
A Pimarane Diterpene and Cytotoxic Angucyclines from a Marine-Derived Micromonospora sp. in Vietnam’s East Sea

Abstract
:
1. Introduction
2. Results and Discussion
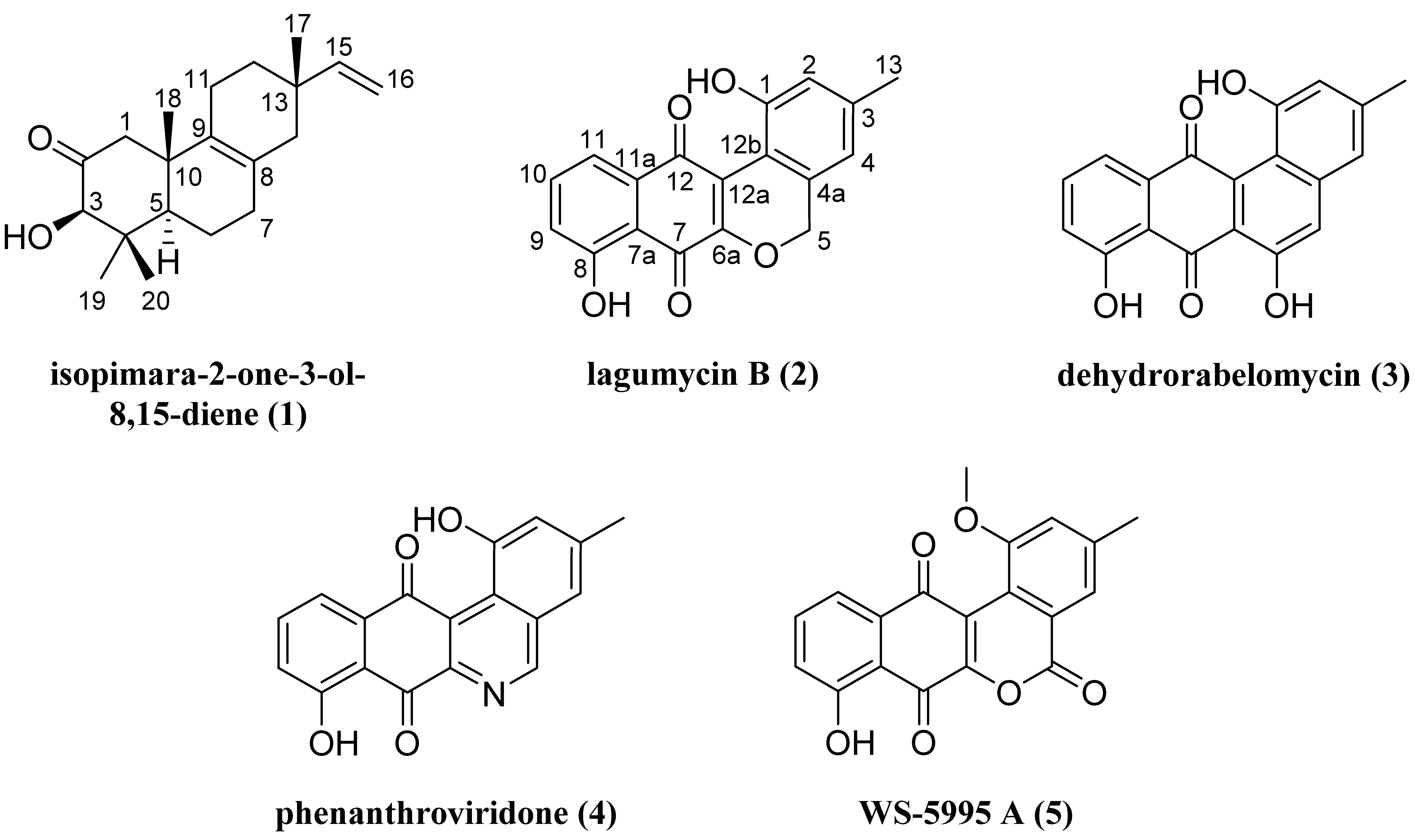
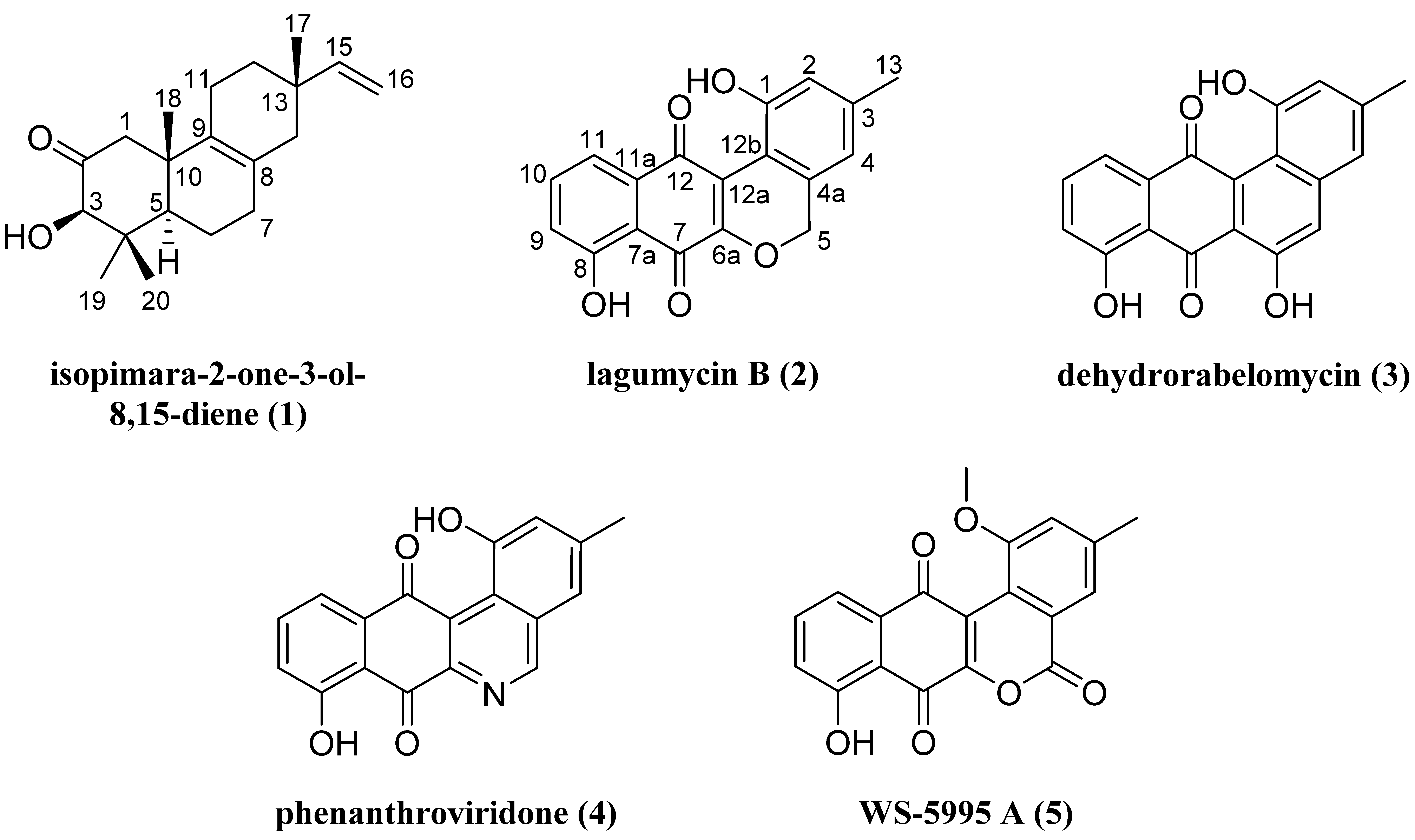
2.1. Structure Elucidation of Isopimara-2-one-3-ol-8,15-diene (1) and Lagumycin B (2)

{kind=link}
{kind=link}
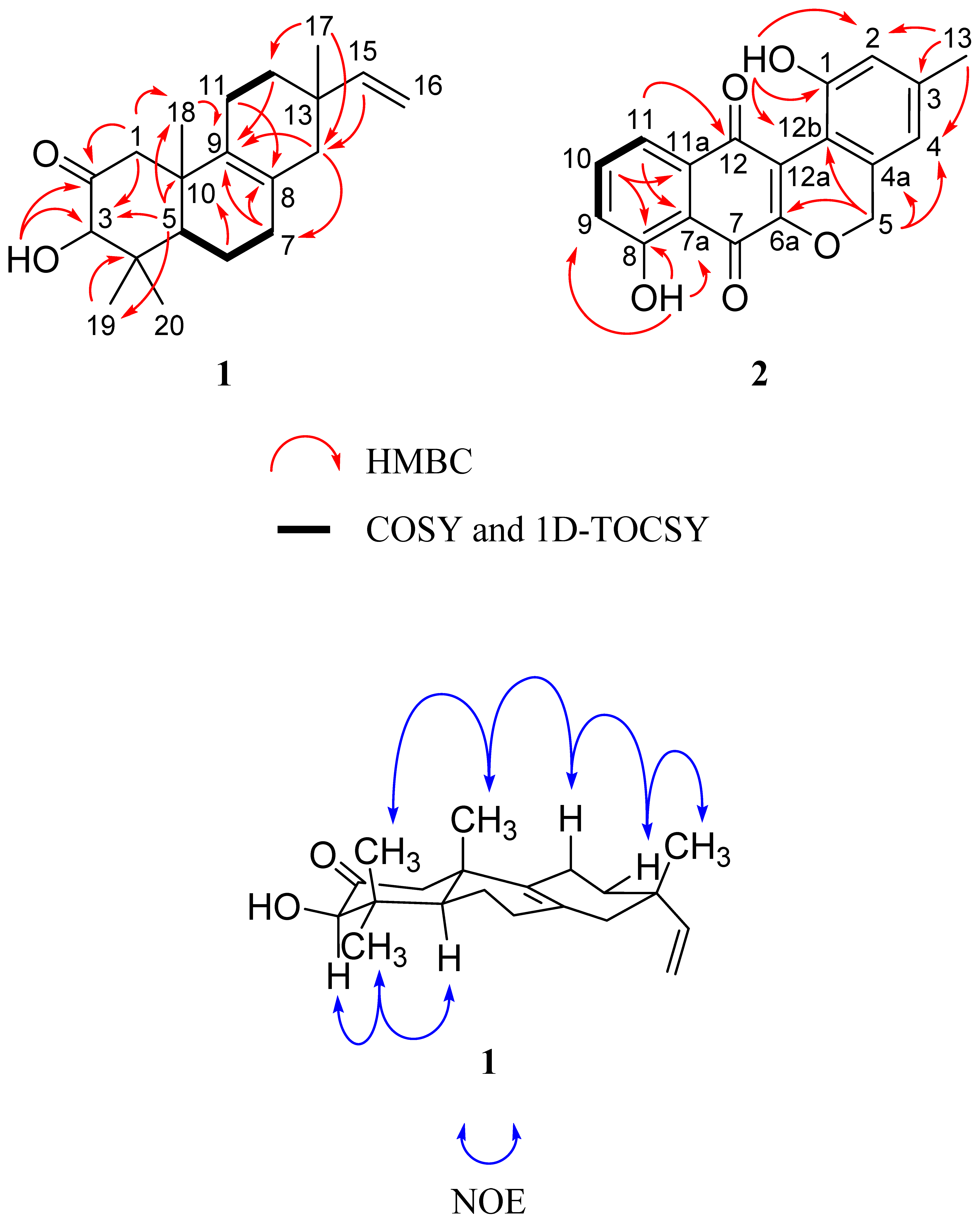{kind=link}
| Position | 13C, Type a | 1H, Mult. (J, Hz) b,c |
|---|---|---|
| 1ax | 49.8, CH2 | 2.25, d (12.3) |
| 1eq | 2.58, d (12.3) | |
| 2 | 211.8,C | |
| 3 | 83.0, CH | 3.91, d (4.0) |
| 3-OH | 3.44, d (4.0) | |
| 4 | 45.3, C | |
| 5 | 50.3, CH | 1.78, m |
| 6 | 18.8, CH2 | 1.58, m |
| 1.80, m | ||
| 7 | 32.3, CH2 | 2.03, m |
| 8 | 126.4, C | |
| 9 | 134.3, C | |
| 10 | 43.8, C | |
| 11ax | 21.4, CH2 | 1.76, m |
| 11eq | 1.88, m | |
| 12eq | 34.8, CH2 | 1.33, m |
| 12ax | 1.53, m | |
| 13 | 35.3, C | |
| 14 | 41.6, CH2 | 1.76, m |
| 1.88, m | ||
| 15 | 145.9, CH | 5.72, dd (17.5, 10.7) |
| 16 | 111.2, CH2 | 4.85, dd (17.5, 1.4) |
| 4.92, dd (10.7, 1.4) | ||
| 17 | 28.3, CH3 | 0.98, s |
| 18 | 20.6, CH3 | 0.93, s |
| 19 | 29.3, CH3 | 1.21, s |
| 20 | 16.5, CH3 | 0.72, s |

| Position | 13C, Type a | 1H, Mult. (J, Hz) b,c |
|---|---|---|
| 1 | 154.5, C | |
| 1-OH | 10.45, s | |
| 2 | 121.1, CH | 6.85, s |
| 3 | 143.7, C | |
| 4 | 118.2, CH | 6.54, s |
| 4a | 130.3, C | |
| 5 | 70.7, CH2 | 5.19, s |
| 6a | 157.0, C | |
| 7 | 183.4, C | |
| 7a | 113.5, C | |
| 8 | 161.7, C | |
| 8-OH | 11.70, s | |
| 9 | 125.2, CH | 7.30, d (8.1) |
| 10 | 137.2, CH | 7.66, t (8.1) |
| 11 | 121.3, CH | 7.81, d (8.1) |
