
Determination of the Chemical Structures of Tandyukisins B–D, Isolated from a Marine Sponge-Derived Fungus

 ,
,
Abstract
:1. Introduction
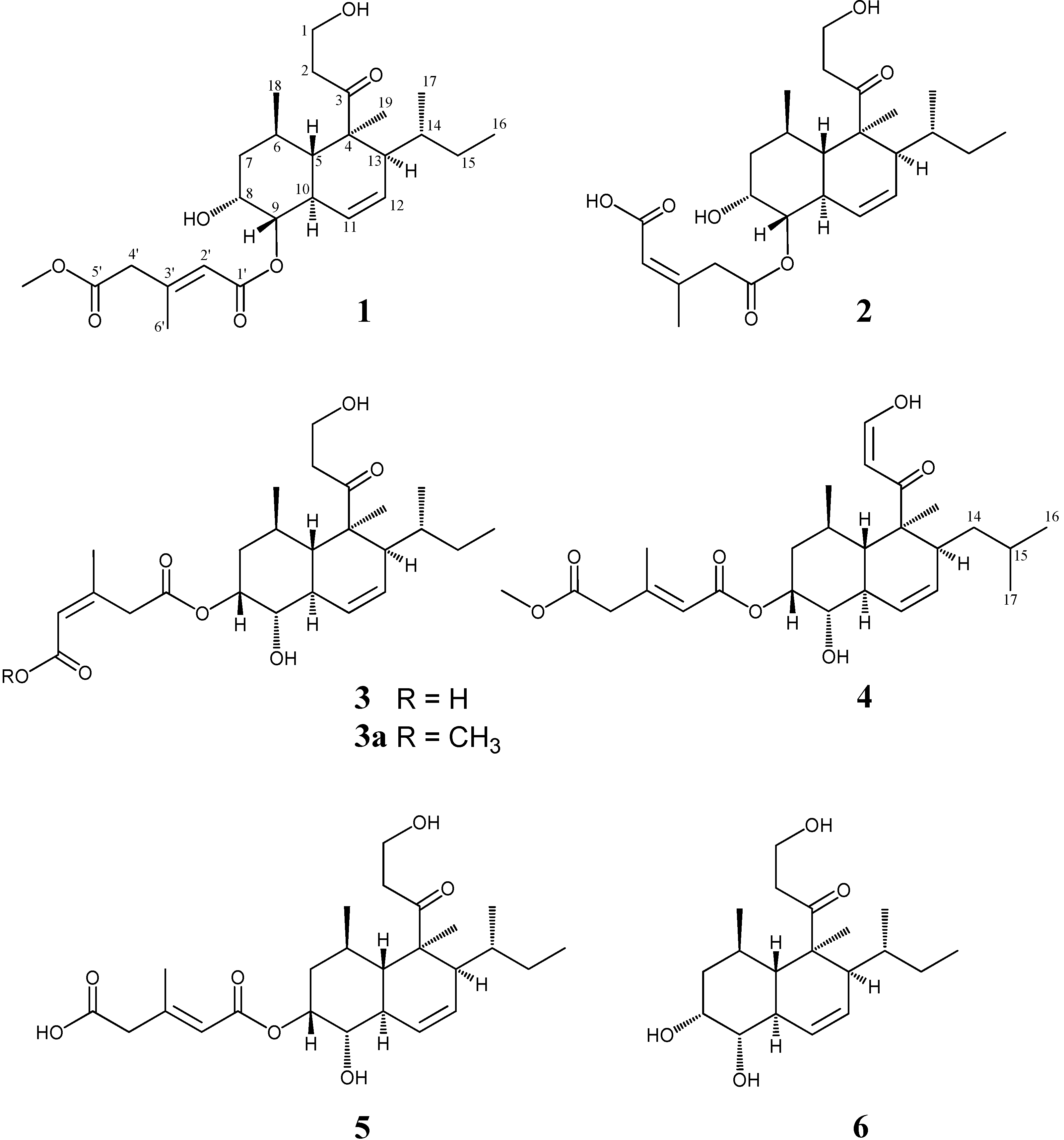
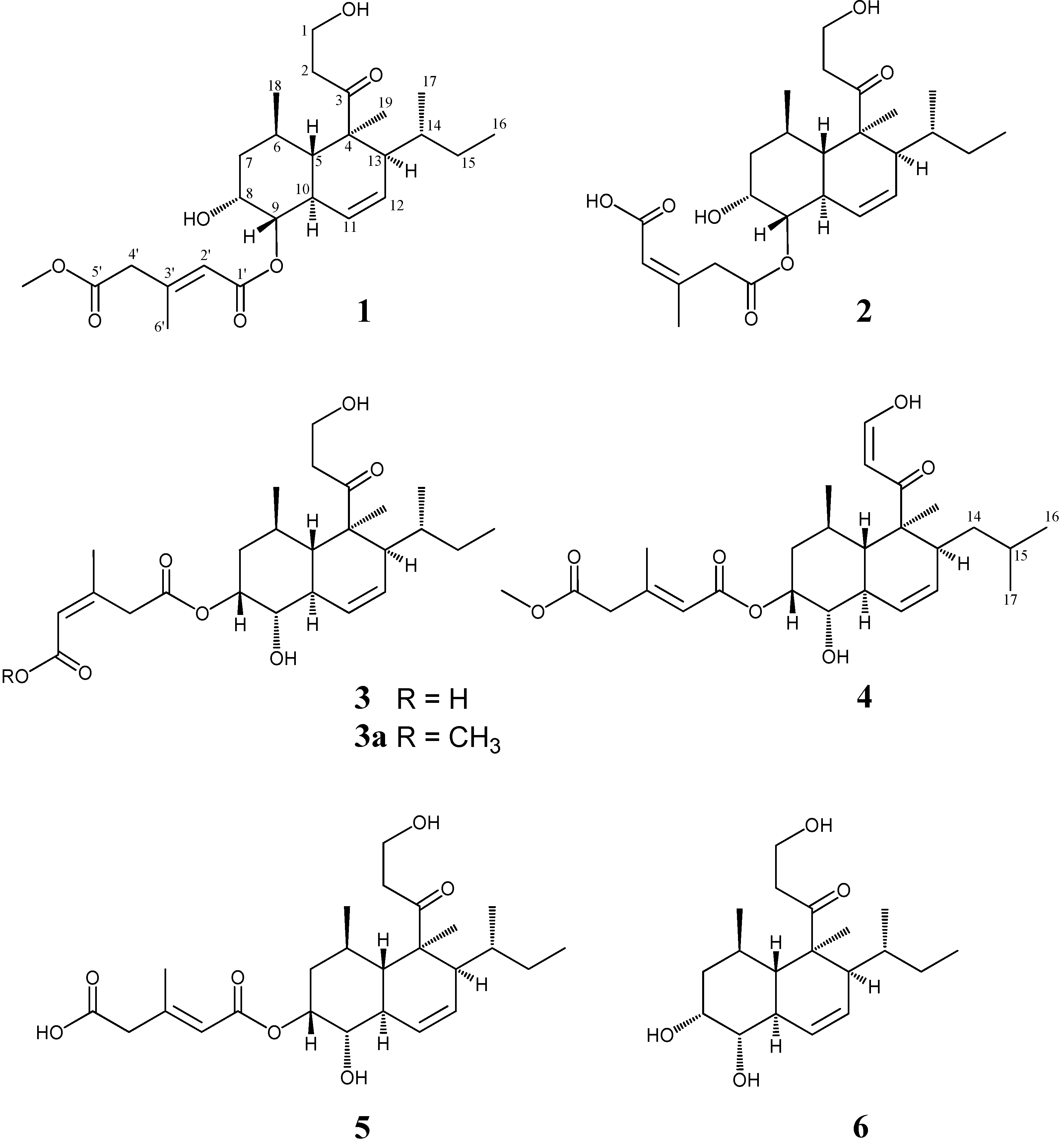
2. Results and Discussion

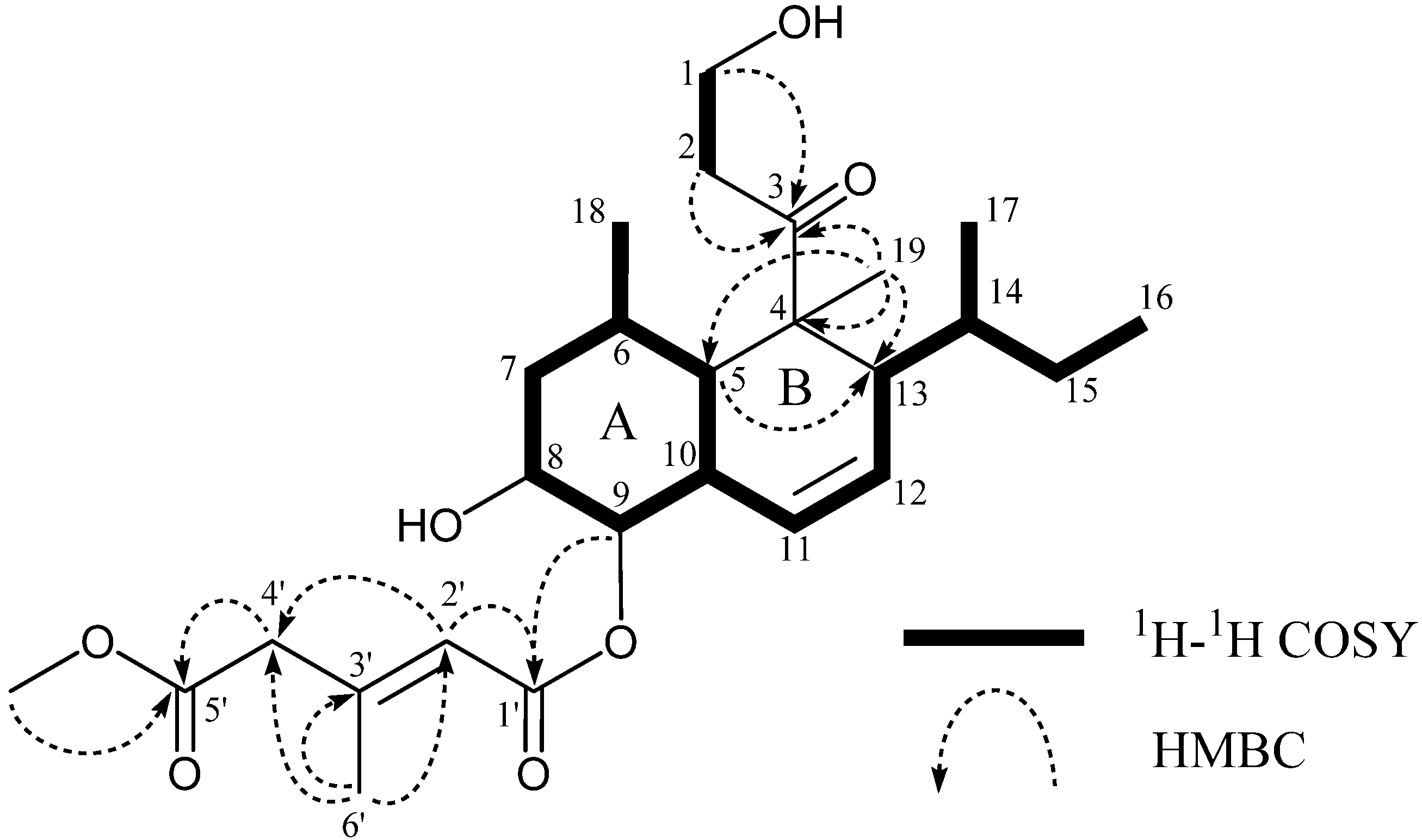
{kind=link}
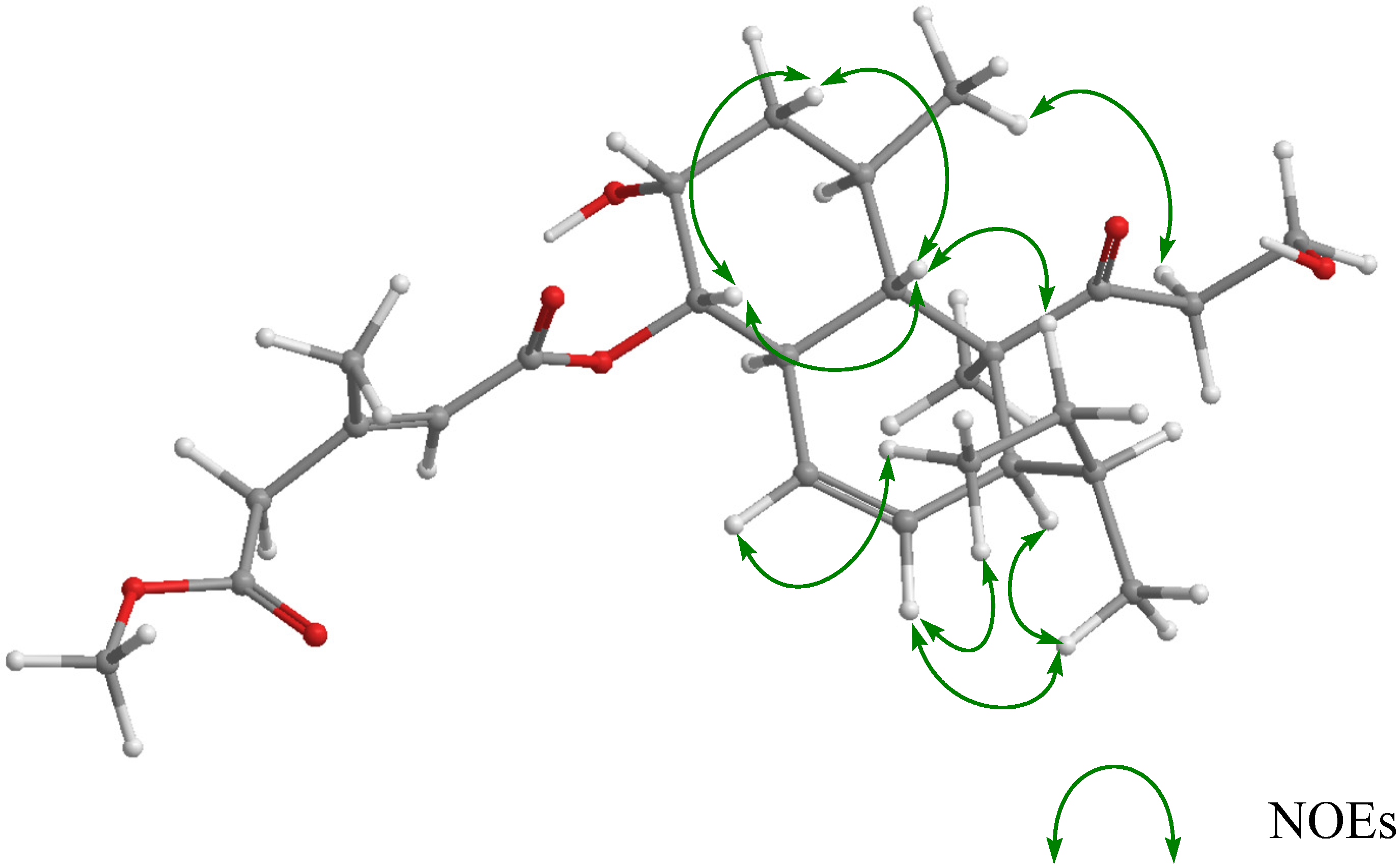
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 5 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δH a | δC | δH a | δC | δH a | δC | δH a | δC | |||||||||||
| 1A | 3.83 | ddd | 58.0 | (t) | 3.84 | ddd | 58.0 | (t) | 3.83 | ddd | 58.0 | (t) | 3.84 | ddd | 58.0 | (t) | ||
| 1B | 3.89 | ddd | 3.90 | ddd | 3.91 | ddd | 3.90 | ddd | ||||||||||
| 2A | 2.66 | ddd | 41.2 | (t) | 2.67 | ddd | 41.1 | (t) | 2.67 | ddd | 41.2 | (t) | 2.67 | ddd | 41.2 | (t) | ||
| 2B | 2.87 | ddd | 2.85 | ddd | 2.86 | ddd | 2.86 | ddd | ||||||||||
| 3 | 215.2 | (s) | 215.2 | (s) | 215.2 | (s) | 215.5 | (s) | ||||||||||
| 4 | 52.5 | (s) | 52.5 | (s) | 52.5 | (s) | 52.5 | (s) | ||||||||||
