Abstract
Antifungal bioactivity-guided fractionation of the organic extract of the sponge Polymastia boletiformis, collected from the west coast of Ireland, led to the isolation of two new sulfated steroid-amino acid conjugates (1 and 2). Extensive 1D and 2D NMR analyses in combination with quantum mechanical calculations of the electronic circular dichroism (ECD) spectra, optical rotation, and 13C chemical shifts were used to establish the chemical structures of 1 and 2. Both compounds exhibited moderate antifungal activity against Cladosporium cucumerinum, while compound 2 was also active against Candida albicans. Marine natural products containing steroidal and amino acid constituents are extremely rare in nature.
1. Introduction
Marine sponges (Porifera) are primitive filter-feeders, yet they represent the richest and best studied sources of novel marine bioactive natural products [1]. Furthermore, sponges are considered to be producers of the highest sterol diversity amongst all animal phyla, presenting molecules with unique functionalization and structures, many with no terrestrial analogues [1,2,3]. The role of sterols in sponges is primarily functional, with sterols as constituents of cell membranes, and a secondary role as metabolic precursors for the production of diverse steroid classes [4,5,6]. Highly functionalized steroids are a growing group of metabolites that exhibit interesting biological and pharmacological properties, including ichthyotoxic, antihistaminic, cytotoxic, and antiviral activities [4,5].
Polymastia boletiformis (Lamarck, 1814, family Polymastiidae, order Hadromerida), is a brightly coloured orange-yellow fistulose sponge that grows on upper rock faces in the sublittoral zone and is widespread on the coasts of Britain and Ireland. Although not extensively studied, the genus Polymastia has been a source of various new fatty acids [7], carotenoids [8], and steroid compounds [9,10], including the antibiotic polymastiamides A–F [11,12]. Polymastiamides are the first examples of natural steroids with a side chain containing an amide bond linking the steroid part to a non-proteinaceous amino acid.
There has been a considerable increase in the frequency of fungal infections during the past decades. Cladosporium cucumerinum has been known as an important phytopathogen that causes scab disease in many commercial vegetables, resulting in significant losses [13]. Candida albicans, one of the most commonly encountered human pathogens, causes a variety of difficult-to-cure mucosal, skin, and systemic infections [14]. The declining number and efficacy of existing antifungal agents, the increased occurrence of systemic fungal infections, and rapid emergence of drug resistance underline the necessity for the discovery of new antifungal drug leads.
In the course of our ongoing investigations within the Beaufort Marine Biodiscovery Research program aimed at identification of novel bioactive metabolites from Irish marine resources [15], we have undertaken a chemical study of the Irish marine sponge P. boletiformis, as it displayed antifungal activity in initial screening studies. This report describes the bioactivity-guided isolation and structure elucidation of two new, minor sulfated steroid-amino acid conjugates, 1, and its 7-methoxy derivative, 2 (Figure 1). Both compounds feature linkage of the non-proteinaceous α-amino acid p-methoxyphenylglycine with 4,24-dimethyl-3-O-sulfocholest-8(14),25(26)-dien-27-oic acid, via an amide bond. The molecular structures of the natural products were established on the basis of 1D and 2D NMR, IR, UV, HRMS, and experimental and calculated ECD, optical rotation, and 13C chemical shift data. The isolated metabolites were evaluated for their antifungal activity and were found to exhibit moderate antifungal activity against C. cucumerinum, while compound 2 was also active against C. albicans.
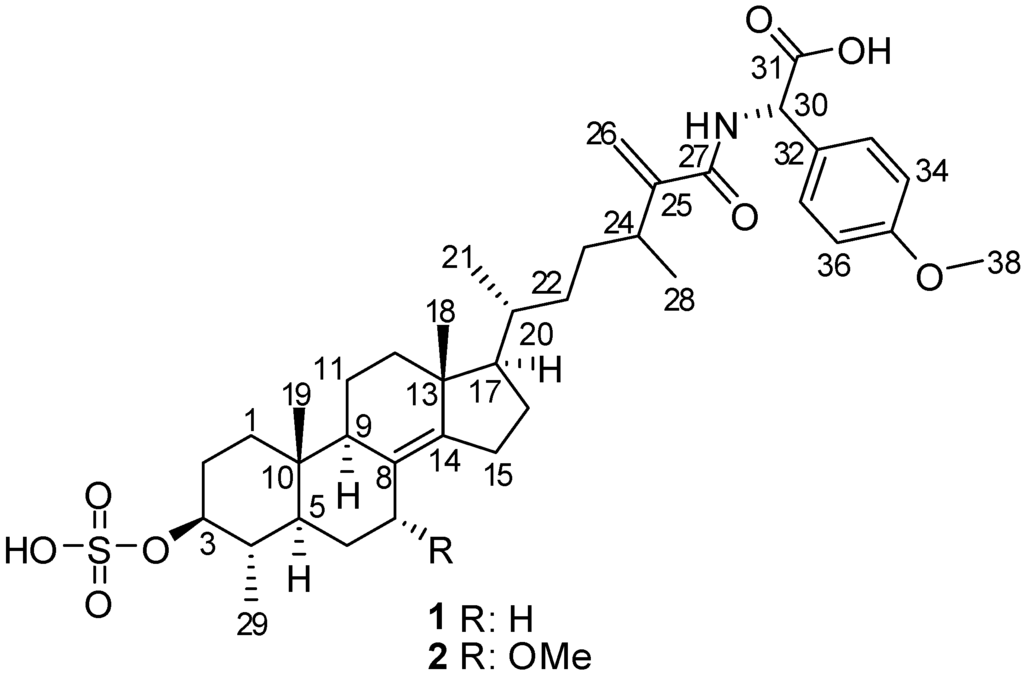
Figure 1.
Structures of compounds 1 and 2.
Figure 1.
Structures of compounds 1 and 2.
2. Results and Discussion
P. boletiformis specimens were collected by SCUBA from Roskeeda, Co. Galway (Ireland) and the CH2Cl2/MeOH extract of the freeze-dried sponge was subjected to a modified Kupchan partitioning scheme to yield n-hexane, CHCl3, and aqueous MeOH subextracts. Bioassay-guided separation of the antifungal aqueous MeOH-soluble fraction, which included a combination of C18 reversed-phase flash column chromatography (RP-FCC), Sephadex LH-20 size-exclusion chromatography, and repeated C18 reversed-phase (RP) HPLC purifications, yielded compounds 1 and 2.
