
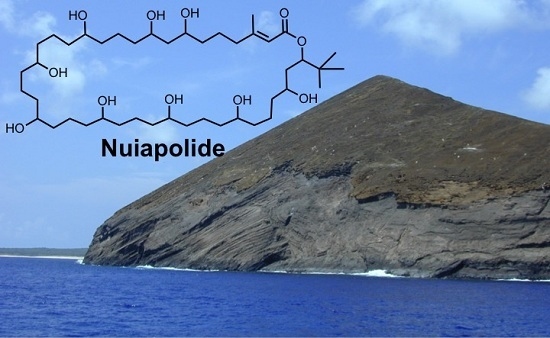
Macrolactone Nuiapolide, Isolated from a Hawaiian Marine Cyanobacterium, Exhibits Anti-Chemotactic Activity


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Evaluation of Hawaiian Marine Natural Product Extracts for Anti-Chemotactic Behavior

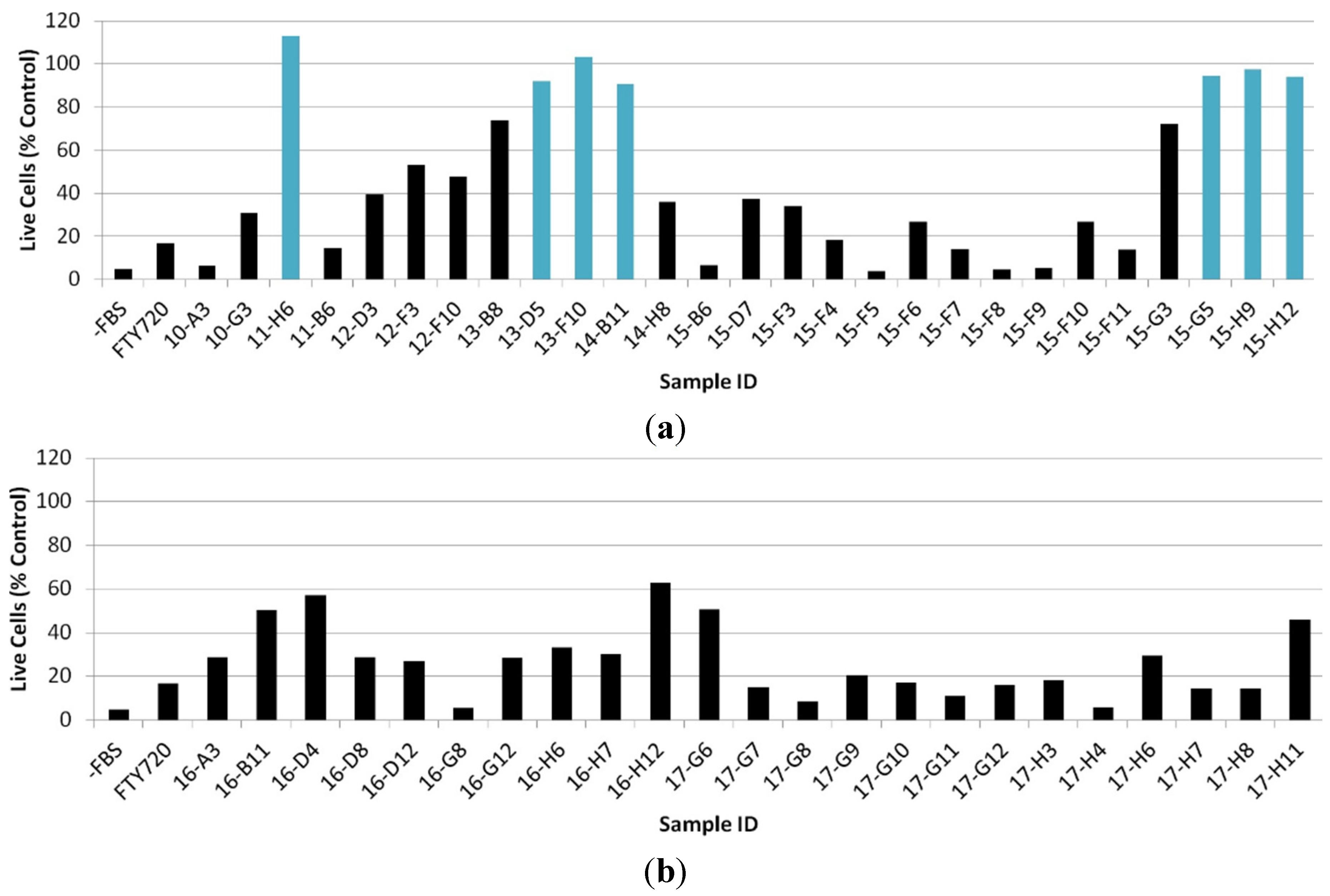
2.2. Cytotoxicity Assay for Active Crude Extracts
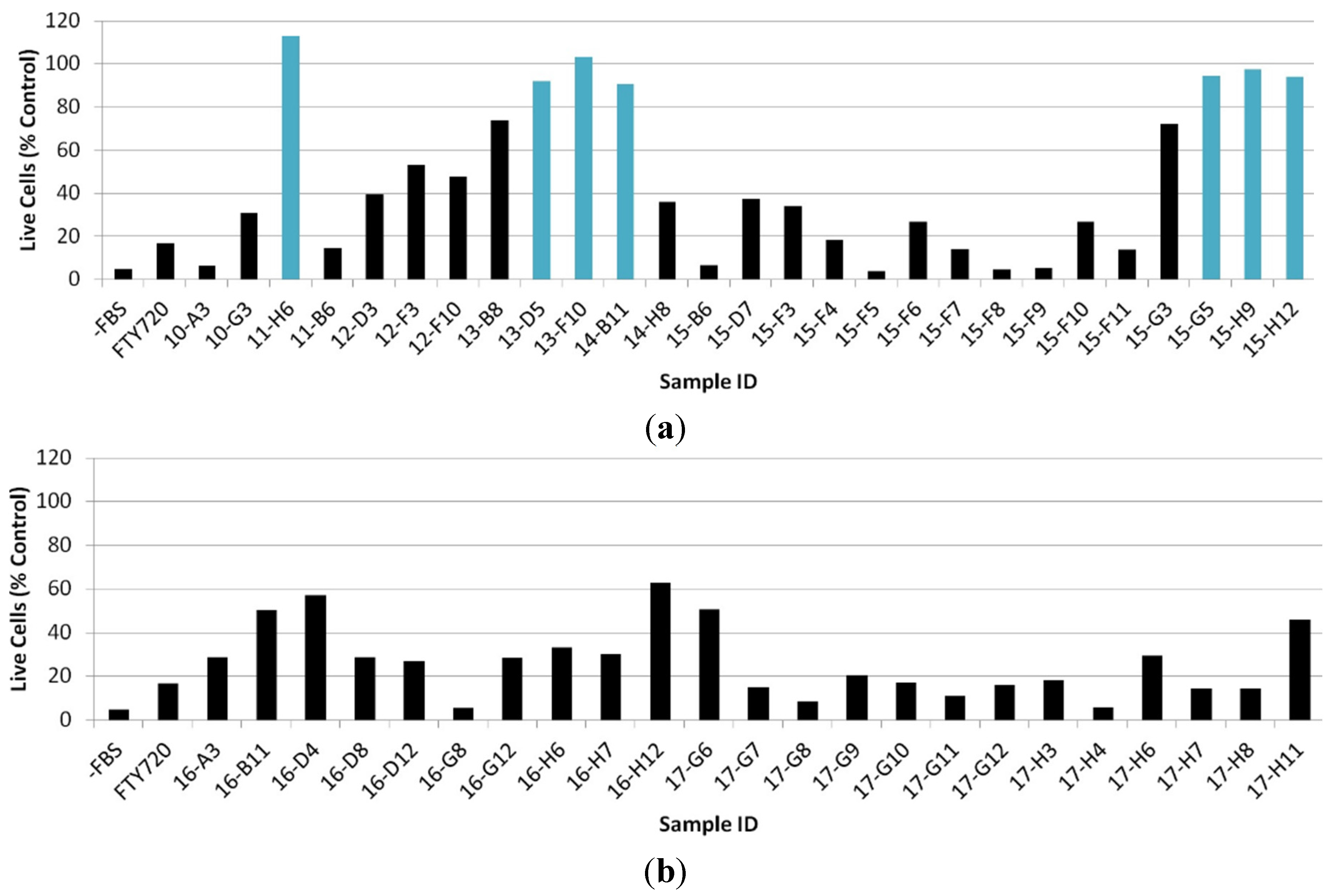
2.3. Evaluation of Fractionated Extracts for Anti-Chemotactic Behavior
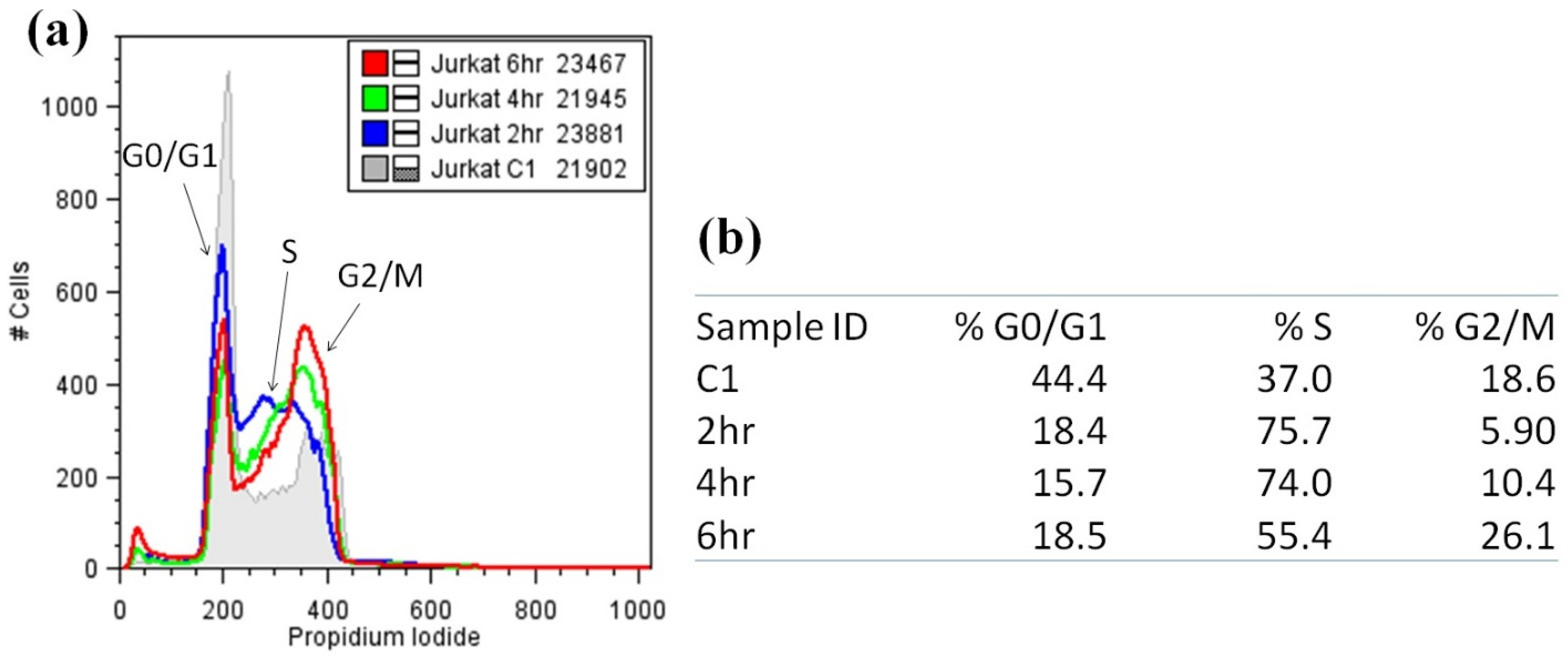
2.4. Cell Cycle Analysis of Nuiapolide (1)


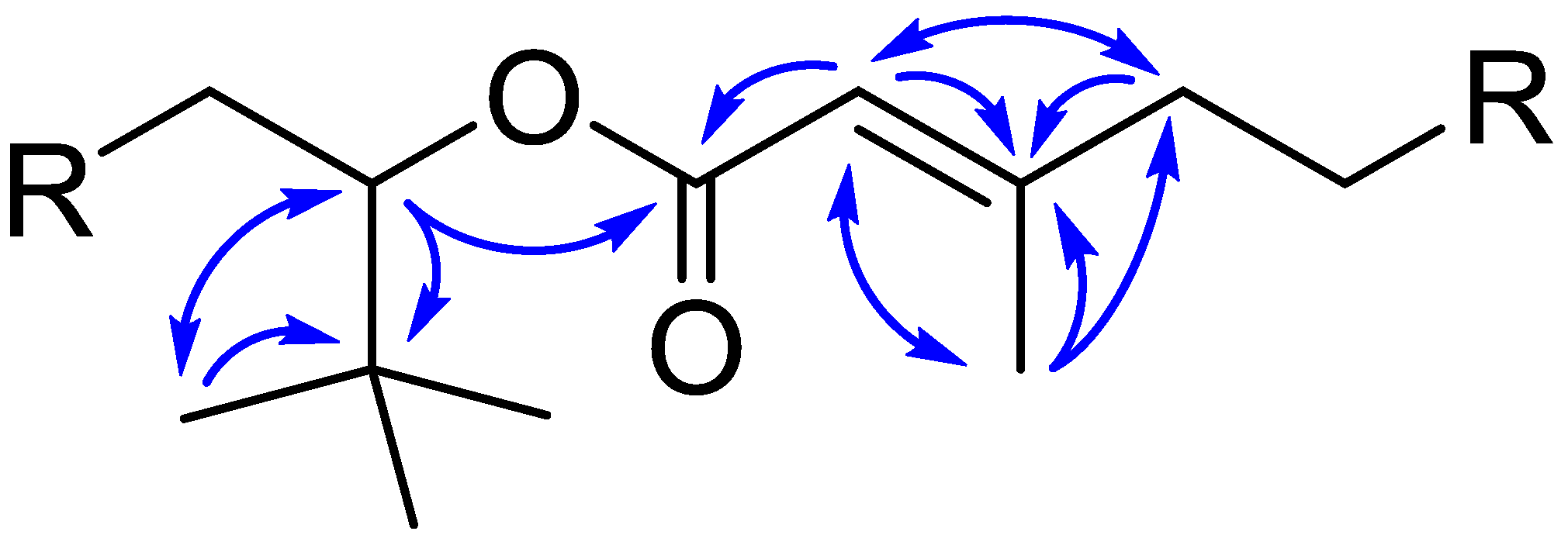
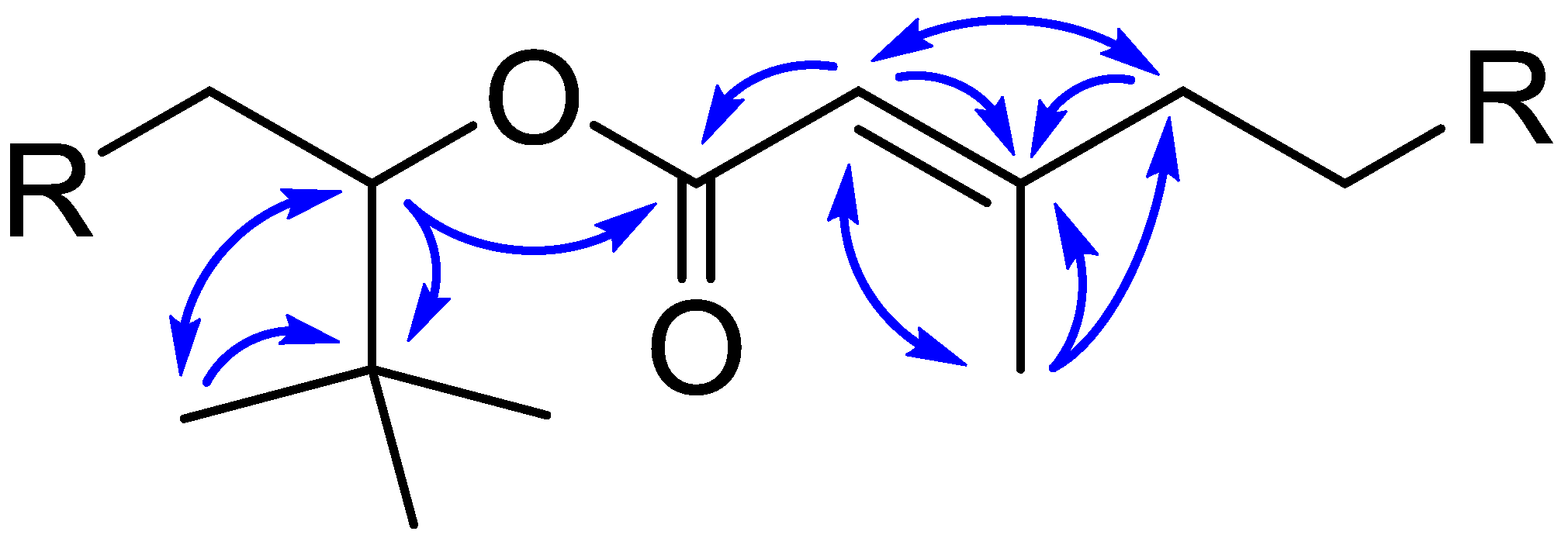
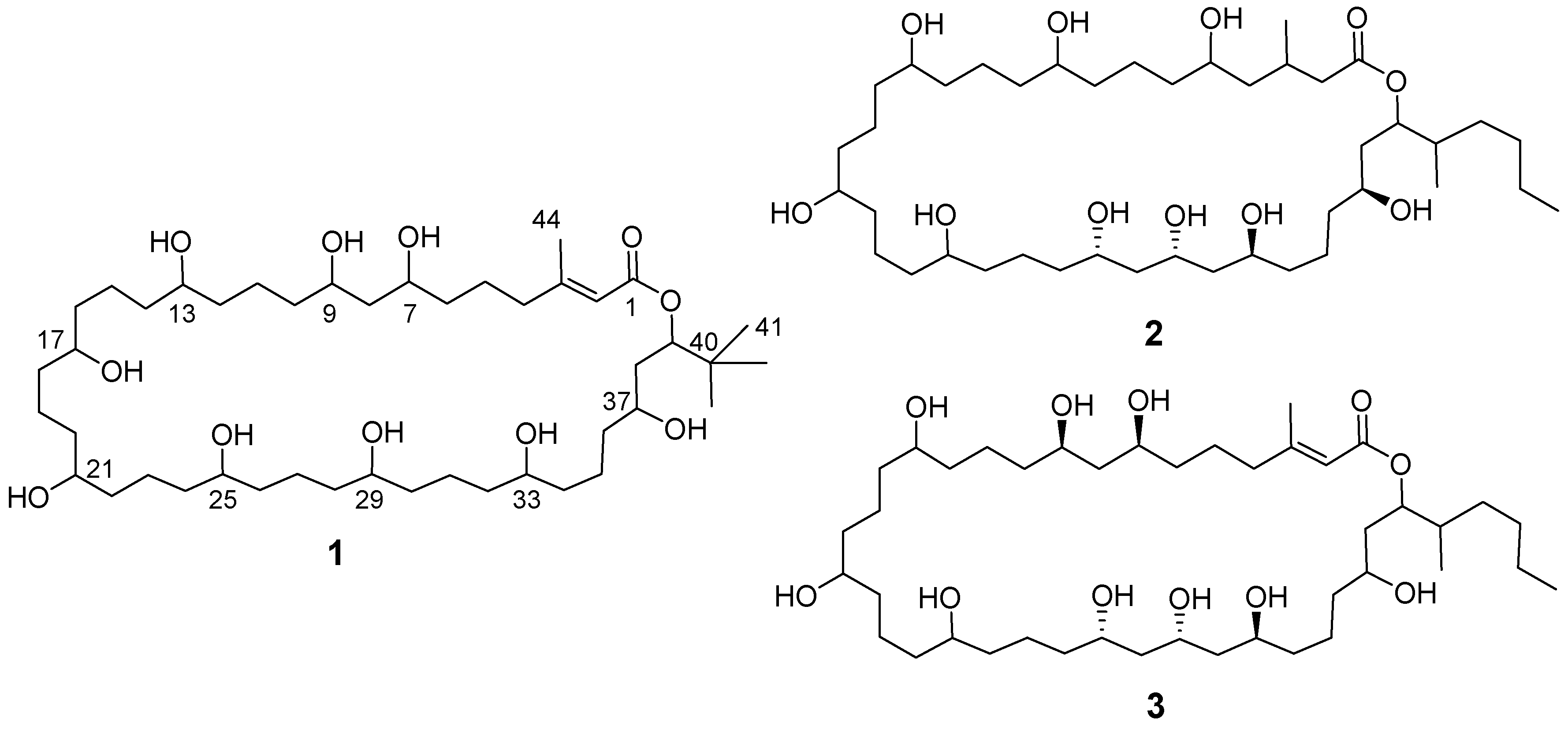
2.5. Structural Analysis of Nuiapolide (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (J in Hz) in MeOH-d4 | δH (J in Hz) in DMSO-d6 | δC in MeOH-d4, Type | δC in DMSO-d6, Type | TOCSY | HSQC-TOCSY b | HMBC c |
|---|---|---|---|---|---|---|---|
| 1 | - | - | 166.90, C | 166.04, C | |||
| 2 | 5.74, s | 5.67, s | 115.73, CH | 116.69, CH | 4, 44, 41–43 | 44 | 1, 3, 4, 44 |
| 3 | - | - | 161.20, C | 160.04, C | |||
| 4 | 2.58, m and 2.83, m | 2.59, m | 32.69, CH2 | 33.11, CH2 | 2, Ha, 5, 6, HC f | 5 | 2, 3, 5 |
| 5 | 1.60, m | 1.49, m | 23.86, CH2 | 24.08, CH2 | 4, H-a, a-OH, HC f | 4, C-i, C-iii | |
| 6 (C-i) d | 1.49, m | 1.31, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | 4, H-a, a-OH, HC f | C-i, C-iii | |