| 11a | 132.2, C | |
| 12 | 186.6, C | |
| 12a | 124.4, C | |
| 12b | 109.9, C | |
| 13 | 21.3, CH3 | 2.33, s |
2.2. Cytotoxicity Evaluation of 1–5
| Compound | Cytotoxicity LC50 (µM) a | |||
|---|---|---|---|---|
| Kuramochi | OVCAR4 | MOSE | MOE | |
| 1 | >33.1 | >33.1 | >33.1 | >33.1 |
| 2 | >32.5 | >32.5 | 9.80 | 10.8 |
| 3 | 6.72 | 11.0 | 3.50 | 28.5 |
| 4 | 1.11 | 4.82 | 2.85 | 6.20 |
| 5 | 18.6 | 127 | >149 | >149 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection and Identification of Actinomycete Strain G039
3.3. Fermentation and Extraction
3.4. Isolation and Characterization of Isopimara-2-one-3-ol-8,15-diene (1) and Lagumycin B (2)
3.5. OVCAR4, Kuramochi, MOSE, MOE, and Vero Cytotoxicity Assays
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: Marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Ma, M.; Rateb, M.E.; Shaaban, K.A.; Yu, Z.; Huang, S.-X.; Zhao, L.-X.; Zhu, X.; Yan, Y.; Peterson, R.M.; et al. Biosynthetic potential-based strain prioritization for natural product discovery: A showcase for diterpenoid-producing actinomycetes. J. Nat. Prod. 2014, 77, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Smanski, M.J.; Peterson, R.M.; Huang, S.-X.; Shen, B. Bacterial diterpene synthases: New opportunities for mechanistic enzymology and engineered biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mullowney, M.W.; Hwang, C.H.; Newsome, A.G.; Wei, X.; Tanouye, U.; Wan, B.; Carlson, S.; Barranis, N.J.; Ó hAinmhire, E.; Chen, W.-L.; et al. Diaza-anthracene antibiotics from a freshwater-derived actinomycete with selective antibacterial activity toward Mycobacterium tuberculosis. ACS Infect. Dis. 2015, 1, 168–174. [Google Scholar] [CrossRef]