Metabolite 1 was obtained as a white amorphous powder with
+45.9 (c 0.03, MeOH). NMR data combined with the [M − H]− ion at m/z 684.3570 in the HRESIMS of 1 suggested a molecular formula of C38H55NO8S (calcd for C38H54NO8S, 684.3576; Δ 0.75 ppm) requiring twelve indices of hydrogen deficiency. The structural characterization of compound 1 was established on the basis of extensive 1D and 2D NMR studies of data acquired in both DMSO-d6 and methanol-d4 (Table 1 and Supplementary Table S1). In DMSO-d6, both 1H and 13C-NMR spectra showed resonances typical of a steroid, displaying shielded resonances for two tertiary methyl groups (δH 0.64, δC 13.7; δH 0.78, δC 18.1) and three secondary methyls (δH 0.86, δC 19.0; δH 0.87, δC 15.5; δH 0.99, δC 19.7). Additional resonances accounting for one oxygenated methine (δH 3.53, δC 80.1), a tetrasubstituted double bond (δC 125.9, 141.4), a terminal olefinic methylene (δH 5.53 and 5.17, δC 113.9 and δC 151.4), a p-disubstituted benzene moiety (δH 6.74, δC 112.7; δH 7.18, δC 127.5; δC 134.9; δC 157.5), two carbonyl groups (δC 166.7 and δC 170.4), and one aromatic methoxy functionality (δH 3.67, δC 55.0) suggested that 1 was a steroid with an atypical, modified side chain. These functional groups also accounted for eight indices of hydrogen deficiency, with the remaining four assembling a typical four-ring steroid core. Furthermore, 1H and 13C-NMR data of 1 (Table 1) suggested the presence of a sulfate group based on deshielded resonances of the H-3/C-3 oxygenated methine (δH 3.53, δC 80.1) relative to a typical hydroxylated methine [16,17]. This was also supported by strong absorptions at 1214 and 1055 cm−1, characteristic of a sulfate functional group, present in the IR spectrum. An amide group was also evident due to the appearance of the resonance for a labile NH (δH 7.78) function in DMSO-d6, in addition to the broad IR absorption band centred at 3416 cm−1 (OH, NH stretch), and the intense bands observed at 1591 and 1548 cm−1. Analysis of the COSY and HMBC data led to the assignment of the ABCD steroid ring system in 1 (Figure 2). The COSY cross peaks between all neighbouring protons from H2-1 to H-7 assisted the assignment of rings A and B. HMBC correlations of angular methyl protons H3-19 with C-1, C-5, C-9, and C-10; of H-6α, Η-7β, and H-9 with C-8; of H-7β with C-9, along with the cross peaks of H-6β and H-9 with C-10; and of H-1β, H-4, Η-6β, H-7β, and H3-29 with C-5, validated the structure of the decalin AB rings. From the COSY spectrum it was possible to differentiate two discrete spin systems in the C and D rings; H-9/H2-11/H2-12 and H2-15/H2-16. HMBC correlations from the H3-18 methyl singlet to C-12, C-13, C-14, and C-17 and from C-13 to H-11β, Η-15β, Η-16α, and H-17 established the connectivity of C-12 and C-17 through the quaternary C-13. Diagnostic HMBC cross peaks between the olefinic C-14 and H-7α, H-9, H-12β, H2-15, and H2-16, along with correlations between C-8 and H-9 and H-15α assembled the ABCD steroidal rings. The COSY spectrum revealed useful information concerning the side chain. COSY correlations among all adjacent protons from C-17 to C-24, along