| 7 (H-a) e (C-iii) d | 3.78, s | 3.58, s | 69.8–70.8, CH | 69.2–70.2, CH | 4, a-OH, H-b, HC f | C-i, 8 | |
| (7)-OH | - | 4.44, d (4.4) or 4.47, d (4.2) | - | - | H-a, HC f | 8, C-i, C-iii | |
| 8 | 1.56, m | 1.37 | 43.51, CH2 | 44.67, CH2 | H-a, a-OH, HC f | C-i, C-ii, C-iii | C-iii |
| 9 (H-a) e (C-iii) d | 3.78, s | 3.58 | 69.8–70.8, CH | 69.2–70.2, CH | 4, a-OH, H-b, HC f | C-i, 8 | |
| (9)-OH | - | 4.44, d (4.4) or 4.47, d (4.2) | - | - | H-a, HC f | 8, C-i, C-iii | |
| 10 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-a, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 11 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 12 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 13 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2, CH | H-a, H-c, HC f | C-i, C-ii | |
| (13)-OH | - | 4.14, d (4.1) | - | - | H-b, HC f | C-i, C-iii | |
| 14 (C-i) c | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 15 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 16 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 17 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2 | H-a, H-c, HC f | C-i, C-ii | |
| (17)-OH | - | 4.14, d (4.1) | - | - | H-b, HC f | C-i, C-iii | |
