
Araguspongine C Induces Autophagic Death in Breast Cancer Cells through Suppression of c-Met and HER2 Receptor Tyrosine Kinase Signaling



Abstract
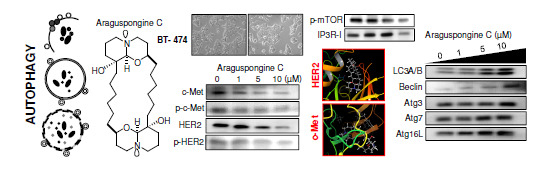
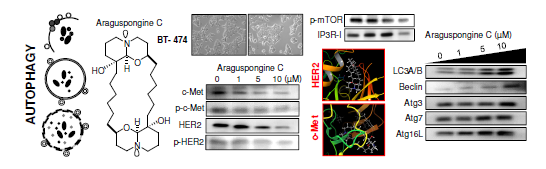
:
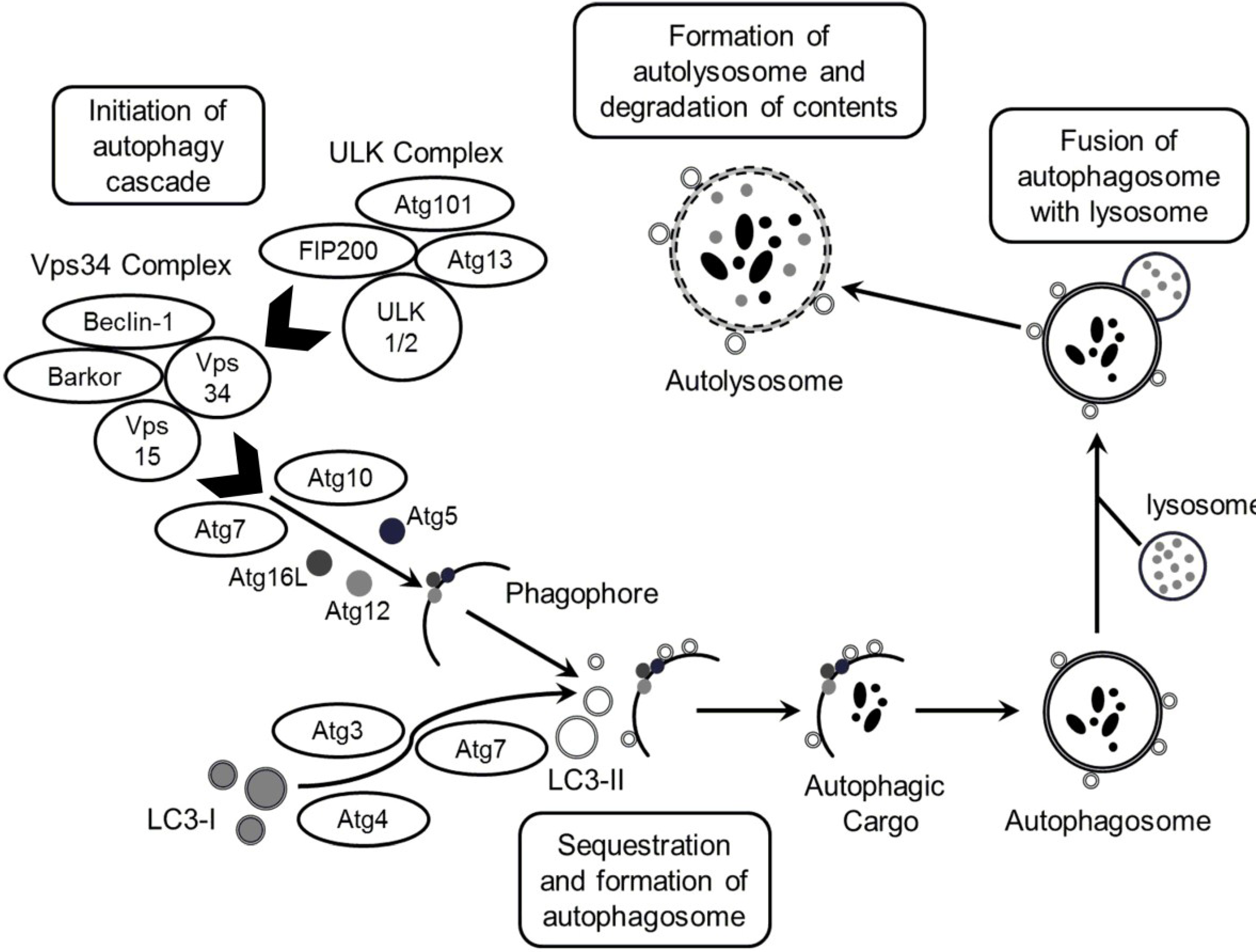
1. Introduction
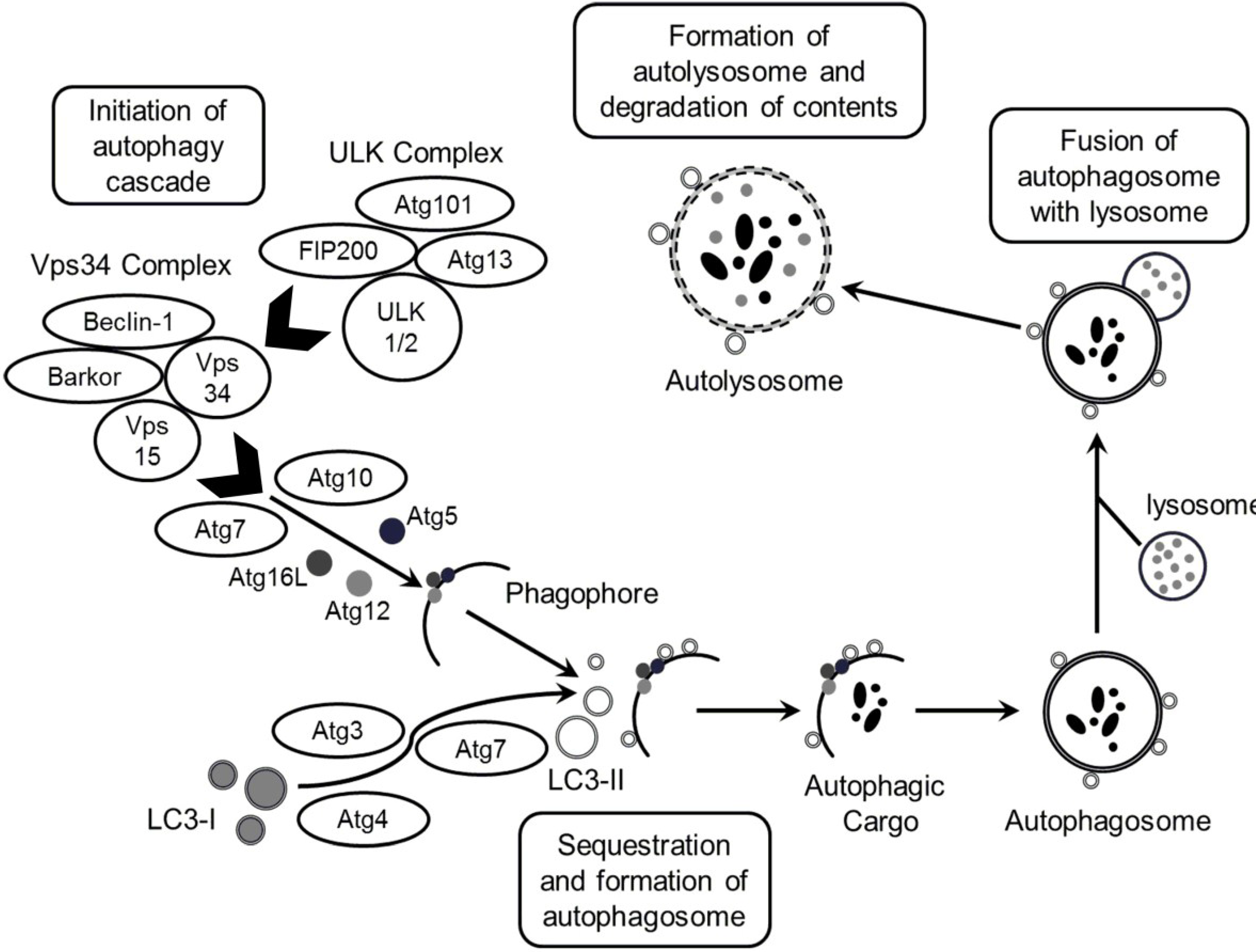
2. Results