| 5 | 2.06 | t | 43.6 | (d) | 2.03 | t | 43.4 | (d) | 1.96 | t | 43.1 | (d) | 1.98 | t | 43.0 | (d) | ||
| 6 | 1.82 | m | 30.3 | (d) | 1.73 | m | 30.5 | (d) | 1.59 | m | 31.4 | (d) | 1.62 | m | 31.5 | (d) | ||
| 7α | 1.86 | dt | 40.7 | (t) | 1.83 | dt | 40.1 | (t) | 1.83 | dt | 39.1 | (t) | 1.87 | dt | 39.0 | (t) | ||
| 7β | 1.55 | td | 1.53 | td | 1.55 | td | 1.56 | td | ||||||||||
| 8 | 4.13 | q | 67.6 | (d) | 4.28 | q | 66.7 | (d) | 5.26 | q | 73.3 | (d) | 5.26 | q | 72.7 | (d) | ||
| 9 | 4.78 | dd | 77.4 | (d) | 4.55 | dd | 78.8 | (d) | 3.48 | dd | 74.2 | (d) | 3.56 | dd | 74.4 | (d) | ||
| 10 | 2.46 | tdd | 36.2 | (d) | 2.47 | brt | 36.0 | (d) | 2.08 | tdd | 40.4 | (d) | 2.12 | tdd | 40.3 | (d) | ||
| 11 | 5.62 | brd | 125.0 | (d) | 5.69 | drd | 125.1 | (d) | 6.06 | dt | 125.9 | (d) | 6.04 | brd | 125.8 | (d) | ||
| 12 | 5.74 | ddd | 124.5 | (d) | 5.67 | dd | 124.5 | (d) | 5.69 | ddd | 123.7 | (d) | 5.70 | ddd | 123.8 | (d) | ||
| 13 | 1.94 | m | 52.4 | (d) | 1.94 | m | 52.3 | (d) | 1.94 | m | 52.4 | (d) | 1.94 | m | 52.4 | (d) | ||