- Mullowney, M.W.; Ó hAinmhire, E.; Shaikh, A.; Wei, X.; Tanouye, U.; Santarsiero, B.D.; Burdette, J.E.; Murphy, B.T. Diazaquinomycins E–G, novel diaza-anthracene analogs from a marine-derived Streptomyces sp. Mar. Drugs 2014, 12, 3574–3586. [Google Scholar] [CrossRef] [PubMed]
- Balk-Bindseil, W.; Laatsch, H. Neue Naturstoffe aus dem Screening Mariner Actinomyceten: Isolierung und Strukturaufklärung der Lagumycine und des Oceamycins. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 1995. [Google Scholar]
- Yamashita, N.; Takashi, H.; Kazuo, S.; Haruo, S. 6-Hydroxytetrangulol, a new CPP32 protease inducer produced by Streptomyces sp. J. Antibiot. 1998, 1, 79–81. [Google Scholar] [CrossRef]
- Liu, W.C.; Parker, L.; Slusarchyk, S.; Greenwood, G.L.; Grahm, S.F.; Meyers, E. Isolation, characterization, and structure of rabelomycin, a new antibiotic. J. Antibiot. 1970, 23, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, G.; Zimmermann, W.; Gruner, J.; Auden, J.A.L. Phenanthridine Derivatives, Process for the Preparation Thereof, and Compositions Containing Them. U.S. Patent 5,093, 3 March 1990. [Google Scholar]
- Gore, M.P.; Gould, S.J.; Weller, D.D. Synthesis of putative intermediates in the biosynthesis of the kinamycin antibiotics: Total synthesis of phenanthroviridin aglycon and related compounds. J. Org. Chem. 1992, 57, 2774–2783. [Google Scholar] [CrossRef]