with the long range coupling of methine H-24 with the sp2 methylene H-26b established the connection of spin system (i) CH17-CH20(CH321)-CH222-CH223-CH24(CH328)-C25(CH226) of the side chain. Two more spin systems were evident within the side chain: (ii) NH-CH30; and (iii) CH33/37-CH34/36. Fragments (i) and (ii) could be connected on the basis of the HMBC correlations between C-27 and both terminal olefinic methylenes H2-26, as well as those between the amide proton NH and C-27, C-30, and C-31. The cross peaks in the HMBC spectrum between H-30 and C-31 clearly established the position of the carboxylic acid group at C-30. Complementary HMBC correlations between C-30 and H-33/37 and between H-30 and the aromatic carbons C-32 and C-33/37 validated the attachment of the p-disubstituted benzene moiety at the end of the side chain, thus connecting fragments (i), (ii), and (iii). Long-range heteronuclear couplings between H3-38 and C-35 secured the position of the methoxy group in the benzene ring and further established the gross structure for metabolite 1. The amide linkage connecting the steroid side chain and d-cysteinolic acid in carolisterols A–C from the starfish Styracaster caroli is another rare example of marine natural products containing a steroidal and an amino acid component [18].

Table 1.
1H (600 MHz) and 13C (150 MHz) NMR chemical shifts of compound 1 and polymastiamide A (polyA) 7a in DMSO-d6, δ in ppm, J values in Hz.
| No. | 1H (δ) m (J) | 13C (δ) m | ||||
|---|---|---|---|---|---|---|
| 1 | polyA | δΗ(1) − δΗ(polyA) | 1 | polyA | δC(1) − δC(polyA) | |
| 1 | 1.59 m
1.03 m | 1.60 m
1.03 m | –0.01
- | 35.8 t | 35.8 t | - |
| 2 | 2.10 m
1.28 m | 2.10 m
1.28 m | -
- | 28.2 t | 28.1 t | +0.1 |
| 3 | 3.53 ddd 10.9, 10.9, 4.4 | 3.53 m | - | 80.1 d | 80.1 d | - |
| 4 | 1.21 m | 1.21 m | - | 37.2 d | 37.1 d | +0.1 |
| 5 | 0.87 m | 0.89 m | −0.02 | 50.7 d | 50.7 d | - |
| 6 | 1.65 m
0.90 m | 1.64 m
0.87 m | +0.01
+0.03 | 24.7 t | 24.7 t | - |
| 7 | 2.32 br. dd 12.6, 3.7
1.65 d | 2.32 m
1.63 m | -
+0.02 | 29.2 t | 29.2 t | - |
| 8 | - | - | - | 125.9 s | 125.8 s | +0.1 |
| 9 | 1.61 m | 1.60 m | +0.01 | 48.7 d | 48.7 d | - |
| 10 | - | - | 36.9 s | 36.8 s | +0.1 | |
| 11 | 1.55 m
1.41 m | 1.55 m
1.40 m | -
+0.01 | 19.5 t | 19.5 t | - |
| 12 | 1.87 ddd 12.2, 3.2, 3.2
1.04 m | 1.85 m
1.85 m | +0.02
−0.81 | 36.9 t | 36.9 t | - |
| 13 | - | - | - | 42.2 s | 42.1 s | +0.1 |
| 14 | - | - | - | 141.4 s | 141.4 s | - |
| 15 | 2.18 br. dd 16.7, 10.2
2.11 m | 2.16 m
2.08 m | +0.02
+0.03 | 25.3 t | 25.3 t | - |
| 16 | 1.71 dddd 13.1, 9.6, 7.3, 2.3
1.28 m | 1.69 m
1.25 m | +0.02
+0.03 | 26.6 t | 26.5 t | +0.1 |
| 17 | 1.02 m | 0.97 m | +0.05 | 56.3 d | 56.3 d | - |
| 18 | 0.78 s | 0.76 s | +0.02 | 18.1 q | 18.0 q | +0.1 |
| 19 | 0.64 s | 0.63 s | +0.01 | 13.7 q | 13.6 q | +0.1 |
| 20 | 1.37 m | 1.34 m | +0.03 | 34.1 d | 33.9 d | +0.2 |
| 21 | 0.86 d 6.6 | 0.83 d 6.5 | +0.03 | 19.0 q | 18.9 q | +0.1 |
| 22 | 1.33 m
0.98 m | 1.32 m
0.96 m | +0.01
+0.02 | 32.5 t | 32.5 t | - |
| 23 | 1.48 dddd 12.4, 12.0, 6.7, 4.3
1.15 dddd 12.0, 10.6, 6.6, 4.8 | 1.46 m
1.13 m | +0.02
+0.02 | 31.3 t | 31.3 t | - |