| 18 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 19 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 20 (C-i) c | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 21 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2, CH | H-a, H-c, HC f | C-i, C-ii | |
| (21)-OH | - | 4.14, d (4.1) | - | - | H-b, HC f | C-i, C-ii | |
| 22 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 23 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 24 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 25 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2, CH | H-a, H-c, HC f | C-i, C-ii | |
| (25)-OH | - | 4.14, d (4.1) | - | - | H-b, HC f | C-i, C-iii | |
| 26 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 27 (C-ii)d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 28 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 29 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2, CH | H-a, H-c, HC f | C-i, C-ii | |
| (29)-OH | - | 4.14, d (4.1) | - | - | H-b, HC f | C-i, C-iii | |
| 30 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 31 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 32 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 33 (H-b) e (C-iii) d | 3.57, s | 3.37, s | 69.8–70.8, CH | 69.2–70.2, CH | H-a, H-c, HC f | C-i, C-ii | |
| (33)-OH | - | 4.14. d (4.1) | - | - | H-b, HC f | C-i, C-iii | |
| 34 (C-i) d | 1.48–1.59, m | 1.24–1.41, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 35 (C-ii) d | 1.31–1.55, m | 1.19–1.37, m | 20.8–21.5, CH2 | 21.4–22.0, CH2 | H-b, HC f | C-i, C-ii, C-iii | C-i, C-iii |