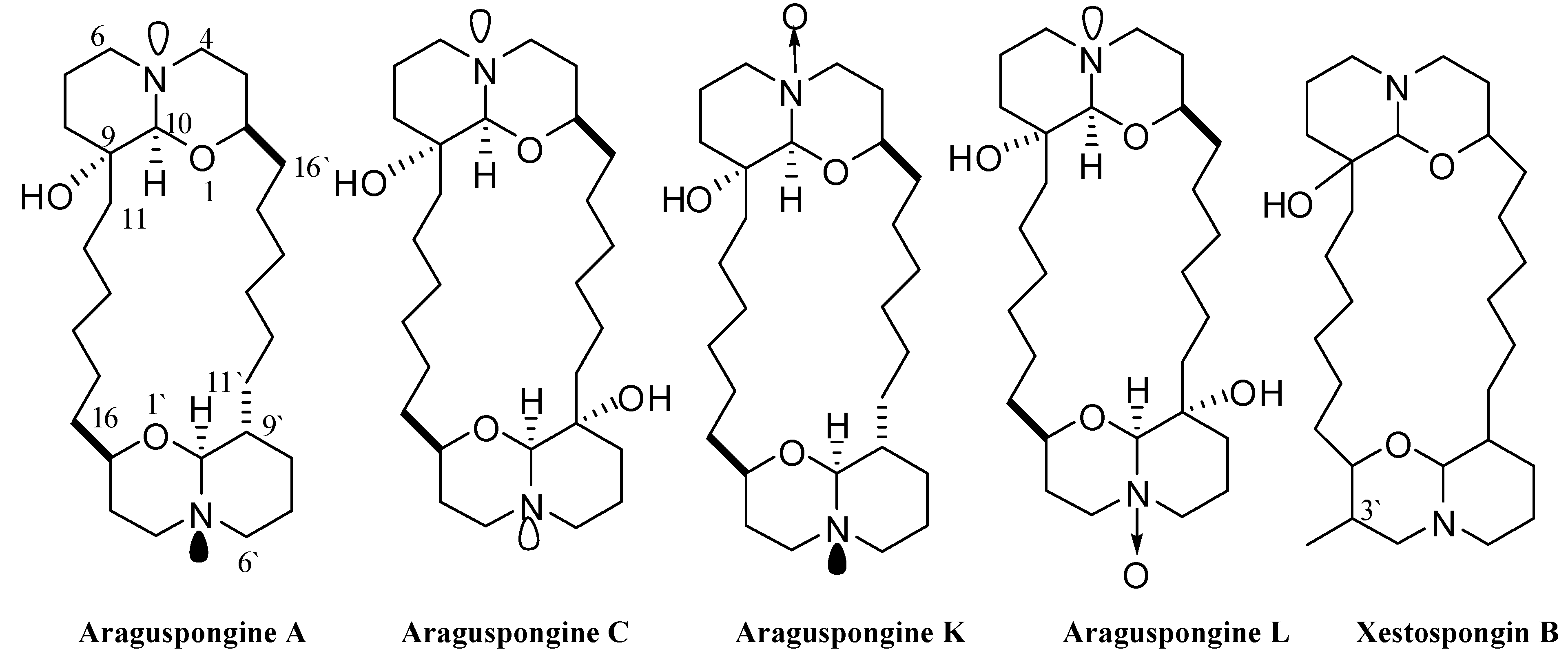
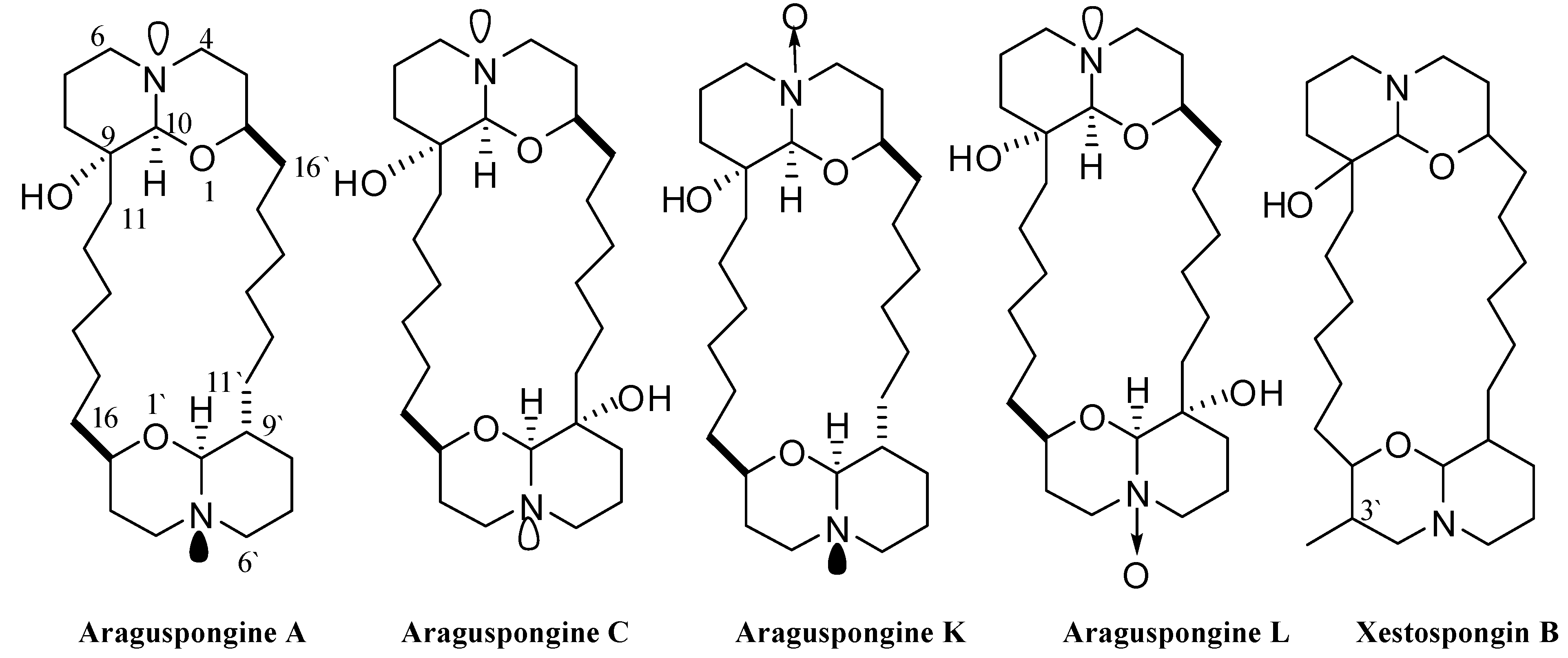
2.1 Chemical Diversity of Tested Oxaquinolizidine Alkaloids and Their Effect on Breast Cancer Cell