| 24 | 2.50 m | 2.54 m | +0.04 | 35.0 d | 34.9 d | +0.1 |
| 25 | - | - | - | 151.4 s | 149.5 s | +1.9 |
| 26 | 5.53 s
5.17 s | 5.61 s
5.21 s | −0.08
−0.04 | 113.9 t | 115.8 t | −1.9 |
| 27 | - | - | - | 166.7 s | 168.5 s | −1.8 |
| 28 | 0.99 d 6.9 | 0.98 d 6.8 | +0.01 | 19.7 q | 19.6 q | +0.1 |
| 29 | 0.87 d 6.3 | 0.87 d 6.2 | - | 15.5 q | 15.5 q | - |
| 30 | 4.60 d 5.8 | 5.35 d 7.6 | −0.75 | 58.2 d | 55.6 d | +2.6 |
| 31 | - | - | - | 170.4 s | 172.2 s | −1.8 |
| 32 | - | - | - | 134.9 s | 129.3 s | +5.6 |
| 33 | 7.18 d 8.6 | 7.31 d 8.7 | −0.13 | 127.5 d | 129.1 d | −1.6 |
| 34 | 6.74 d 8.6 | 6.88 d 8.7 | −0.14 | 112.7 d | 113.6 d | −0.9 |
| 35 | - | - | - | 157.5 s | 158.8 s | −1.3 |
| 36 | 6.74 d 8.6 | 6.88 d 8.7 | −0.14 | 112.7 d | 113.6 d | −0.9 |
| 37 | 7.18 d 8.6 | 7.31 d 8.7 | −0.13 | 127.5 d | 129.1 d | −1.6 |
| 38 | 3.67 s | 3.72 s | −0.05 | 55.0 q | 55.1 q | −0.1 |
| NH | 7.78 d 5.8 | 8.48 d 7.5 | −0.70 | - | - | |
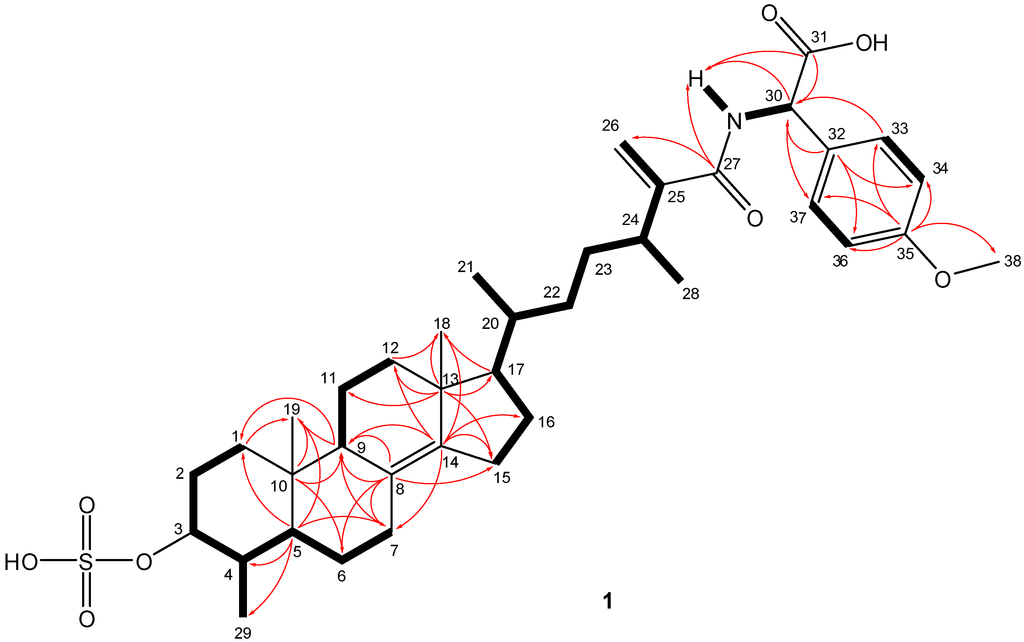
Figure 2.
Key HMBC (arrows) and COSY (bold line) correlations observed in 1.
Figure 2.
Key HMBC (arrows) and COSY (bold line) correlations observed in 1.
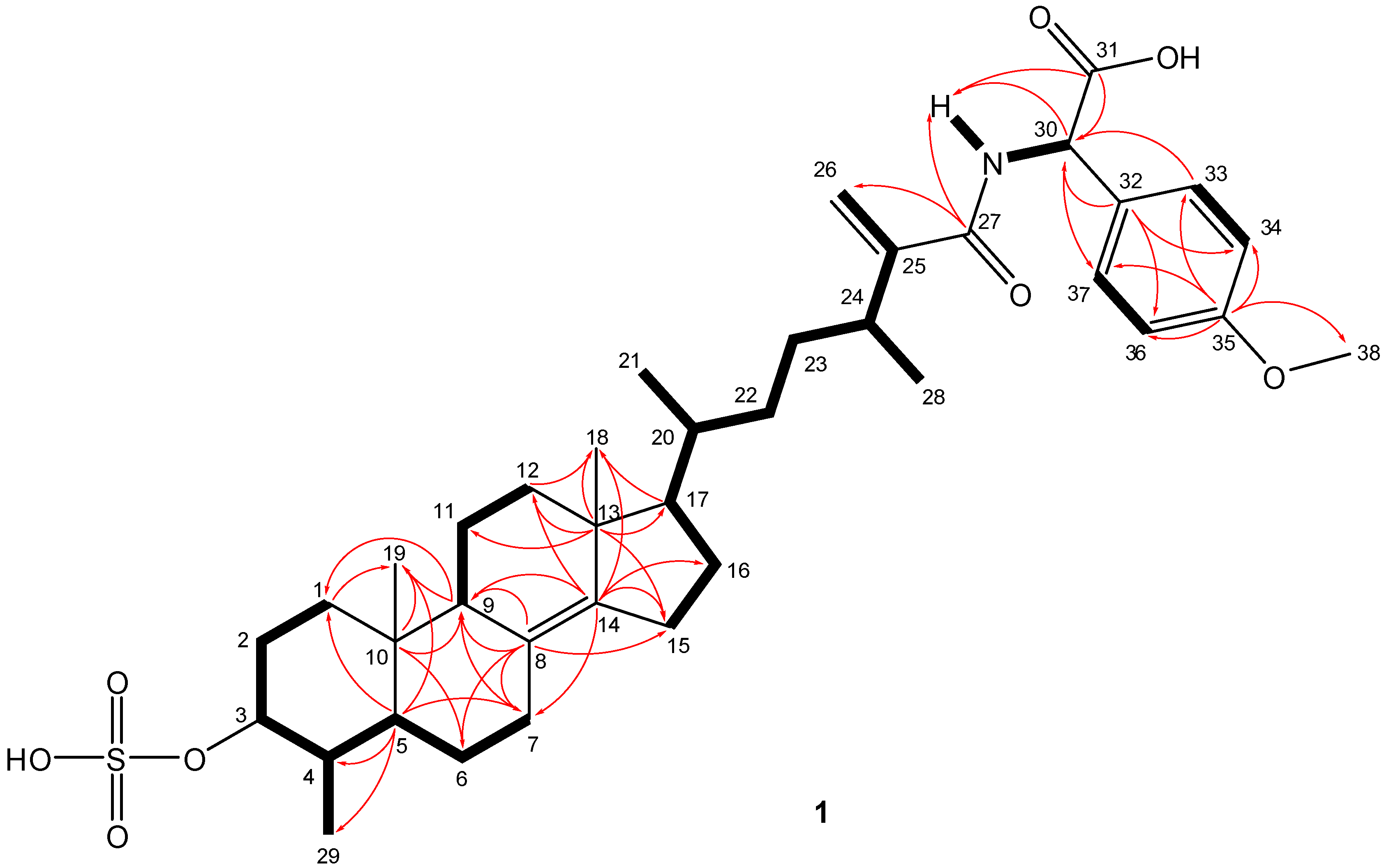
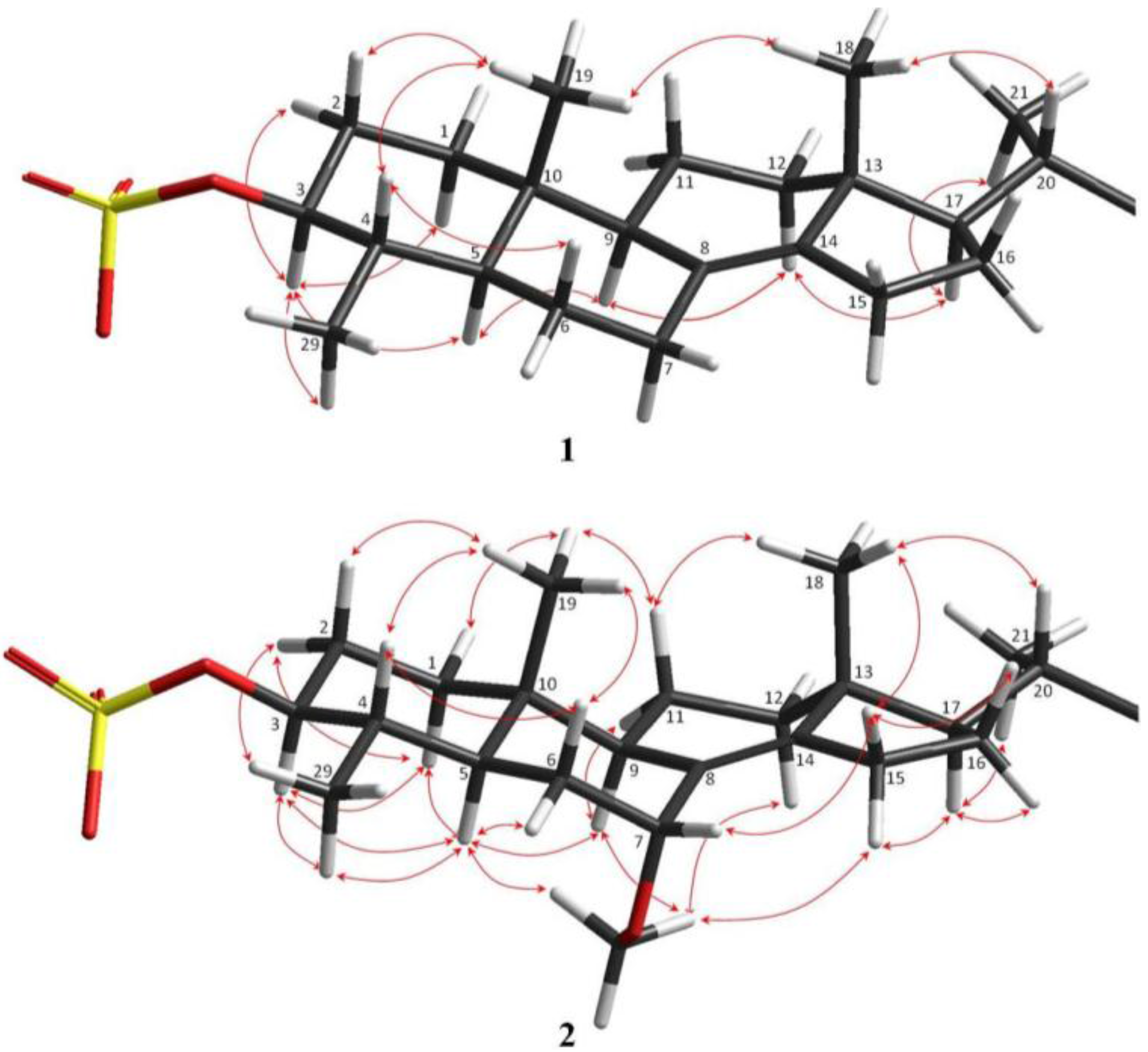
The relative configurations of the steroidal ring stereogenic centres of 1 were deduced from NOE studies and J values in the 1H NMR spectra. The NOESY data indicated that 1 contained the common 5α/10β/9α/13β steroid nucleus (Figure 3). Key NOE correlations of angular methyl protons H3-19 with Η-4, as well as cross peaks of H-3 with Η-5 and H3-29, and of H-5 with H-9 established the trans-fusion of the AB decalin ring system, indicating that H-4 and H3-19 were cofacial and β-oriented, while H3-29, H-3, H-5, and H-9 were cofacially α-oriented. H-3 was designated axial based on a large vicinal coupling constant (J = 10.9 Hz), and the NOE observed between H-3 and H-5. The NOE correlations from Η3-18 to both H3-19 and H-20, and an additional cross peak between H-17 and H-21 established the relative configurations of the remaining stereocentres of rings C and D, and the β-orientation of the side chain. However, the NOESY data did not allow definition of the relative configurations of the C-24 and C-30 stereogenic centres of the freely rotating side chain.
Comparison of the gross NMR data of 1 with those reported for polymastiamide A obtained from a P. boletiformis specimen from Norway by Kong and Anderson in 1993 [11] indicated similar planar structures. The NMR data in DMSO-d6 (Table 1) displayed similar 1H and 13C chemical shifts, for all carbons and protons in the rigid steroid portion (ABCD rings). Notable differences were observed in the chemical shifts of the carbons and protons in the proximity of the chiral environment of C-24 (C-24 to C-27) and C-30 (especially C-30 to C-32). Both 1H and 13C-NMR chemical shift values of 1 were solvent dependent (Table 1, Supplementary Table S1). For example, in methanol-d4, H-30 appeared at δH 5.25 in 1, comparable to that of polymastiamide A (δH 5.35) in DMSO-d6. The above-mentioned data pointed out the necessity for a detailed investigation of the absolute configuration at stereocentres C-24 and C-30.
