lipopolysaccharide-activated murine macrophage RAW 264.7 cells. J. Agric. Food Chem. 2015, 63, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Zhu, L.H.; Jing, X.N.; Zeng, Z.F.; Liang, Y.R.; Xu, A.D.; Liu, J.; Xiao, S.H.; Yang, L.H.; Shi, Q.Y.; Guo, L.; Tao, E.X. Rifampicin improves neuronal apoptosis in LPS-stimulated co-cultured BV2 cells through inhibition of the TLR-4 pathway. Mol. Med. Rep. 2014, 10, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.M.; Sharma, S. Sinomenine inhibits microglial activation by Abeta and confers neuroprotection. J. Neuroinflammation 2011, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef] [PubMed]
- He, G.L.; Liu, Y.; Li, M.; Chen, C.H.; Gao, P.; Yu, Z.P.; Yang, X.S. The amelioration of phagocytic ability in microglial cells by curcumin through the inhibition of EMF-induced pro-inflammatory responses. J. Neuroinflammation 2014, 11, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Kurachi, M.; Nakashima, T.; Kim, D.; Yamaguchi, K.; Oda, T.; Iwamoto, Y.; Muramatsu, T. Structure-activity relationship of alginate oligosaccharides in the induction of cytokine production from RAW264.7 cells. FEBS Lett. 2005, 579, 4423–4429. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wu, Y.; Huang, H.; He, C.; Li, W.; Wang, H.; Chen, H.; Yin, Y. Biochanin A attenuates LPS-induced pro-inflammatory responses and inhibits the activation of the MAPK pathway in BV2 microglial cells. Int. J. Mol. Med. 2015, 35, 391–398. [Google Scholar] [PubMed]
- Essa, M.M.; Vijayan, R.K.; Castellano-Gonzalez, G.; Memon, M.A.; Braidy, N.; Guillemin, G.J. Neuroprotective Effect of Natural Products Against Alzheimer’s Disease. Neurochem. Res. 2012, 37, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Li, Y.H.; Cheng, W.J.; Zhou, L.; Shen, Y.; Wang, Y. Neuroprotective effects of Lycium barbarum polysaccharides in lipopolysaccharide-induced BV2 microglial cells. Mol. Med. Rep. 2013, 7, 1977–1981. [Google Scholar] [PubMed]
- Zhu, Y.Y.; Bickford, P.C.; Sanberg, P.; Giunta, B.; Tan, J. Blueberry opposes beta-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activation protein kinase. Rejuv. Res. 2008, 11, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.-Y.; Kim, G.-Y.; Choi, I.-W.; Kim, N.D.; Nam, T.-J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of NF-κB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Xin, X.L.; Gan, L.; Nie, Q.; Geng, M.Y. Determination of the accessibility of acidic oligosaccharide sugar chain to blood-brain barrier using surface plasmon resonance. Biol. Pharm. Bull. 2006, 29, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hu, J.F.; Li, J.; Yang, Z.; Xin, X.L.; Wang, J.; Ding, J.; Geng, M.Y. Effect of acidic oligosaccharide sugar chain on scopolamine-induced memory impairment in rats and its related mechanisms. Neurosci. Lett. 2005, 374, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain. Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Emmanouil, M.; Taoufik, E.; Tseveleki, V.; Vamvakas, S.-S.; Probert, L. A Role for Neuronal NF-κB in Suppressing Neuroinflammation and Promoting Neuroprotection in the CNS. In Advances in TNF Family Research; Wallach, D., Kovalenko, A., Feldmann, M., Eds.; Springer: New York, NY, USA, 2011; Volume 691, pp. 575–581. [Google Scholar]
- Cherry, J.; Olschowka, J.; O’Banion, M. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Halle, M.; Lampron, A.; Theriault, P.; Prefontaine, P.; Filali, M.; Tribout-Jover, P.; Lanteigne, A.M.; Jodoin, R.; Cluff, C.; et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. USA 2013, 110, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.L.; Kan, E.M.; Lu, J.; Hao, A.; Dheen, S.T.; Kaur, C.; Ling, E.-A. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: Role of TLR4 in hypoxic microglia. J. Neuroinflammation 2013, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhou, H.Y.; Xiong, B.; He, Y.; Yeung, E.S. Pericellular Matrix Enhances Retention and Cellular Uptake of Nanoparticles. J. Am. Chem. Soc. 2012, 134, 13404–13409. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.; Shi, X.-Y.; Bi, D.-C.; Fang, W.-S.; Wei, G.-B.; Xu, X. Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid. Mar. Drugs 2015, 13, 5828-5846. https://doi.org/10.3390/md13095828
Zhou R, Shi X-Y, Bi D-C, Fang W-S, Wei G-B, Xu X. Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid. Marine Drugs. 2015; 13(9):5828-5846. https://doi.org/10.3390/md13095828
Chicago/Turabian StyleZhou, Rui, Xu-Yang Shi, De-Cheng Bi, Wei-Shan Fang, Gao-Bin Wei, and Xu Xu. 2015. "Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid" Marine Drugs 13, no. 9: 5828-5846. https://doi.org/10.3390/md13095828
APA StyleZhou, R., Shi, X.-Y., Bi, D.-C., Fang, W.-S., Wei, G.-B., & Xu, X. (2015). Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid. Marine Drugs, 13(9), 5828-5846. https://doi.org/10.3390/md13095828