
A New Meroditerpene and a New Tryptoquivaline Analog from the Algicolous Fungus Neosartorya takakii KUFC 7898


 , , ,
, , ,  and
and 
Abstract
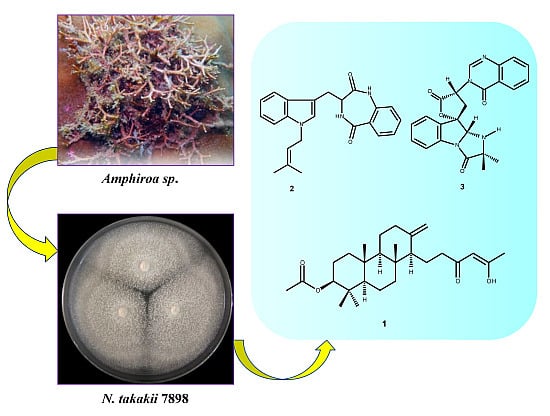
:
1. Introduction
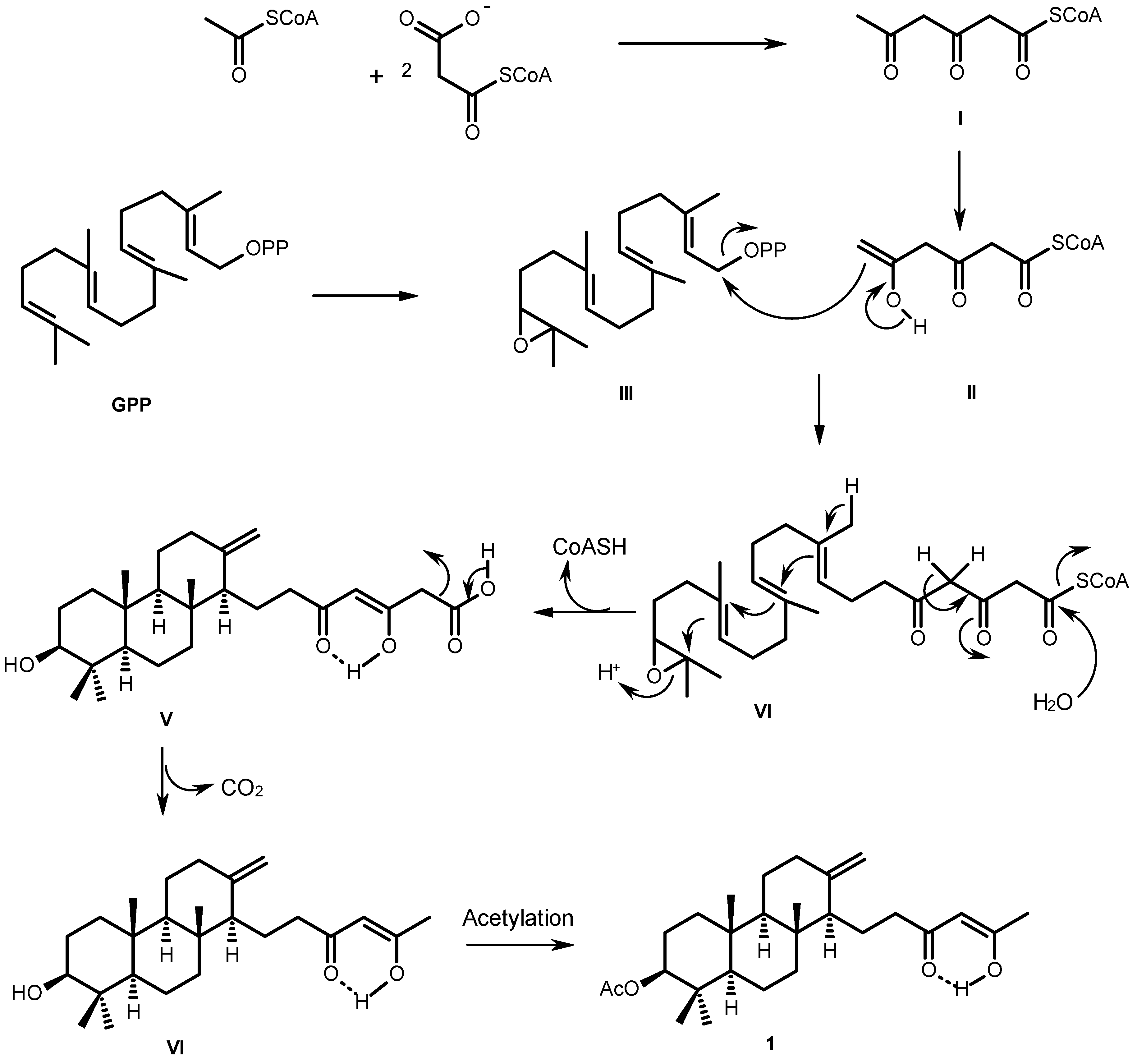
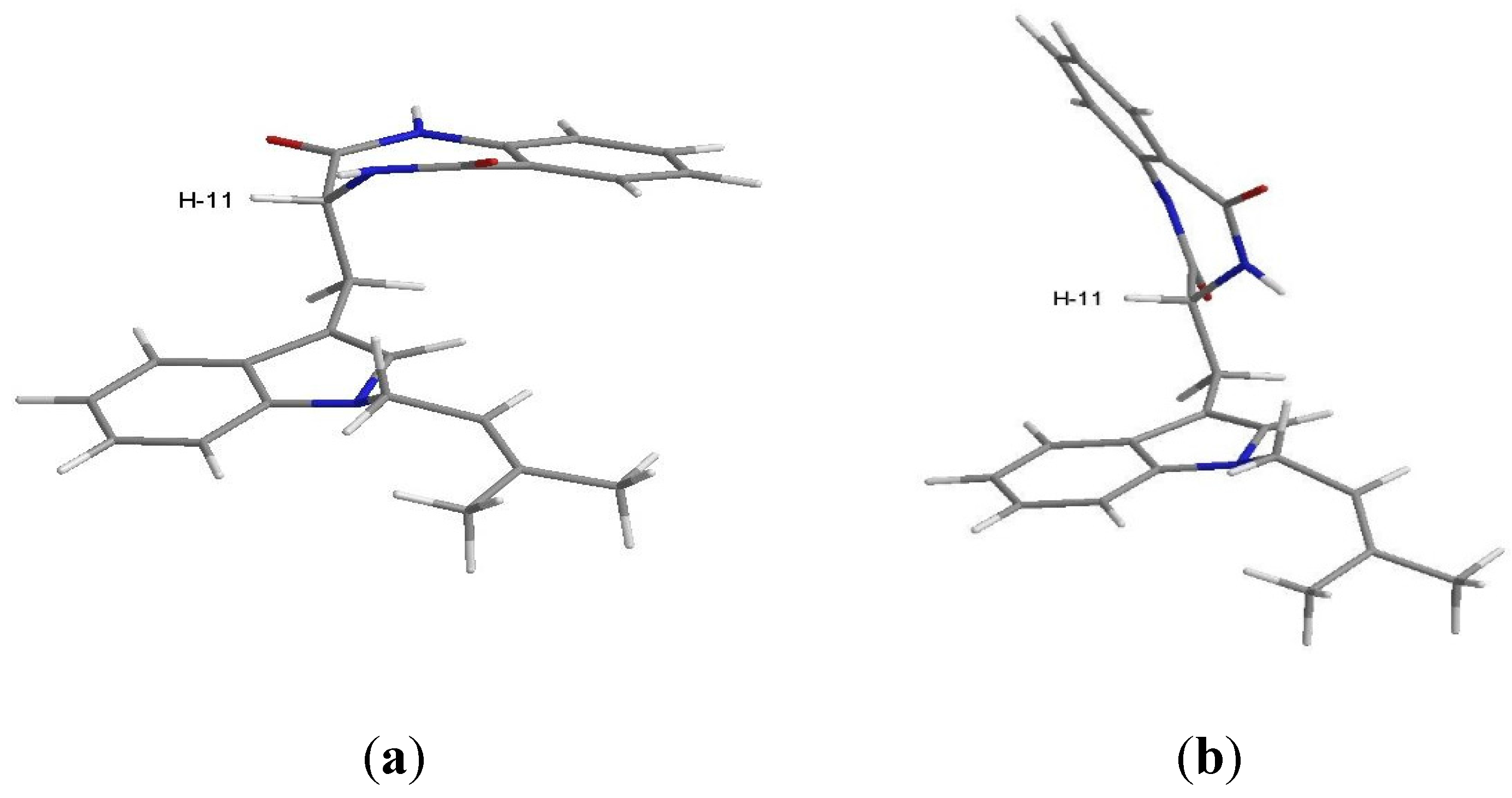
2. Results and Discussion
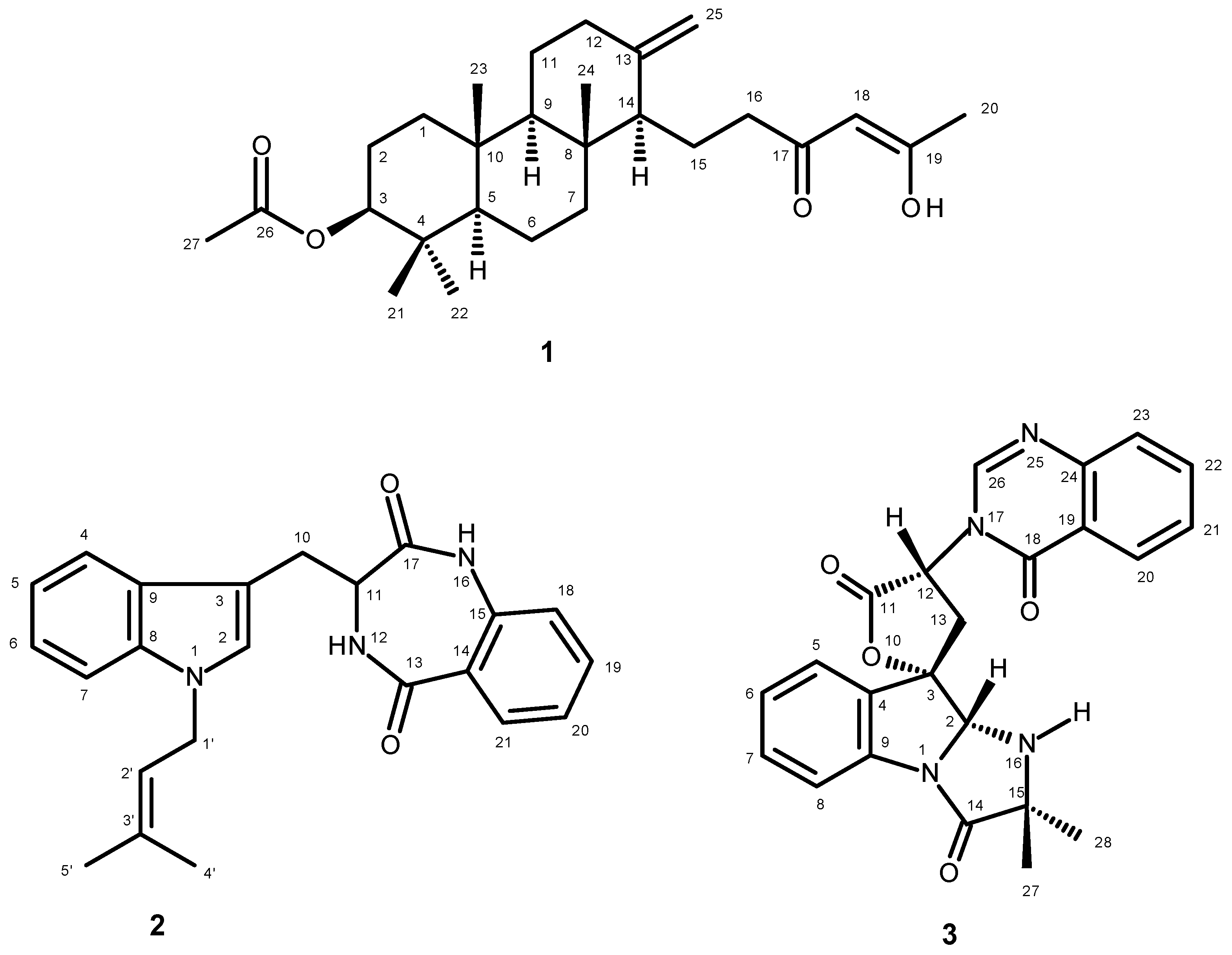

{kind=link}
{kind=link}
{kind=link}
{kind=link}
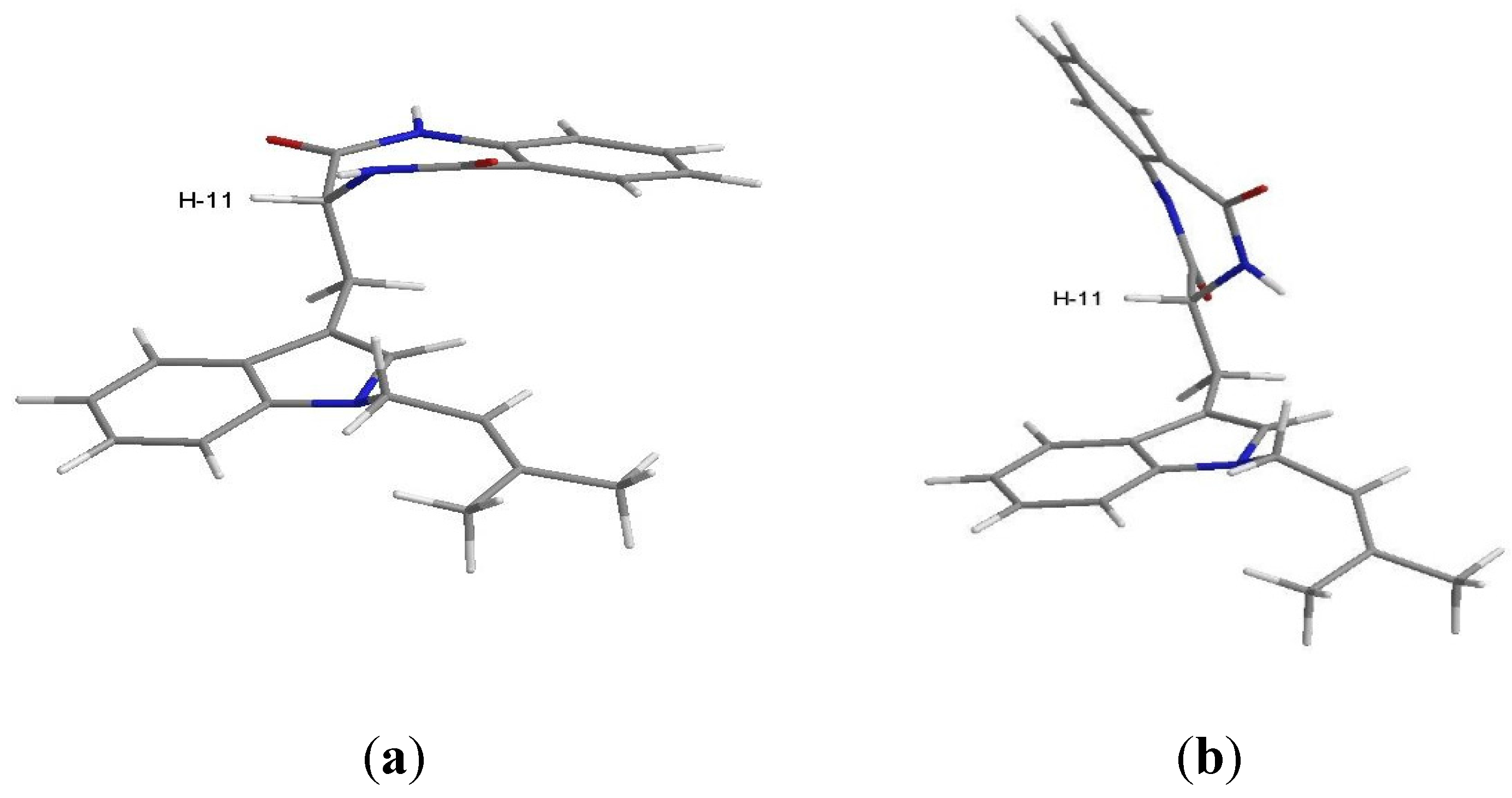{kind=link}
{kind=link}
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 38.2, CH2 | 1.05, m | H-2 | |
| 2 | 23.3, CH2 | 1.65, m | H-1, 3 | |
| 1.33, dd (12.9, 4.2) | H-1, 3 | C-4 | ||
| 3 | 80.8, CH | 4.48, dd (10.9, 4.6) | H-2 | C-1, 4, 21, 22 |
| 4 | 37.8, C | - | ||
| 5 | 55.4, CH | 0.91, dd (12.0, 2.2) | H-6 | |
| 6 | 18.7, CH2 | 1.62, m | H-5 | |
| 1.14, m | ||||
| 7 | 40.5, CH2 | 1.18, dd (12.5, 3.6) | ||
| 1.88, m | ||||
| 8 | 39.8, C | - | ||
| 9 | 59.8, CH | 1.02, dd (12.3, 2.6) | ||
| 10 | 37.4, C | - | ||
| 11 | 23.6, CH2 | 1.70, m | ||
| 12 | 38.0, CH2 | 2.38, m | ||
| 1.92, m | C-14, 25 | |||
| 13 | 147.7, C | - | ||