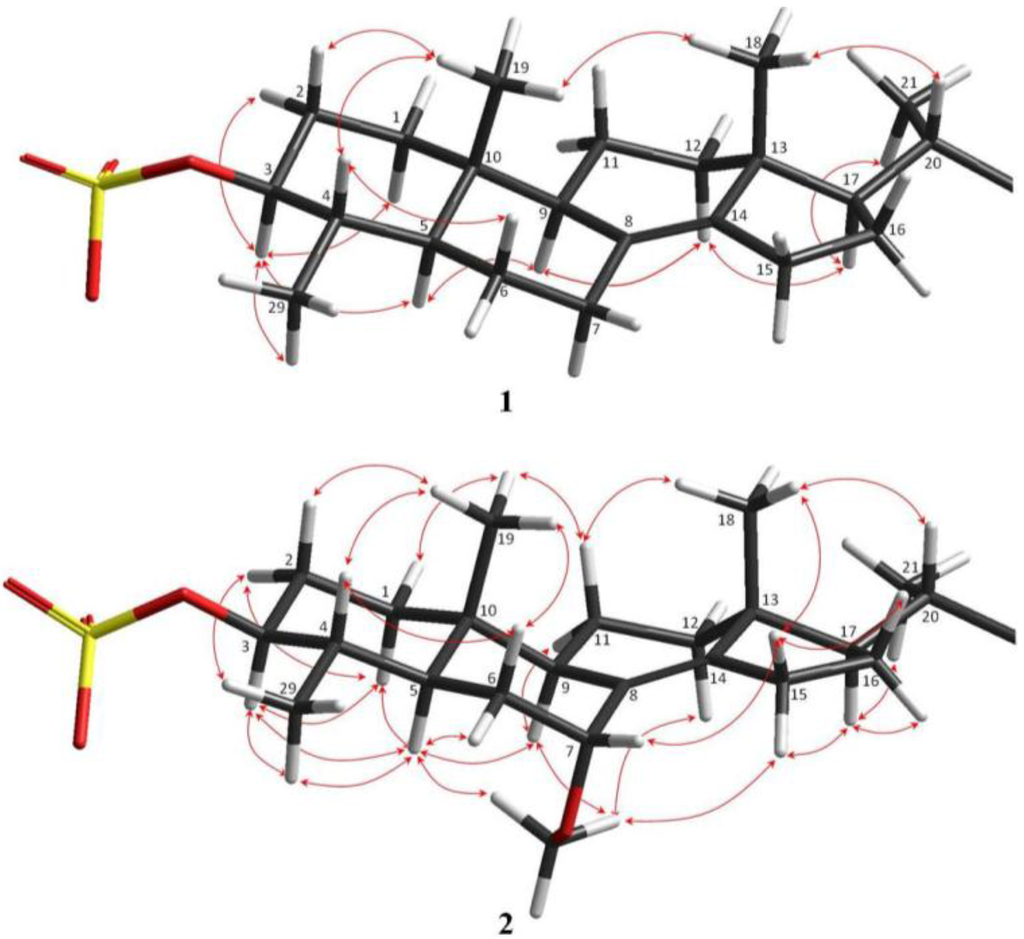
Figure 3.
Relative configurations and key NOESY correlations observed within the steroid portion of 1 and 2.
Figure 3.
Relative configurations and key NOESY correlations observed within the steroid portion of 1 and 2.
Suitable crystals of 1 could not be obtained. The absolute configuration of the side chain was therefore investigated by comparing the experimental electronic circular dichroism (ECD) spectrum of 1 to the ECD spectra predicted from quantum mechanical calculations for the four possible diastereomers. The ECD spectra [19,20,21,22] were simulated to assign the absolute configuration (AC) of 1 by using the Gaussian 09 software packages [23]. The configurations of the 4-methoxyphenylglycine moiety and C-24 were assigned as S- or R-, denoted as (24R,30R)-, (24R,30S)-, (24S,30R)-, and (24S,30S)-1, respectively. The OPLS-2005 force field in MacroModel [24] was employed to perform the conformational random search with an energy window of 130 kJ/mol, and yielded a total of 591 conformers for (24S,30R)-1 and 546 conformers for (24S,30S)-1 (Supplementary Figure S1). Fourteen of the (24S,30S)-1 (energy cut-off, 20 kJ/mol) and 44 of the (24S,30R)-1 conformers (energy cut-off, 15 kJ/mol) were included for the geometric optimization, followed by harmonic vibrational frequency computation to confirm these as energy minima. Nine of the (24S,30S)-1 conformers were relocated at the B3LYP/6-31G** level in the gas phase, among which conformer 7 (Supplementary Table S3 and Figure S2) was calculated to dominantly occupy 97.5% of the conformational itinerary. The ECD spectrum of the predominant conformer was simulated at the B3LYP/6-31G** level in the gas phase and matched very well with the experimental spectrum (Figure 4). Conformer 1 of (24S,30R)-1 occupies 96.7% of the conformational itinerary and its computed ECD spectrum was opposite to the experimental spectrum (Figure 4). Optimized geometries of the predominant conformers of (24S,30S)- and (24S,30R)-1 show strong intramolecular hydrogen bonding between the hydroxy group of the carboxylic moiety and the C-3 sulfate group. The distance of (C3-O-SO2-)O…H(-O-C31) was optimized as 1.519 and 1.469 Å for (24S,30S)- and (24S,30R)-1, respectively (Supplementary Figure S2). The C25-C27(O)-N-C30-C31(O)-O-H…O-S-O “backbone” in (24S,30S)-1 is coplanar, thus delocalized (π1), with the delocalized 4-methoxyphenyl group (π2) perpendicular to the plane. Molecular orbital analysis of (24S,30S)-1 indicated the experimentally observed positive Cotton effect (CE) at 234 nm corresponds to the electronic transitions at 230.9 and 230.2 nm (Supplementary Table S4) from Orb182 (πC-8=C-14, Supplementary Figure S3) to Orb187 (π*2), and from Orb183 (π2) to Orb188 (π*2). The negative CE at 213 nm results predominantly from the transition at 201 nm from Orb182 (πC-8=C-14) to Orb189 (π*C-8=C-14), and the transitions at 216 and 213 nm from Orb180 to Orb187, and from Orb181 to Orb188. The similar CEs result from the electronic transitions between π1, π2, πC-8=C-14, and the unoccupied orbitals, but did not permit definition of the absolute configuration at C-24. This is confirmed by the finding that the calculated ECD spectra of (24R,30S)- and (24R,30R)-1 are similar to those of (24S,30S)- and (24S,30R)-1, respectively (Figure 4). An attempt to differentiate these pairs by calculation of their specific rotations was also unsuccessful. The calculated specific rotations of (24S,30R)- and (24R,30R)-1 are −62.9 and −73.5 at the B3LYP/6-311++G**//B3LYP/6-31G** level in the gas phase, and those of (24R,30S)- and (24S,30S)-1 are 96.4 and 96.2, respectively [25].