- Ikushima, H.; Iguchi, E.; Kohsaka, M.; Aoki, H.; Imanaka, H. Streptomyces auranticolor sp. nov., a new anticoccidial antibiotics producer. J. Antibiot. 1980, 33, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Okamoto, M.; Tanaka, H.; Ohe, O.; Kohsaka, M.; Aoki, H.; Imanaka, H. New anticoccidial antibiotics, WS-5995 A and B: I. Isolation and characterization. J. Antibiot. 1980, 33, 1107–1113. [Google Scholar] [PubMed]
- Crews, P.; Rodríguez, J.; Jaspars, M. Organic structure Analysis, 2nd ed.; Oxford University Press: New York, NJ, USA, 2010; p. 48. [Google Scholar]
- Moffitt, W.; Woodward, R.B.; Moscowitz, A.; Klyne, W.; Djerassi, C. Structure and the optical rotatory dispersion of saturated ketones. J. Am. Chem. Soc. 1961, 83, 4013–4018. [Google Scholar] [CrossRef]
- Dawson, B.A.; Girard, M.; Kindack, D.; Fillion, J.; Awang, D.V.C. 13C NMR of lapachol and some related naphthoquinones. Magn. Reson. Chem. 1989, 27, 1176–1177. [Google Scholar] [CrossRef]
- Yamashita, M.; Kaneko, M.; Tokuda, H.; Nishimura, K.; Kumeda, Y.; Iida, A. Synthesis and evaluation of bioactive naphthoquinones from the Brazilian medicinal plant, Tabebuia avellanedae. Bioorg. Med. Chem. 2009, 17, 6286–6291. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lombó, F.; Abdelfattah, M.S.; Braña, A.F.; Salas, J.A.; Rohr, J.; Méndez, C. Elucidation of oxygenation steps during oviedomycin biosynthesis and generation of derivatives with increased antitumor activity. ChemBioChem 2009, 10, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N. Manufacture of Dehydrorabelomycin with Streptomyces Species and Use of the Antibiotic Agent as Antitumor Agent. JP 10201494 A, 4 August 1998. [Google Scholar]