| 14 | 1.12 | m | 37.2 | (d) | 1.12 | m | 37.2 | (d) | 1.12 | m | 37.2 | (d) | 1.12 | m | 37.2 | (d) | ||
| 15A | 0.74 | m | 24.4 | (t) | 0.72 | m | 24.5 | (t) | 0.74 | m | 24.4 | (t) | 0.74 | m | 24.4 | (t) | ||
| 15B | 1.50 | m | 1.47 | m | 1.47 | m | 1.47 | m | ||||||||||
| 16 | 0.76 | t | 12.5 | (q) | 0.75 | t | 12.5 | (q) | 0.76 | t | 12.5 | (q) | 0.76 | t | 12.5 | (q) | ||
| 17 | 0.92 | d | 19.1 | (q) | 0.92 | d | 19.2 | (q) | 0.93 | d | 19.2 | (q) | 0.93 | d | 19.3 | (q) | ||
| 18 | 0.60 | d | 22.3 | (q) | 0.59 | d | 22.3 | (q) | 0.59 | d | 22.3 | (q) | 0.59 | d | 22.2 | (q) | ||
| 19 | 1.26 | s | 19.4 | (q) | 1.26 | s | 19.3 | (q) | 1.25 | s | 19.3 | (q) | 1.26 | s | 19.4 | (q) | ||
| 1′ | 164.9 | (s) | 170.0 | (s) | 168.9 | (s) | 166.2 | (s) | ||||||||||
| 2′A | 5.88 | s | 118.9 | (d) | 3.29 | d | 39.7 | (t) | 5.92 | s | 118.1 | (d) | 5.88 | s | 119.7 | (d) | ||
| 2′B | 3.77 | d | ||||||||||||||||
| 3′ | 152.0 | (s) | 153.4 | (s) | 153.9 | (s) | 151.7 | (s) | ||||||||||
| 4′A | 3.19 | s | 45.7 | (t) | 5.94 | s | 118.5 | (d) | 3.54 | d | 39.6 | (t) | 3.19 | s | 45.4 | (t) | ||