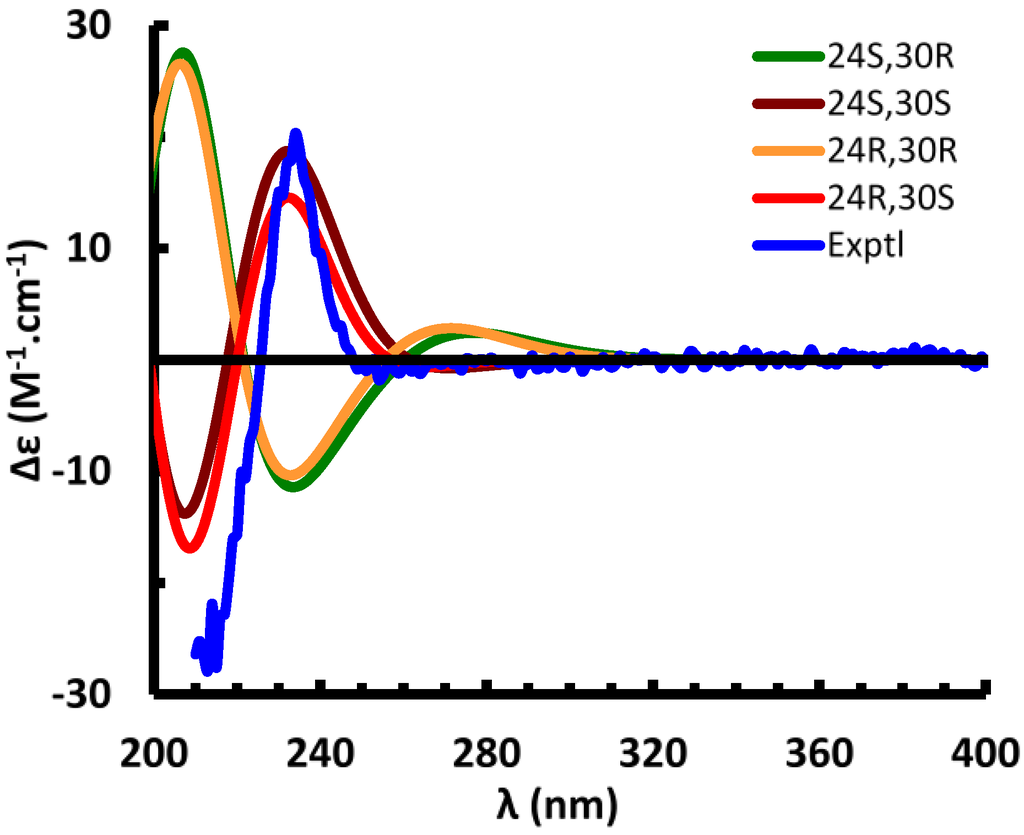
Figure 4.
Experimental and calculated electronic circular dichroism (ECD) spectra of compound 1.
Figure 4.
Experimental and calculated electronic circular dichroism (ECD) spectra of compound 1.
The comparison of quantum chemical calculated 13C NMR chemical shifts with the experimental data was considered as a useful tool to assist the assignment of absolute configuration of chiral molecules [26]. However, due to the large size of compound 1, the computation of 13C NMR shielding constants of the above conformers failed when using the GIAO technique at the mPW1PW91-SCRF (PCM, MeOH)/6-311++G** level of theory [27]. The calculated 13C NMR chemical shifts of (24R,30S)-1 and (24S,30S)-1 at the B3LYP/6-31G** level in the gas phase show very high similarity, differing less than 2.4 ppm and roughly match the experimental data (Supplementary Table S5). Even though the calculated chemical shifts of C-28 differ by 4.7 ppm, both match the experimental value. The noticeable difference is that the calculated chemical shifts of C-24 differ by 5.3 ppm, whereas that in (24S,30S)-1 was calculated as 43.1 ppm that was closer to the experimental value of 35.0 ppm than that in (24R,30S)-1 which was computed as 48.4 ppm, implying that most likely the absolute configuration at C-24 is (24S).
In summary, the ECD study provided unambiguous confirmation of the (30S) absolute configuration, but could not differentiate between (24R) and (24S) configurations. Calculation of the specific rotations of the four diastereomers similarly failed to differentiate the (24R) and (24S) configurations. The (30S)-configuration matches with the reported assignment for polymastiamide A that resulted from hydrolysis of the amino acid residue (p-hydroxyphenylglycine) from the desulfated methyl ester of the compound by Marfey’s method [11]. The l-configuration of the amino acid residue in polymastiamide A dictated (30S) configuration at C-30 while the configuration of C-24 remained unidentified [11]. Compound 1 and polymastiamide A have similar specific rotations (
+45.9 for 1, (
+67.4 for polymastiamide A), both acquired in MeOH. Based on these data we conclude that 1 and polymastiamide A differ at the C-24 stereogenic centre; however, the C-24 absolute configuration remains undefined in both compounds.
The molecular formula for the optically active (+33.8, c 0.04, MeOH) metabolite 2, C39H57NO9S, was derived from NMR data and the HRESIMS ion at m/z 714.3674 ([M − H]−, calcd for C39H56NO9S, 714.3681; Δ 1.07 ppm). Comparison of the molecular formula of 2 with that of 1 revealed that 2 contained a methoxymethine rather than a methylene functionality. The 1H and 13C-NMR resonances in both DMSO-d6 and methanol-d4, fully assigned through COSY, HSQC, and HMBC experiments (Table 2 and Supplementary Table S2) indicated that the only difference between the two molecules was the presence of a C-7 methoxy group in 2 (δH 3.94, δC 73.3). The COSY couplings from H-7 to both H-6α and H-6β, and the HMBC correlations from H-7 to C-5, C-6, C-9 and C-14, and from C-7 to H-6α, H-9 and C7-OMe confirmed the position of the methoxy group at C-7. Once the planar structure of 2 was defined, its relative configuration was established on the basis of 2D NMR NOESY to be the same as that of 1 (Figure 3). The NOESY correlations between H-7 and H-6β, between H-6β and H3-19, and between C7-OCH3 and H-5 and H-9 confirmed the α-axial orientation of the methoxy group. The similarity of the experimental ECD spectra of compounds 1 and 2 indicates they have the same absolute configurations for all stereogenic centres, with the exception of the (7R)-configuration of 2.
Table 2.
1H (600 MHz) and 13C (150 MHz) NMR chemical shifts of compound 2 in DMSO-d6, δ in ppm, J values in Hz.
| No. | 1H (δ) m (J) | 13C (δ) m |
|---|---|---|
| 1 | 1.56 m
1.02 m | 35.6 t |
| 2 | 2.09 dm 12.2
1.28 m | 28.1 t |
| 3 | 3.50 ddd 11.2, 10.0, 4.8 | 80.2 d |
| 4 | 1.20 m | 36.8 d |
| 5 | 1.22 m | 44.1 d |
| 6 | 1.83 m
1.07 m | 30.4 t |
| 7 | 3.94 br. s | 73.3 d |
| 8 | - | 124.5 s |
| 9 | 1.87 m | 43.4 d |
| 10 | - | 37.1 s |
| 11 | 1.56 m
1.36 m | 19.8 t |
| 12 | 1.89 m
1.04 m | 36.6 t |
| 13 | - | 42.8 s |
| 14 | - | 147.9 s |
| 15 | 2.37 ddd 17.6, 9.5, 8.7
2.21 br. dd 17.6, 12.4 | 25.0 t |
| 16 | 1.76 m
1.31 m | 26.4 t |
| 17 | 1.05 m | 56.6 d |
| 18 | 0.80 s | 17.4 q |
| 19 | 0.63 s | 12.9 q |
| 20 | 1.38 m | 34.1 d |
| 21 | 0.89 d 6.4 | 19.0 q |
| 22 | 1.36 m
0.97 m | 32.5 t |
| 23 | 1.49 m
1.17 m | 31.5 t |
| 24 | 2.50 m | 35.0 d |
| 25 | - | 151.3 s |
| 26 | 5.53 s
5.17 br. s | 114.0 t |
| 27 | - | 166.7 s |
| 28 | 1.00 d 6.9 | 19.0 q |
| 29 | 0.84 d 5.8 | 15.4 q |
| 30 | 4.57 d 5.0 | 58.2 d |
| 31 | - | 170.5 s |
| 32 | - | 134.9 s |
| 33 | 7.18 d 8.4 | 127.5 d |
| 34 | 6.74 d 8.4 | 112.7 d |
| 35 | - | 157.5 s |
| 36 | 6.74 d 8.4 | 112.7 d |
| 37 | 7.18 d 8.4 | 127.5 d |
| 38 | 3.69 s | 55.0 q |
| 39 | 3.03 s | 53.5 q |
| NH | 7.78 d 5.0 | - |
Compounds 1 and 2 were evaluated for their antifungal activity against Cladosporium cucumerinum and Candida albicans, and for their antibacterial activity against Staphylococcus aureus. The assays demonstrated that 1 and 2 showed moderate inhibitory effects on C. cucumerinum at 60 and 30 μg/disc, respectively. The observed diameters of their inhibition zones were 8.0 and 10.1 mm, respectively. Compound 2 was also found to possess significant activity at 100 μg/disc against C. albicans, displaying an inhibition zone diameter of 9.8 mm. The diameters of inhibition zones of the positive controls used in the assays, CuSO4 and nystatin, were 16.8 and 25.8 mm, respectively. No significant activity was demonstrated against S. aureus. Polymastiamide A was reported by Kong et al., to be active against both human pathogens S. aureus and C. albicans [11].