- Zhang, W.; Liu, Z.; Li, S.; Lu, Y.; Chen, Y.; Zhang, H.; Zhang, G.; Zhu, Y.; Zhang, G.; Zhang, W.; et al. Fluostatins I–K from the South China Sea-derived Micromonospora rosaria SCSIO N160. J. Nat. Prod. 2012, 75, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Perchellet, E.M.; Sperfslage, B.J.; Qabaja, G.; Jones, G.B.; Perchellet, J.P. Quinone isomers of the WS-5995 antibiotics: Synthetic antitumor agents that inhibit macromolecule synthesis, block nucleoside transport, induce DNA fragmentation, and decrease the growth and viability of L1210 leukemic cells more effectively than ellagic acid and genistein in vitro. Anticancer Drugs 2001, 12, 401–417. [Google Scholar] [PubMed]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef] [PubMed]
- Seaton, P.J.; Gould, S.J. Kinamycin biosynthesis. Derivation by excision of an acetate unit from a single-chain decaketide intermediate. J. Am. Chem. Soc. 1987, 109, 5282–5284. [Google Scholar] [CrossRef]
- Cone, M.C.; Hassan, A.M.; Gore, M.P.; Gould, S.J.; Borders, D.B.; Alluri, M.R. Detection of phenanthroviridin aglycon in a UV mutant of Streptomyces murayamaensis. J. Org. Chem. 1994, 59, 1923–1924. [Google Scholar] [CrossRef]
- Tibrewal, N.; Pahari, P.; Wang, G.; Kharel, M.K.; Morris, C.; Downey, T.; Hou, Y.; Bugni, T.S.; Rohr, J. Baeyer–Villiger C–C bond cleavage reaction in gilvocarcin and jadomycin biosynthesis. J. Am. Chem. Soc. 2012, 134, 18181–18184. [Google Scholar] [CrossRef] [PubMed]