| 14 | 56.4, CH | 1.59, m | H-15 | |
| 15 | 19.6, CH2 | 1.86, m | H-14, 16 | C-13 |
| 16 | 37.2, CH2 | 2.08, m | H-15 | |
| 17 | 194.7, CO | - | ||
| 18 | 99.9, CH | 5.45, s | C-16, 17, 19, 20 | |
| 19 | 191.1, C | - | ||
| 20 | 24.9, CH3 | 2.05, s | C-18, 19 | |
| 21 | 16.3, CH3 | 0.83, s | C-3, 4, 5, 22 | |
| 22 | 28.0, CH3 | 0.86, s | C-3, 4, 5, 21 | |
| 23 | 16.4, CH3 | 0.84, s | C-1, 5, 9, 10 | |
| 24 | 15.3, CH3 | 0.69, s | C-7, 8, 9, 14 | |
| 25a | 106.4, CH2 | 4.84, brs | C-12, 14 | |
| b | 4.50, brs | C-12, 13, 14 | ||
| 26 | 171.0, CO | - | ||
| 27 | 21.3, CH3 | 2.05, s | C-26 | |
| OH-19 | 15.47, s |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 2 | 127.3, CH | 7.15, s | C-3, 8, 9 | |
| 3 | 108.0, C | - | ||
| 4 | 118.4, CH | 7.54, d (7.8) | H-5 | C-3, 6, 8 |
| 5 | 119.2, CH | 7.08, ddd (7.8, 7.8, 0.7) | H-4, 6 | C-7, 9 |
| 6 | 121.7, CH | 7.20, ddd (7.8, 7.8, 0.7) | H-5, 7 | C-4, 8 |
| 7 | 109.9, CH | 7.31, d (7.8) | H-6 | C-5, 9 |
| 8 | 136.3, C | - | ||
| 9 | 127.9, C | - | ||
| 10 | 22.4, CH2 | 3.57, dd (15.2, 5.5) | H-11 | C-2, 3, 17 |