Viability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) ± SEM |
|---|---|
| Araguspongine A | 9.3 ± 3.2 |
| Araguspongine C | 15.2 ± 2.1 |
| Araguspongine K | 29.5 ± 3.8 |
| Araguspongine L | 35.6 ± 3.7 |
| Xestospongin B | 52.5 ± 4.5 |
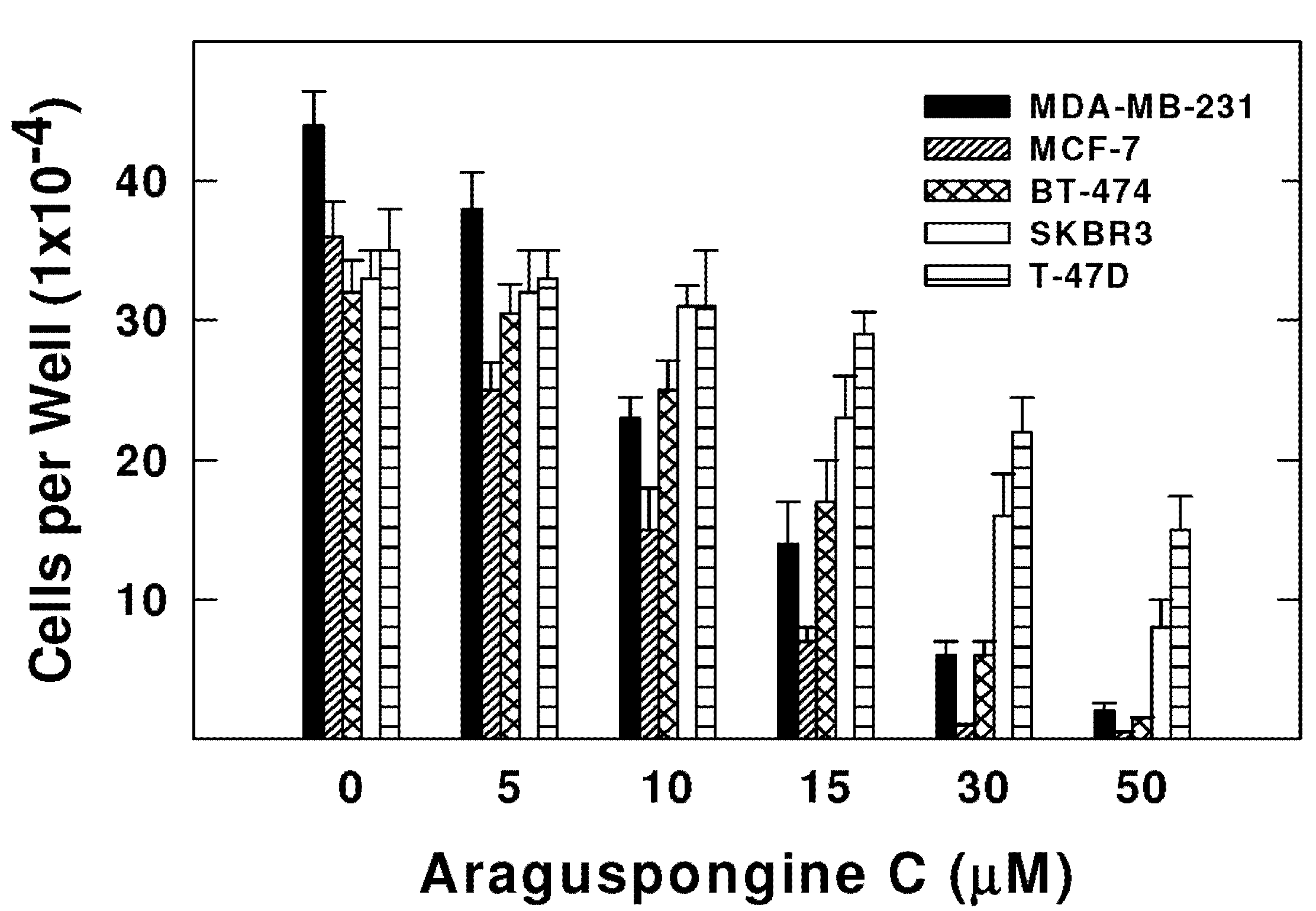
2.2. Effects of Araguspongine C on Viability, Morphology, and Colony