- Cone, M.C.; Seaton, P.J.; Halley, K.A.; Gould, S.J. New products related to kinamycin from Streptomyces murayamaensis. I. Taxonomy, production, isolation, and biological properties. J. Antibiot. 1989, 42, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.Y.; Qu, Z.; Lin, H.P.; Li, L.; Hong, K. Micromonospora haikouensis sp. nov., isolated from mangrove soil. Antonie Leeuwenhoek 2012, 101, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Ó hAinmhire, E.; Quartuccio, S.M.; Cheng, W.; Ahmed, R.A.; King, S.M.; Burdette, J.E. Mutation or loss of p53 differentially modifies TGFβ action in ovarian cancer. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullowney, M.W.; Ó hAinmhire, E.; Tanouye, U.; Burdette, J.E.; Pham, V.C.; Murphy, B.T. A Pimarane Diterpene and Cytotoxic Angucyclines from a Marine-Derived Micromonospora sp. in Vietnam’s East Sea. Mar. Drugs 2015, 13, 5815-5827. https://doi.org/10.3390/md13095815
Mullowney MW, Ó hAinmhire E, Tanouye U, Burdette JE, Pham VC, Murphy BT. A Pimarane Diterpene and Cytotoxic Angucyclines from a Marine-Derived Micromonospora sp. in Vietnam’s East Sea. Marine Drugs. 2015; 13(9):5815-5827. https://doi.org/10.3390/md13095815
Chicago/Turabian StyleMullowney, Michael W., Eoghainín Ó hAinmhire, Urszula Tanouye, Joanna E. Burdette, Van Cuong Pham, and Brian T. Murphy. 2015. "A Pimarane Diterpene and Cytotoxic Angucyclines from a Marine-Derived Micromonospora sp. in Vietnam’s East Sea" Marine Drugs 13, no. 9: 5815-5827. https://doi.org/10.3390/md13095815
APA StyleMullowney, M. W., Ó hAinmhire, E., Tanouye, U., Burdette, J. E., Pham, V. C., & Murphy, B. T. (2015). A Pimarane Diterpene and Cytotoxic Angucyclines from a Marine-Derived Micromonospora sp. in Vietnam’s East Sea. Marine Drugs, 13(9), 5815-5827. https://doi.org/10.3390/md13095815