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured at the sodium D line (589.3 nm), at 20 °C on a Unipol L1000 Schmidt + Haensch polarimeter with a 10 cm cell. UV spectra were acquired in spectroscopic grade MeOH or CHCl3 on a Varian, Cary 100 UV-Vis spectrophotometer. IR spectra were obtained using a Perkin Elmer 400 FT-IR spectrometer with ATR attached. NMR spectra were recorded using a Jeol 400 MHz, a Varian 500 MHz, or an Agilent 600 MHz spectrometer with a cryo probe. Chemical shifts are given on the δ (ppm) scale using TMS as internal standard, and J values in Hz. High resolution mass spectrometric data were acquired on an Agilent 1290 Infinity-QTOF 6540 UPLC/MS system, with electrospray ionization (ESI) in the negative ion mode. C18 silica gel (40–63 μm; Merck, Tullagreen, Ireland), and Sephadex LH-20 (GE Healthcare, Cork, Ireland) were used for column chromatography (CC). Thin layer chromatography (TLC) was performed with Kieselgel 60 F254 aluminium support plates (Merck, Tullagreen, Ireland) and spots were detected after spraying with 6% vanillin and 15% H2SO4 in MeOH reagents and charring. HPLC separations were conducted using an Agilent 1260 model equipped with a diode array detector, either on semi-preparative (250 × 10 mm, 5 μm) or on analytical (150 × 4.6 mm, 5 μm) Kaseisorb ODS2000 HPLC columns. The chromatography was monitored at 254 nm. All solvents were of HPLC grade and were purchased from Sigma Aldrich (Arklow, Ireland).
3.2. Biological Material
Specimens of Polymastia boletiformis were collected by SCUBA diving in Roskeeda, Co. Galway, Ireland, at a depth of 6–9 m in August 2009. A voucher specimen has been deposited at the Department of Zoology, Ryan Institute, School of Natural Sciences, National University of Ireland, Galway, under reference MIIG30041.
3.3. Extraction and Isolation
Polymastia boletiformis was initially freeze-dried (9.1 g dry weight) and then exhaustively extracted with CH2Cl2 (3 × 500 mL) and MeOH (3 × 500 mL) at room temperature. The combined extracts were concentrated under reduced pressure at 35 °C to give a dark brown residue (1.44 g) that was subjected to a modified Kupchan partition. Briefly, the crude extract was partitioned between 10% aqueous MeOH (500 mL) and n-hexane (3 × 500 mL). The water concentration was increased to 35%, before extracting three times with CHCl3 (3 × 500 mL). Evaporation of the solvent under reduced pressure at 35 °C afforded the n-hexane (0.33 g), CHCl3 (0.06 g), and aqueous MeOH (1.04 g) subextracts. The aqueous subextract (W) was fractionated by RP-FCC (Lichroprep RP18, 40–63 μm), eluting with gradient mixtures of water and MeOH of reducing polarity to afford nine fractions. Antifungal activity against C. cucumerinum was detected in fractions 6 (W6) and 7 (W7), eluted with 60 and 70% MeOH in water, respectively. Fraction W7 (7 mg) afforded compound 1 (1.0 mg) after repeated purifications with RP-HPLC performed on a 5 μm, 150 × 4.6 mm Kaseisorb ODS2000 column. A binary eluent consisting of H2O (solvent A) and MeCN (solvent B) delivered at 1.2 mL/min was used with the following gradient profile: 0 min, 20% B; 10 min, 40% B. Fraction W6 (50 mg) was chromatographed on a Sephadex LH-20 column eluted with MeOH to give a fraction containing steroid-like compounds (28.8 mg). Final purification of 2 (1.3 mg) was achieved by repeated semi-preparative RP-HPLC of the latter fraction on a 5 μm, 250 × 10 mm Kaseisorb ODS2000 column, using a gradient binary solvent system, consisting of H2O (solvent A) and MeCN (solvent B), at a flow rate of 1.25 mL/min: 0 min, 5% B; 10 min, 20% B; 55 min, 40% B.
Compound 1. White amorphous powder;
+45.9, (c 0.03, MeOH); UV
(log ε): 287 (2.80); IR (film) νmax 3416, 1591, 1548, 1214, 1055 and 1033 cm−1; NMR data (DMSO-d6, methanol-d4), see Table 1 and Supplementary Table S1; HRESIMS m/z 684.3570 [M − H]−, (calcd for C38H54NO8S, 684.3576; Δ 0.75 ppm).
Compound 2. White amorphous powder;
+33.8 (c 0.04, MeOH); UV
nm (log ε): 289 (2.87), 239 (2.30); IR (film) νmax cm−1 3401, 1600, 1511, 1214, 1062 and 1033 cm−1; NMR data (DMSO-d6, CD3OD), see Table 2 and Supplementary Table S2; HRESIMS m/z 714.3674 [M − H]−, (calcd for C39H56NO9S, 714.3681; Δ 1.07 ppm).
3.4. Disc Diffusion Assay against C. cucumerinum
Freeze-dried ampoules of spores from the plant fungus C. cucumerinum (IMI 299104) were obtained from the Centre for Agriculture and Biosciences International UK and cultures of the fungus were maintained to harvest spores. These spores were subsequently inoculated onto potato dextrose agar petri dishes upon which antibiotic disks (6 mm) impregnated with MeOH solutions of the fractions or isolated compounds (100 μg/disc for fractions, and 30 and 60 μg/disc for pure compounds) were placed. Plates were incubated at 25 °C and observed at 24 and 48 h [28]. The positive control was CuSO4 (3000 μg on disk) and the negative control was the solvent, MeOH.
3.5. Disc Diffusion Assay against C. albicans and S. aureus
About 20 mL of sterilized nutrient agar medium was poured into each sterile petri plate and allowed to solidify. Inoculums of the test pathogens C. albicans (CBS 562 strain) and S. aureus (8325-4 MSSA strain) were prepared using a McFarland 0.5 standard and left at RT for 15 min. The inoculums were evenly spread over the appropriate media by using a sterile cotton swab and incubated for 15 min. Sterile discs (6 mm) impregnated with 30 μL (100 μg/disc) of the compounds dissolved in MeOH and the controls were placed on the surface of the media using sterile forceps. MeOH was used as the negative control, while nystatin (33.3 μg/disc) and ampicillin (10 μg/disc) were used as positive controls for C. albicans and S. aureus, respectively. Both C. albicans (incubated at 30 °C) and S. aureus (incubated at 37 °C) were observed at 24 h for development of inhibition zones.