| 36 (C-i) d | 1.42, m | 1.28, m | 36.2–37.4, CH2 | 37.2–38.1, CH2 | H-b, HC f | 37, C-i, C-ii, C-iii | C-i, C-ii, C-iii |
| 37 (H-c) e | 3.42, m | 3.25, m | 67.58, CH | 67.25, CH | H-b, c-OH, 39 | C-i | |
| (37)-OH | - | 4.15, d (5.58) | - | - | 39, H-b, HC f | 38, C-i, C-iii | |
| 38 | 1.58, m | 1.44, m | 37.55, CH2 | 38.56, CH2 | H-c, c-OH, HC f | 37, 39 | C-i, C-iii |
| 39 | 5.02, dd (9.7, 2.5) | 4.94, dd (9.7, 2.5) | 76.82, CH | 76.91, CH | H-c, c-OH, 41–43, 44, HC f | 38 | 1, 37, 38, 40, 41–43 |
| 40 | - | - | 33.91, C | 34.60, C | |||
| 41–43 | 0.93, s | 0.85, s | 25.00, CH3 | 26.27, CH3 | 2, 39 | 39, 40 | |
| 44 | 1.96, s | 1.87, s | 23.89, CH3 | 24.91, CH3 | 39 | 2, 5 | 2, 3, 4 |
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Reagents and Chemicals
3.3. Sample Preparation for Assay Analysis
3.3.1. Details on Cyanobacteria Biomass
3.3.2. DNA Extraction and Sequencing
3.3.3. Scaled-Up Extraction of the Active Cyanobacterial Species
3.3.4. Bioassay-Linked Fractionation of Methanol and Dichloromethane Extracts of the Cyanobacterium 071905-NII-01
3.3.5. Isolation of Nuiapolide (1)
3.3.6. Structural Analysis of Nuiapolide (1)
3.4. Chemotaxis Assay with Jurkat Cells
3.5. Cytotoxicity Assay with Jurkat Cells
3.6. Cell Cycle Analysis by Flow Cytometry