| 4′B | 3.73 | d | ||||||||||||||||
| 5′ | 170.2 | (s) | 168.9 | (s) | 169.8 | (s) | 173.5 | (s) | ||||||||||
| 6′ | 2.27 | s | 19.2 | (q) | 2.08 | s | 27.4 | (q) | 2.05 | s | 26.9 | (q) | 2.27 | s | 19.1 | (q) | ||
| 5′-OCH3 | 3.73 | s | 52.2 | (q) | ||||||||||||||
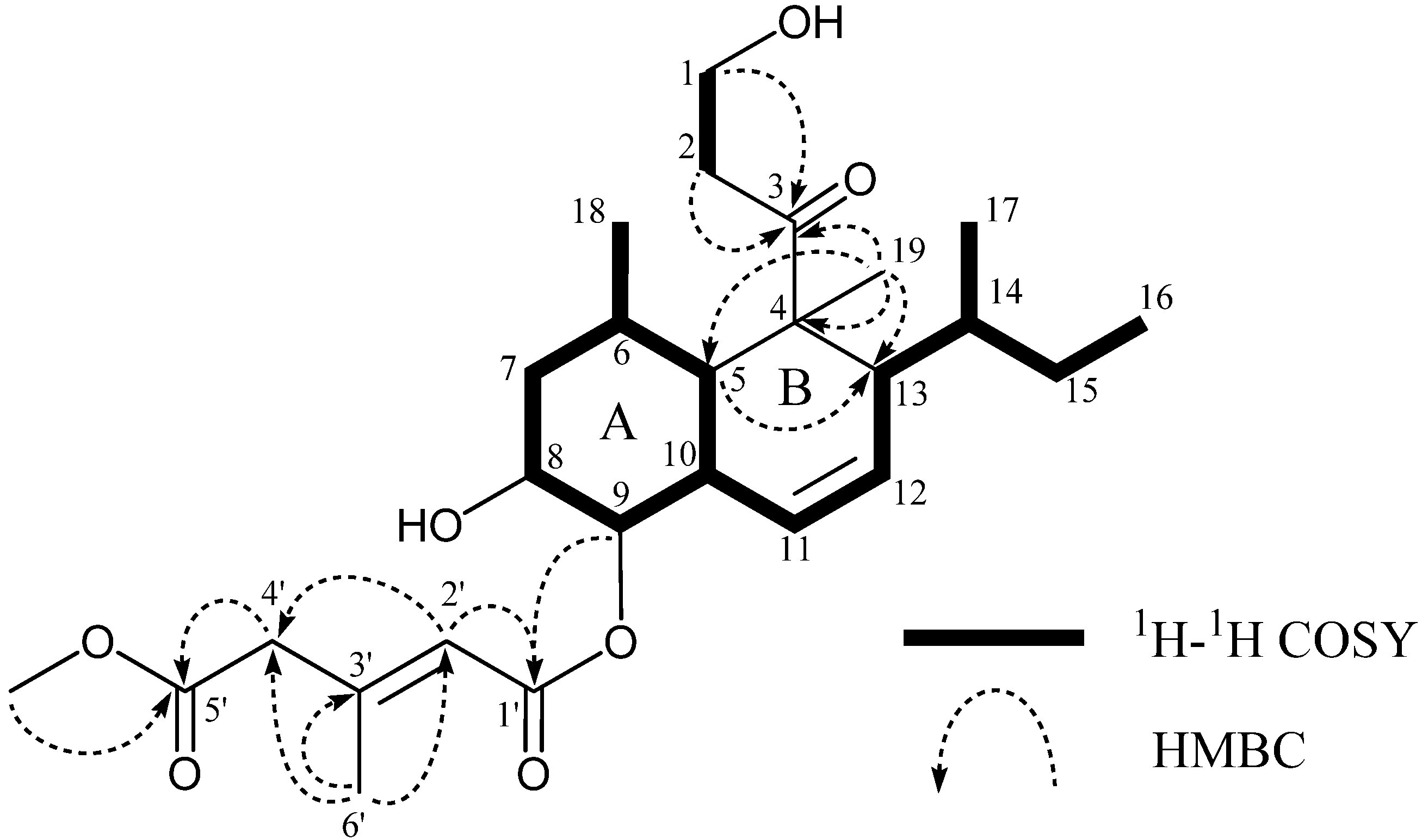
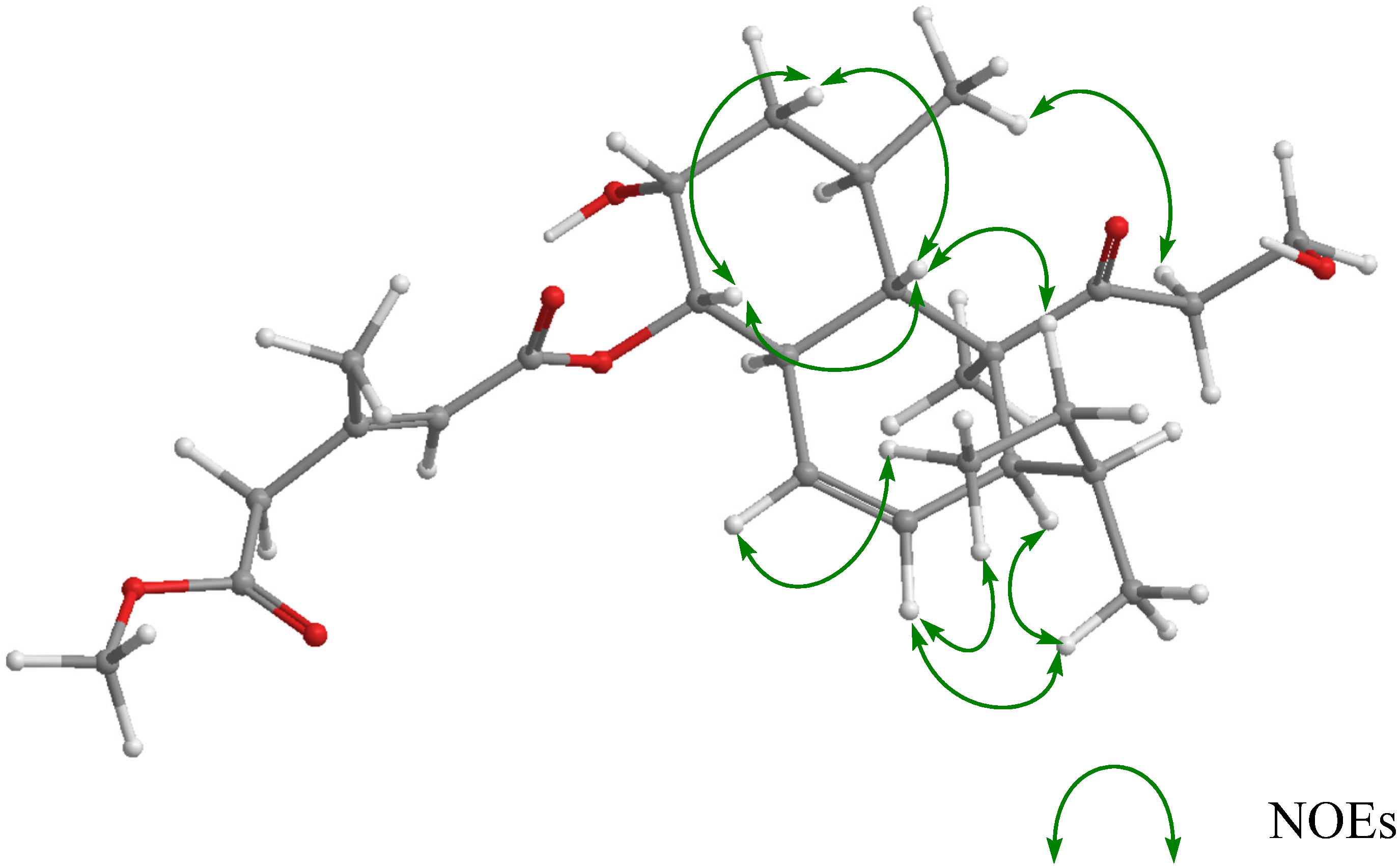
| Sample | 1 | 2 | 3 |
|---|---|---|---|
| MG-MID a | −4.01 | −4.04 | −4.01 |
| Delta b | 0.35 | 0.52 | 0.53 |
| Range c | 0.36 | 0.56 | 0.54 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Culturing and Isolation of Metabolites
3.4. Chemical Transformation
3.4.1. The Hydrolysis of 1–3
3.4.2. The Methylation of 3
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yamada, T.; Kitada, H.; Kajimoto, T.; Numata, A.; Tanaka, R. The relationship between the CD cotton effect and the absolute configuration of FD-838 and its seven stereoisomers. J. Org. Chem. 2010, 75, 4146–4153. [Google Scholar]