| 3.26, dd (15.2, 8.3) | H-11 | C-2, 3, 17 | ||
| 11 | 52.4, CH | 4.12, dt (8.3, 5.5) | H-10, NH-12 | |
| 13 | 168.9, CO | - | ||
| 14 | 125.5, C | - | ||
| 15 | 135.7, C | - | ||
| 17 | 172.0, CO | |||
| 18 | 121.0, CH | 7.06, d (8.0) | H-19 | C-14, 20 |
| 19 | 133.1, CH | 7.50, ddd (8.0, 8.0, 1.5) | H-18, 20 | C-15, 21 |
| 20 | 125.2, CH | 7.24, dd (8.0, 8.0) | H-19, 21 | C-14, 18 |
| 21 | 131.4, CH | 7.91, dd (8.0, 1.5) | H-20 | C-13, 19, 15 |
| 1′ | 44.2, CH2 | 4.63, d (6.8) | H-2′ | C-2, 2′, 3′ |
| 2′ | 119.9, CH | 5.35, m | H-1′, 4′, 5′ | |
| 3′ | 136.4, C | - | ||
| 4′ | 25.6, CH3 | 1.74, s | H-1′, 2′ | C-2′, 3′, 5′ |
| 5′ | 18.1, CH3 | 1.80, s | H-1′, 2′ | C-2′, 3′, 5′ |
| NH-12 | 7.03, d (5.5) | H-11 | ||
| NH-16 | 9.03, s | C-11, 14 |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 2 | 82.0, CH | 5.55, d (8.4) | NH-16 | C-13, 14 |
| 3 | 84.7, C | - | ||
| 4 | 132.0, C | - | ||
| 5 | 125.7, CH | 7.71, d (7.3) | H-6 | C-7, 9 |
| 6 | 125.7, CH | 7.38, ddd (7.5, 7.5, 1.2) | H-5, 7 | C-4, 8 |
| 7 | 131.6, CH | 7.57, ddd (8.1, 7.7, 1.2) | H-6, 8 | C-5, 9 |