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Attinger, P. La médicine mésopotamienne. J. Med. Cuneif. 2008, 11–12, 1–96. [Google Scholar]
- Nunn, J. Ancient Egyptian Medicine; University of Oklahoma Press: Norman, OK, USA, 2002. [Google Scholar]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar] [PubMed]
- Hong, F. History of medicine in China. McGill J. Med. 2004, 8, 7984. [Google Scholar]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenza. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar]
- Houbraken, J.; Frisvad, J.C.; Samson, R.A. Fleming’s penicillin producing strain is not Penicillium chrysogenum but P. rubens. IMA Fungus 2011, 2, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Keam, S.J. Trabectedin: A review of its use in the management of soft tissue sarcoma and ovarian cancer. Drugs 2007, 67, 2257–2276. [Google Scholar] [CrossRef] [PubMed]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Wadhams, G.H.; Armitage, J.P. Making sense of it all: Bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 2004, 5, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Keshikar, N.D.; Weyand, I. Mechanisms of sperm chemotaxis. Annu. Rev. Physiol. 2008, 70, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Sidik, S.M.; Mahmud, R.; Stanslas, J. Molecular targets in the discovery and development of novel antimetastatic agents: Current progress and future prospects. Clin. Exp. Pharmacol. Physiol. 2013, 40, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Strader, C.R.; Pearce, C.J.; Oberlies, N.H. Fingolimod (FTY720): A recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 2011, 74, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Boyden, S.V. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.; Schwenk, H.; Bornkamm, G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer 1977, 19, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.D.; Ji, X.J.; Cong, Z.X.; Zhu, J.H.; Zhou, Y. FTY720 for cancer therapy (review). Oncol. Rep. 2013, 30, 2571–2578. [Google Scholar] [PubMed]
- Qin, X.; Yue, Z.; Sun, B.; Yang, W.; Xie, J.; Ni, E.; Feng, Y.; Mahmood, R.; Zhang, Y.; Yue, L. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. Br. J. Pharmacol. 2013, 168, 1294–1312. [Google Scholar] [CrossRef] [PubMed]
- Schafer, K.A. The cell cycle: A review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Bockstaele, D.R.V.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, J.B.; Molinski, T.F. Caylobolide A, a unique 36-membered macrolactone from a Bahamian Lyngbya majuscula. Org. Lett. 2002, 4, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Paul, V.J.; Luesch, H. Caylobolide B, a macrolactone from symplostatin 1-producing marine cyanobacteria Phormidium spp. from Florida. J. Nat. Prod. 2010, 73, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Nübel, U.; Garcia-Pichel, F.; Muyzer, G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 1997, 63, 3327–3332. [Google Scholar] [PubMed]