- Yamada, T.; Kikuchi, T.; Tanaka, R.; Numata, A. Halichoblelides B and C, potent cytotoxic macrolides from a Streptomyces species separated from a marine fish. Tetrahedron Lett. 2012, 53, 2842–2846. [Google Scholar]
- Nakanishi, K.; Doi, M.; Usami, Y.; Amagata, T.; Minoura, K.; Tanaka, R.; Numata, A.; Yamada, T. Anthcolorins A–F, novel cytotoxic metabolites from a sea urchin-derived Aspergillus versicolor. Tetrahedron 2013, 69, 4617–4623. [Google Scholar]
- Yamada, T.; Mizutani, Y.; Umebayashi, Y.; Inno, N.; Kawashima, M.; Kikuchi, T.; Tanaka, R. Tandyukisin, a novel ketoaldehyde decalin derivative, produced by a marine sponge-derived Trichoderma harzianum. Tetrahedron Lett. 2014, 54, 662–664. [Google Scholar]
- Kobayashi, M.; Uehara, H.; Matsunami, K.; Aoki, S.; Kitagawa, I. Trichoharzin, a new polyketide produced by the imperfect fungus Trichoderma harzianum separated from the marine sponge Micale cecilia. Tetrahedron Lett. 1993, 34, 7925–7928. [Google Scholar]
- Nakadate, S.; Nozawa, K.; Horie, H.; Fujii, Y.; Nagai, M.; Hosoe, T.; Kawai, K.; Yaguchi, T.; Fukushima, K. Eujavanicols A–C, decalin derivatives from Eupenicillium javanicum. J. Nat. Prod. 2007, 70, 1510–1512. [Google Scholar] [PubMed]