| 8 | 116.2, CH | 7.49, d (7.2) | H-7 | C-4, 6 |
| 9 | 139.8, C | - | ||
| 11 | 170.7, CO | - | ||
| 12 | 56.9, CH | 5.58, dd (10.8, 9.1 | H-13 | C-3, 11, 13, 18, 26 |
| 13 | 31.6, CH2 | 2.86, dd (12.9, 9.1) | H-12 | C-2, 4, 11, 12 |
| 3.45, dd (12.9, 11.2) | H-12 | C-2, 3, 4, 12 | ||
| 14 | 176.0, CO | - | ||
| 15 | 64.6, C | - | ||
| 16 | - | 3.76, d (8.4) | H-2 | C-2, 3, 14, 15, 26, 27 |
| 18 | 159.6, CO | - | ||
| 19 | 121.4, C | - | ||
| 20 | 126.1, CH | 8.23, dd (8.0, 1.2) | H-21 | C-18, 22, 24 |
| 21 | 127.6, CH | 7.63, ddd (7.6, 7.6, 1.0) | H-20, 22 | C-19, 23 |
| 22 | 135.0, CH | 7.92, ddd (8.2, 8.2, 1.5) | H-21, 23 | C-20, 24 |
| 23 | 127.3, CH | 7.76, d (7.7) | H-22 | C-19, 21 |
| 24 | 147.5, C | - | ||
| 26 | 147.4, CH | 8.49, s | C-12, 18, 24 | |
| 27 | 26.5, CH3 | 1.45, s | C-14, 15, 28 | |
| 28 | 26.9, CH3 | 1.24, s | C-14, 15, 27 |
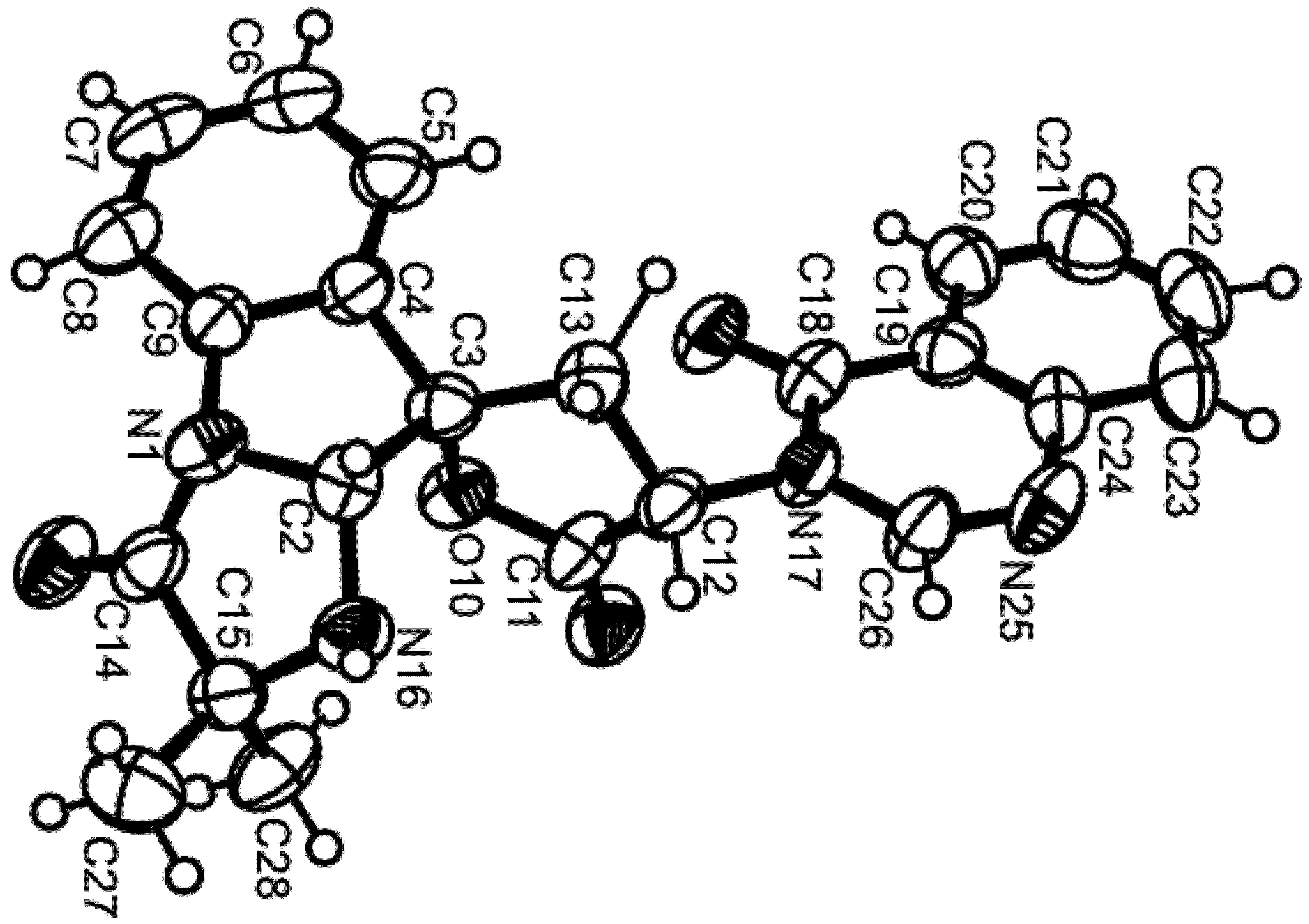
3. Experimental Section
3.1. General Procedure
3.2. Extraction and Isolation
3.2.1. Satorenol (1)
3.2.2. Takakiamide (2)
3.2.3. Tryptoquivaline U (3)
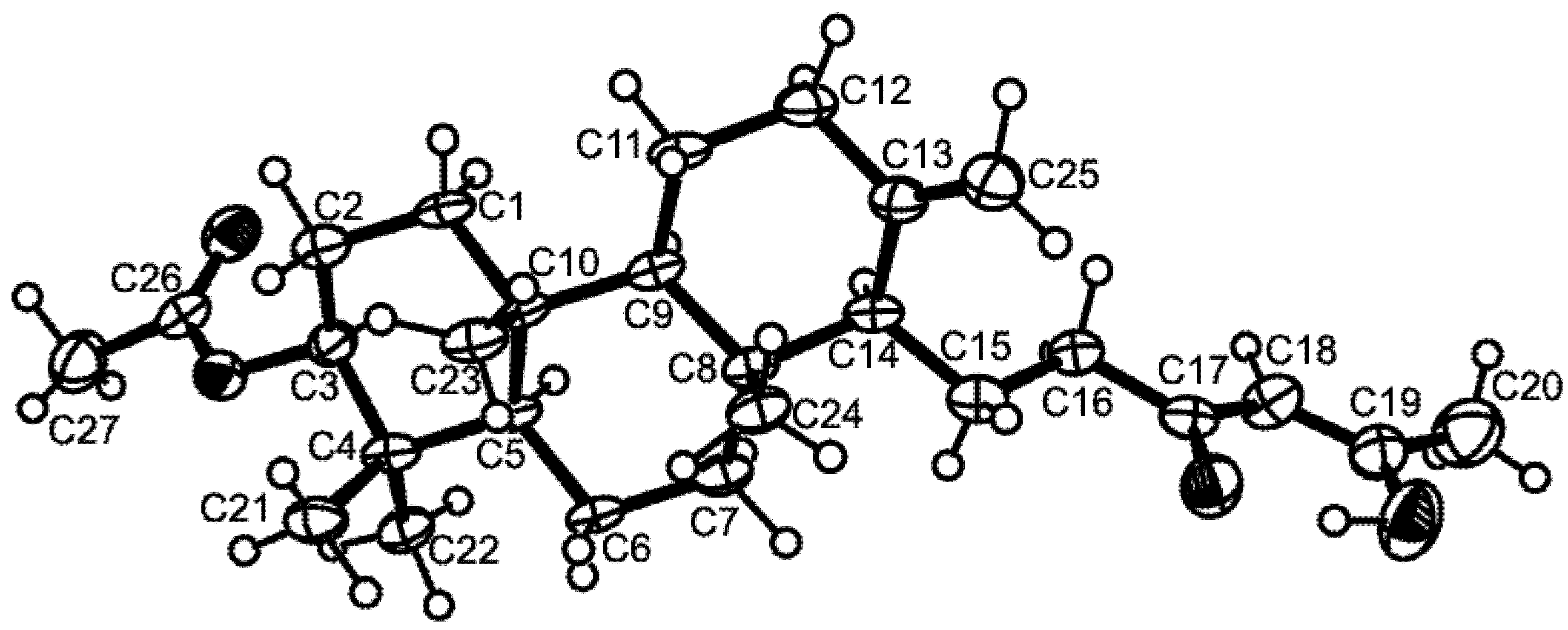