4. Conclusions
In our continuing investigation aimed at the biological screening and isolation of bioactive metabolites from Irish marine organisms, two new sulfated steroid–amino acid conjugates, 1 and its 7-methoxy derivative 2, were isolated from the Irish marine sponge Polymastia boletiformis. Marine natural products containing steroidal and amino acid components are rare and have previously been encountered only in polymastiamides from the same sponge collected from Norway [11,12], and in carolisterols from the starfish Styracaster caroli [18]. The discovery of 1 and 2 presents a unique case, as 1 was found to share the same planar structure with polymastiamide A [11], yet the two compounds differ significantly in both proton and carbon resonances of the side chain atoms, especially around the C-24 and C-30 stereogenic centres. ECD and optical rotation calculations have been applied to 1 and permitted definition of absolute configuration, except for at the C-24 stereogenic centre. Metabolites 1 and 2 exhibited moderate antifungal activity against the plant pathogen C. cucumerinum, while 2 was also found active against C. albicans. The latter activity was also reported for polymastiamide A [11].
Supplementary Files
Supplementary File 1Acknowledgments
The Beaufort Marine Research Award is carried out under the Sea Change Strategy and the Strategy for Science Technology and Innovation (2006–2013), with the support of the Marine Institute, funded under the Marine Research Sub-Programme of the National Development Plan 2007–2013. Part of this work was financially supported by the US Department of Agriculture, Agricultural Research Service Specific Cooperative Agreement (No. 58-6408-2-0009). We also thank the Mississippi Center for Supercomputing Research (MCSR) for the computational facilities.
Author Contributions
V.S. Extraction of biological material, isolation and structure elucidation of metabolites, and manuscript preparation; M.R. Pilot extraction and antifungal assay against C. cucumerinum; S.S. Antifungal and antibacterial assays against C. albicans and S. aureus; Y.D., C.M.C. and D.F. ECD analysis of the compounds and manuscript editing; C.W. and G.M. Collection and identification of the sponge sample; D.T. is the principal investigator who designed and directed the project, as well as is the main editor of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Y.M.; Djerassi, C. Steroids from sponges. Tetrahedron 1974, 30, 4095–4103. [Google Scholar] [CrossRef]
- Djerassi, C.; Silva, C.J. Biosynthetic studies of marine lipids. 41. Sponge sterols: Origin and biosynthesis. Acc. Chem. Res. 1991, 24, 371–378. [Google Scholar] [CrossRef]
- Aiello, A.; Fattorusso, E.; Menna, M. Steroids from sponges: Recent reports. Steroids 1999, 64, 687–714. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M.V.; Minale, L.; Riccio, R. Polyoxygenated steroids of marine origin. Chem. Rev. 1993, 93, 1839–1895. [Google Scholar] [CrossRef]
- Kerr, R.G.; Baker, B.J. Marine sterols. Nat. Prod. Rep. 1991, 8, 465–497. [Google Scholar] [CrossRef]
- Ayanoglu, E.; Kurtz, K.; Kornprobst, J.M.; Djerassi, C. New natural 2-acetoxy fatty acids using chemical ionization and electron impact mass spectrometry. Lipids 1985, 20, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, S.; Englert, G.; Bergquist, P.; Liaaen-Jensen, S. Carotenoids of the sponge Polymastia granulosa (Hadromerida). Bulletin des Sociétés Chimiques Belges 1986, 95, 801–814. [Google Scholar] [CrossRef]
- Xu, S.H.; Zeng, L.M. The identification of two new sterols from marine organism. Chin. Chem. Lett. 2000, 11, 531–534. [Google Scholar]
- Xu, S.H.; Zeng, L.M. Study on the chemical constituents of marine sponge Polymastia robusta. Chin. J. Org. Chem. 2001, 21, 45–48. [Google Scholar]
- Kong, F.; Andersen, R.J. Polymastiamide A, a novel steroid/amino acid conjugate isolated from the norwegian marine sponge Polymastia boletiformis (Lamarck, 1815). J. Org. Chem. 1993, 58, 6924–6927. [Google Scholar] [CrossRef]
- Kong, F.; Andersen, R.J. Polymastiamides B–F, novel steroid/amino acid conjugates isolated from the norwegian marine sponge Polymastia boletiformis. J. Nat. Prod. 1996, 59, 379–385. [Google Scholar] [CrossRef]
- Hostettmann, K.; Potterat, O. Strategy for the isolation and analysis of antifungal, molluscicidal, and larvicidal agents from tropical plants. In Phytochemicals for Pest Control; American Chemical Society Symposium Series: Washington, DC, USA, 1997; Volume 658, pp. 13–26. [Google Scholar]
- Nielsen, K.; Heitman, J. Sex and virulence of human pathogenic fungi. Adv. Genet. 2007, 57, 143–173. [Google Scholar] [PubMed]
- Rae, M.; Folch, H.; Moniz, M.B.J.; Wolff, C.W.; McCormack, G.P.; Rindi, F.; Johnson, M.P. Marine bioactivity in irish waters. Phytochem. Rev. 2013, 12, 555–565. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Sennett, S.H.; Kelly-Borges, M.; Bryant, R.W. Ophirapstanol trisulfate, a new biologically active steroid sulfate from the deep water marine sponge Topsentia ophiraphidites. J. Nat. Prod. 1994, 57, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Umeyama, A.; Adachi, K.; Ito, S.; Arihara, S. New 24-isopropylcholesterol and 24-isopropenylcholesterol sulfate from the marine sponge Epipolasis species. J. Nat. Prod. 2000, 63, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- De Riccardis, F.; Minale, L.; Riccio, R.; Iorizzi, M.; Debitus, C.; Duhet, D.; Monniot, C. A novel group of polyhydroxycholanic acid derivatives from the deep water starfish Styracaster caroli. Tetrahedron Lett. 1993, 34, 4381–4384. [Google Scholar] [CrossRef]
- Ding, Y.; Li, X.-C.; Ferreira, D. Theoretical calculation of electronic circular dichroism of the rotationally restricted 3,8′-biflavonoid morelloflavone. J. Org. Chem. 2007, 72, 9010–9017. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.-C.; Ferreira, D. Theoretical calculation of electronic circular dichroism of a hexahydroxydiphenoyl-containing flavanone glycoside. J. Nat. Prod. 2009, 72, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.-C.; Ferreira, D. 4-arylflavan-3-ols as proanthocyanidin models: Absolute configuration via density functional calculation of electronic circular dichroism. J. Nat. Prod. 2010, 73, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-W.; Ding, Y.; Zhang, J.-J.; Liu, X.; Yang, L.-X.; Li, X.-N.; Ferreira, D.; Walker, L.A.; Xu, G. New acylphloroglucinol derivatives with diverse architectures from Hypericum henryi. Org. Lett. 2014, 16, 2434–2437. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- MacroModel; Version 9.9; Schrödinger LLC: New York, NY, USA, 2011.
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Calculation of optical rotation using density functional theory. J. Phys. Chem. A 2001, 105, 5356–5371. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).