- Smetanina, O.F.; Kalinovskii, A.I.; Kicha, A.A.; Yurchenko, A.N.; Pivkin, M.V.; Kuznetsova, T.A. Dehydrodecalin derivative from marine isolate of the fungus Wardomyces inflatus. Chem. Nat. Compd. 2009, 45, 753–755. [Google Scholar]
- Ichihara, A.; Oikawa, H.; Hayashi, K.; Sakamura, S. Structures of betaenones A and B, novel phytotoxins from Phoma betae Fr. J. Am. Chem. Soc. 1983, 105, 2907–2908. [Google Scholar]
- Barash, I.; Pupkin, G.; Netzer, D.; Kashman, Y. A novel enolic β-ketoaldehyde phytotoxin by Stemphylium botryosum f. sp. Lycopersici. Partial chemical and biological characterization. Plant Physiol. 1982, 69, 23–27. [Google Scholar] [PubMed]
- Kobayashi, H.; Sunaga, R.; Furihara, K.; Morisaki, N.; Iwasaki, S. Isolation and structures of an antifungal antibiotic, fusarietin A, and related compounds produced by a Fusarium sp. J. Antibiot. 1995, 48, 42–52. [Google Scholar] [PubMed]
- Parish, C.A.; Cruz, M.; Smith, S.K.; Zink, D.; Baxter, J.; Tucker-Samaras, S.; Collado, J.; Platas, G.; Bills, G.; Diez, M.T.; et al. Antisense-guided isolation and structure elucidation of pannomycin, a substituted cis-decalin from Geomyces pannorum. J. Nat. Prod. 2009, 72, 59–62. [Google Scholar] [PubMed]
- Otto, D.H.; Gregory, L.; Helms Tracy, T.J.; Guy, H.H. Structure elucidation of australifungin, a potent inhibitor of sphinganine N-acyltransferase in sphingolipid biosynthesis from Sporormiella australis. J. Org. Chem. 1995, 60, 1772–1776. [Google Scholar]
- Yamori, T.; Matsunaga, A.; Sato, S.; Yamazaki, K.; Komi, A.; Ishizu, K.; Mita, I.; Edatsugi, H.; Matsuba, Y.; Takezawa, K.; et al. Potent antitumor activity of MS-247, a novel DNA minor groove binder, evaluated by an in vitro and in vivo human cancer cell line panel. Cancer Res. 1999, 59, 4042–4049. [Google Scholar] [PubMed]
- Yamori, T. Panel of human cancer cell lines provides valuable database for drug discovery and bioinformatics. Cancer Chemother. Pharmacol. 2003, 52 (Suppl. 1), S74–S79. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, T.; Umebayashi, Y.; Kawashima, M.; Sugiura, Y.; Kikuchi, T.; Tanaka, R. Determination of the Chemical Structures of Tandyukisins B–D, Isolated from a Marine Sponge-Derived Fungus. Mar. Drugs 2015, 13, 3231-3240. https://doi.org/10.3390/md13053231
Yamada T, Umebayashi Y, Kawashima M, Sugiura Y, Kikuchi T, Tanaka R. Determination of the Chemical Structures of Tandyukisins B–D, Isolated from a Marine Sponge-Derived Fungus. Marine Drugs. 2015; 13(5):3231-3240. https://doi.org/10.3390/md13053231
Chicago/Turabian StyleYamada, Takeshi, Yoshihide Umebayashi, Maiko Kawashima, Yuma Sugiura, Takashi Kikuchi, and Reiko Tanaka. 2015. "Determination of the Chemical Structures of Tandyukisins B–D, Isolated from a Marine Sponge-Derived Fungus" Marine Drugs 13, no. 5: 3231-3240. https://doi.org/10.3390/md13053231
APA StyleYamada, T., Umebayashi, Y., Kawashima, M., Sugiura, Y., Kikuchi, T., & Tanaka, R. (2015). Determination of the Chemical Structures of Tandyukisins B–D, Isolated from a Marine Sponge-Derived Fungus. Marine Drugs, 13(5), 3231-3240. https://doi.org/10.3390/md13053231