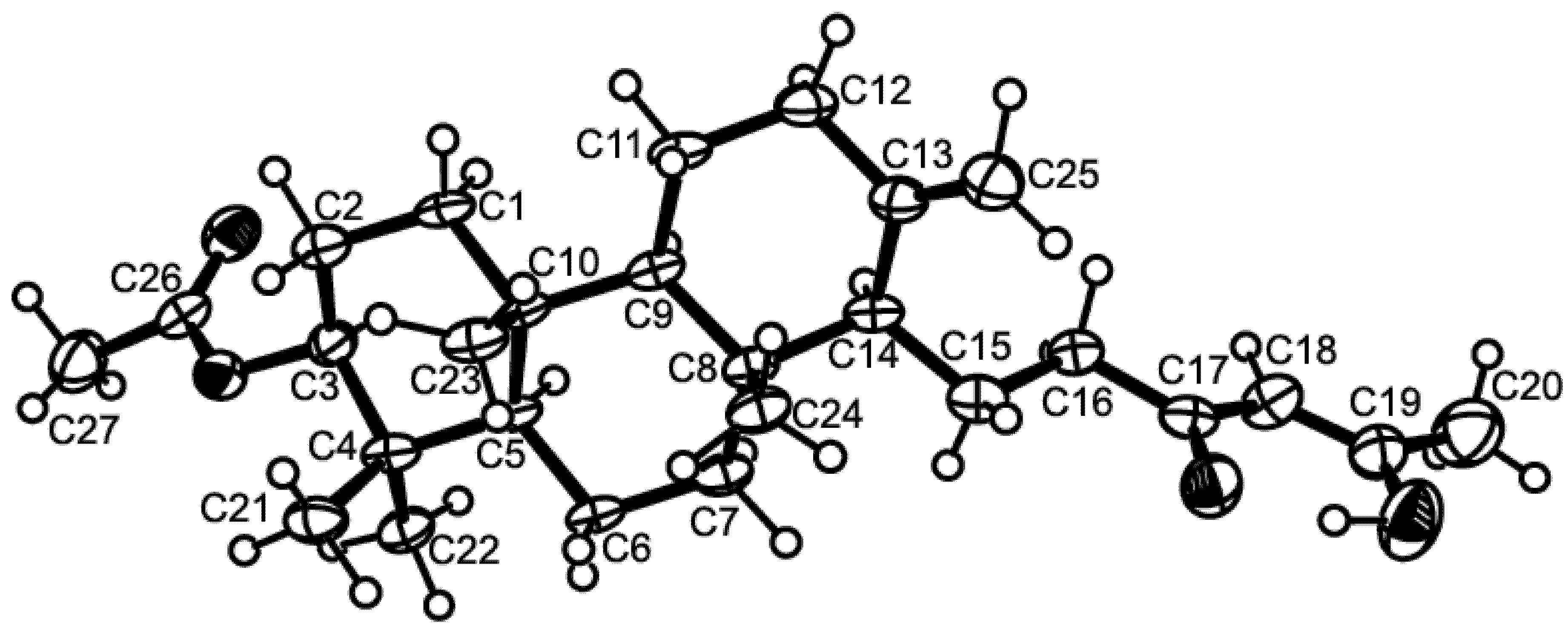
3.3. X-Ray Crystal Structure of Sartorenol (1)
3.4. X-Ray Crystal Structure of Tryptoquivaline U (3)
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Damare, S.; Singh, P.; Raghukumar, S. Biotechnology of Marine Fungi. In Biology of Marine Fungi; Raghukumar, C., Ed.; Springer: Heidelberg, Germany, 2012; pp. 277–297. [Google Scholar]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Ali, M.S.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R. Marine microorganisms: A new biomedical resource. In Marine Biotechnology; Attaway, D.H., Zaborsky, O.R., Eds.; Plenum Press: New York, NY, USA, 1993; Volume 1, pp. 2419–2457. [Google Scholar]
- Eamvijarn, A.; Gomes, N.M.; Dethoup, T.; Buaruang, J.; Manoch, L.; Silva, A.; Pedro, M.; Marini, I.; Roussis, V.; Kijjoa, A. Bioactive meroditerpenes and indole alkaloids from the soil fungus Neosartorya fischeri (KUFC 6344), and the marine-derived fungi Neosartorya laciniosa (KUFC 7896) and Neosartorya tsunodae (KUFC 9213). Tetrahedron 2013, 69, 8583–8591. [Google Scholar] [CrossRef]