- Engene, N.; Paul, V.J.; Byrum, T.; Gerwick, W.H.; Thor, A.; Ellisman, M.H. Five chemically rich species of tropical marine cyanobacteria of the genus Okeania gen. nov. (Oscillatoriales, Cyanoprokaryota). J. Phycol. 2013, 49, 1095–1106. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Juan, G. Unit 7.5 DNA content measurement for DNA ploidy and cell cycle analysis. In Current Protocols in Cytometry; Chambers, K., Ed.; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Castillo, G.; Demoulin, V.V. Madangolide and laingolide A, two novel macrolides from Lyngbya bouillonii (Cyanobacteria). J. Nat. Prod. 1999, 62, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Braekman, J.-C.; Daloze, D.; Hoffmann, L.; Demoulin, V. Laingolide, a novel 15-membered macrolide from Lyngbya bouillonii (Cyanophyceae). Tetrahedron. Lett. 1996, 37, 7519–7520. [Google Scholar] [CrossRef]
- Salvador-Reyes, L.A.; Sneed, J.; Paul, V.J.; Luesch, H. Amantelides A and B, polyhydroxylated macrolides with differential broad-spectrum cytotoxicity from a Guamanian marine cyanobacterium. J. Nat. Prod. 2015, 78, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.-L.; Linington, R.G.; Balunas, M.J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T.S.; Kyle, D.E.; et al. Bastimolide A, a potent antimalarial polyhydroxy macrolide from the marine cyanobacterium Okeania hirsuta. J. Org. Chem. 2015, 80, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, S.; Williams, H.; Cagle, D.; Karanovich, K.; Horgen, F.D.; III, R.S.; Watanabe, C.M.H. Macrolactone Nuiapolide, Isolated from a Hawaiian Marine Cyanobacterium, Exhibits Anti-Chemotactic Activity. Mar. Drugs 2015, 13, 6274-6290. https://doi.org/10.3390/md13106274
Mori S, Williams H, Cagle D, Karanovich K, Horgen FD, III RS, Watanabe CMH. Macrolactone Nuiapolide, Isolated from a Hawaiian Marine Cyanobacterium, Exhibits Anti-Chemotactic Activity. Marine Drugs. 2015; 13(10):6274-6290. https://doi.org/10.3390/md13106274
Chicago/Turabian StyleMori, Shogo, Howard Williams, Davey Cagle, Kristopher Karanovich, F. David Horgen, Roger Smith III, and Coran M. H. Watanabe. 2015. "Macrolactone Nuiapolide, Isolated from a Hawaiian Marine Cyanobacterium, Exhibits Anti-Chemotactic Activity" Marine Drugs 13, no. 10: 6274-6290. https://doi.org/10.3390/md13106274
APA StyleMori, S., Williams, H., Cagle, D., Karanovich, K., Horgen, F. D., III, R. S., & Watanabe, C. M. H. (2015). Macrolactone Nuiapolide, Isolated from a Hawaiian Marine Cyanobacterium, Exhibits Anti-Chemotactic Activity. Marine Drugs, 13(10), 6274-6290. https://doi.org/10.3390/md13106274