- Gomes, N.M.; Bessa, L.J.; Buttachon, S.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. Antibacterial and Antibiofilm Activities of Tryptoquivalines and Meroditerpenes Isolated from the Marine-Derived Fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the Soil Fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Kanokmedhakul, K.; Kanokmedhakul, S.; Suwannatrai, R.; Soytong, K.; Prabpai, S.; Kongsaeree, P. Bioactive meroterpenoids and alkaloids from the fungus Eurotium chevalieri. Tetrahedron 2011, 67, 5461–5468. [Google Scholar] [CrossRef]
- Buttachon, S.; Chandrapatya, A.; Manoch, L.; Silva, A.; Gales, L.; Bruyére, C.; Kiss, R.; Kijjoa, A. Sartorymensin, a new indole alkaloid, and new analogues of tryptoquivaline and fiscalins produced by Neosartorya siamensis (KUFC 6349). Tetrahedron 2012, 68, 3253–3262. [Google Scholar] [CrossRef]
- Shimada, A.; Kusano, M.; Takeuchi, S.; Fujioka, S.; Inokuchi, T.; Kimura, Y. Aspterric acid and 6-hydroxymellein, inhibitors of pollen development in Arabidopsis thaliana, produced by Aspergillus terreus. Z. Naturforsch. 2002, 57, 459–464. [Google Scholar] [CrossRef]
- PubChem Substance. Available online: http://pubchem.ncbi.nlm.nih.gov/substance/185030170 (accessed on 25 April 2015).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- McLean, R.J.C.; Pearson, L.S., III; Fuqua, C. A simple screening protocol for the identification of quorum signal antagonists. J. Microbiol. Methods 2004, 58, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Abliz, P.; Fukushima, K.; Okada, K.; Gusmão, N.B. Neosartorya takakii, a new species from soil in Brazil. Mycoscience 2001, 42, 91–95. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Horie, Y.; Abliz, P.; Gonoi, T.; Yaguchi, T. Aspergillus huiyaniae sp. nov., a teleomorphic species in sect. Fumigati isolated from desert soil in China. Mycoscience 2014, 55, 213–220. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for Basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zin, W.W.M.; Buttachon, S.; Buaruang, J.; Gales, L.; Pereira, J.A.; Pinto, M.M.M.; Silva, A.M.S.; Kijjoa, A. A New Meroditerpene and a New Tryptoquivaline Analog from the Algicolous Fungus Neosartorya takakii KUFC 7898. Mar. Drugs 2015, 13, 3776-3790. https://doi.org/10.3390/md13063776
Zin WWM, Buttachon S, Buaruang J, Gales L, Pereira JA, Pinto MMM, Silva AMS, Kijjoa A. A New Meroditerpene and a New Tryptoquivaline Analog from the Algicolous Fungus Neosartorya takakii KUFC 7898. Marine Drugs. 2015; 13(6):3776-3790. https://doi.org/10.3390/md13063776
Chicago/Turabian StyleZin, War War May, Suradet Buttachon, Jamrearn Buaruang, Luís Gales, José A. Pereira, Madalena M. M. Pinto, Artur M. S. Silva, and Anake Kijjoa. 2015. "A New Meroditerpene and a New Tryptoquivaline Analog from the Algicolous Fungus Neosartorya takakii KUFC 7898" Marine Drugs 13, no. 6: 3776-3790. https://doi.org/10.3390/md13063776
APA StyleZin, W. W. M., Buttachon, S., Buaruang, J., Gales, L., Pereira, J. A., Pinto, M. M. M., Silva, A. M. S., & Kijjoa, A. (2015). A New Meroditerpene and a New Tryptoquivaline Analog from the Algicolous Fungus Neosartorya takakii KUFC 7898. Marine Drugs, 13(6), 3776-3790. https://doi.org/10.3390/md13063776