Formation of Breast Cancer Cells
| Cell Line | IC50 (µM) ± SEM |
|---|---|
| MDA-MB-231 | 10.1 ± 2.3 |
| MCF-7 | 8.5 ± 1.6 |
| BT-474 | 15.2 ± 2.1 |
| SKBR3 | 18.3 ± 2.5 |
| T-47D | 46.1 ± 4.8 |
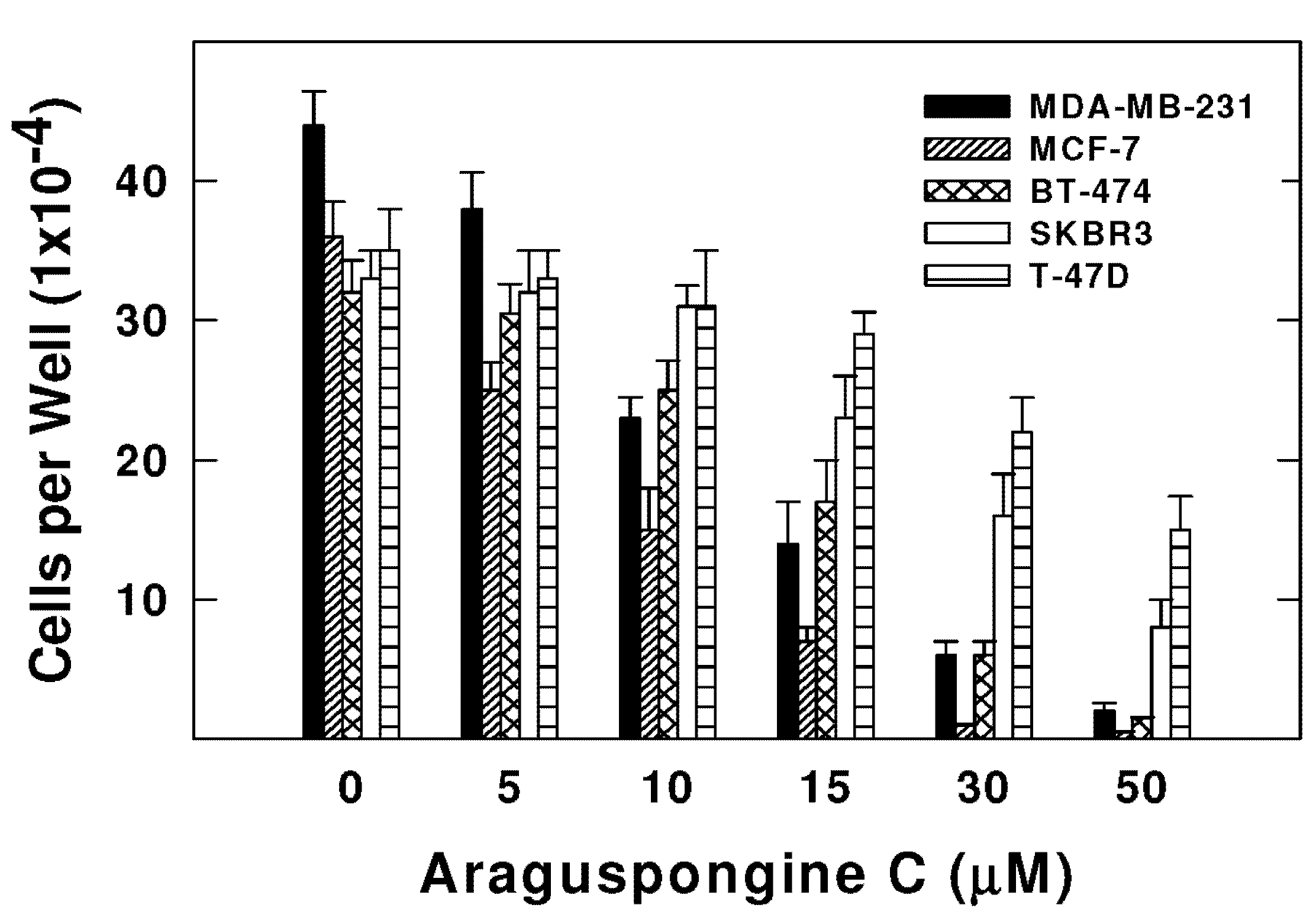

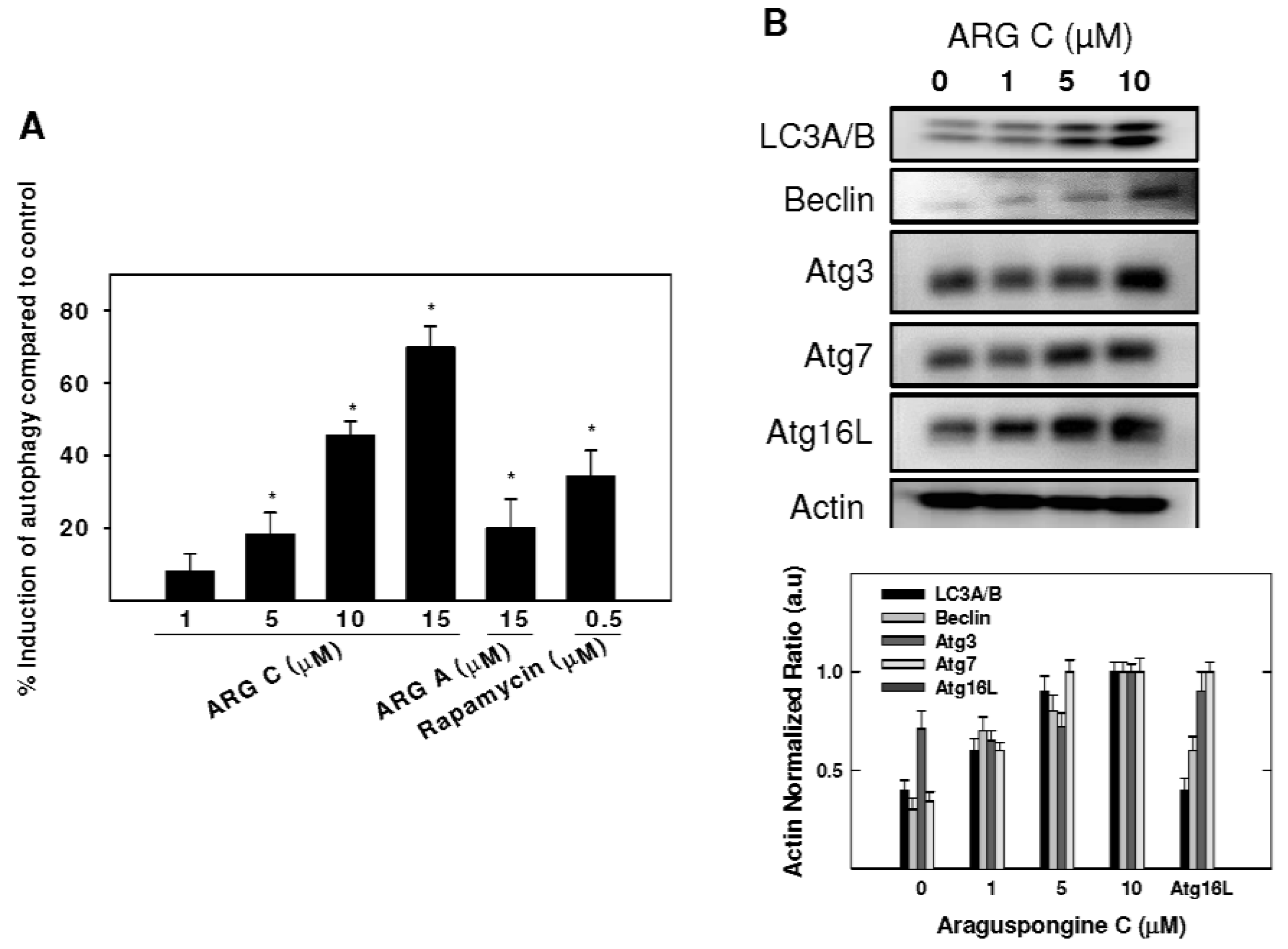
2.3. Autophagic Activity of Araguspongine C in BT-474 Breast Cancer Cells

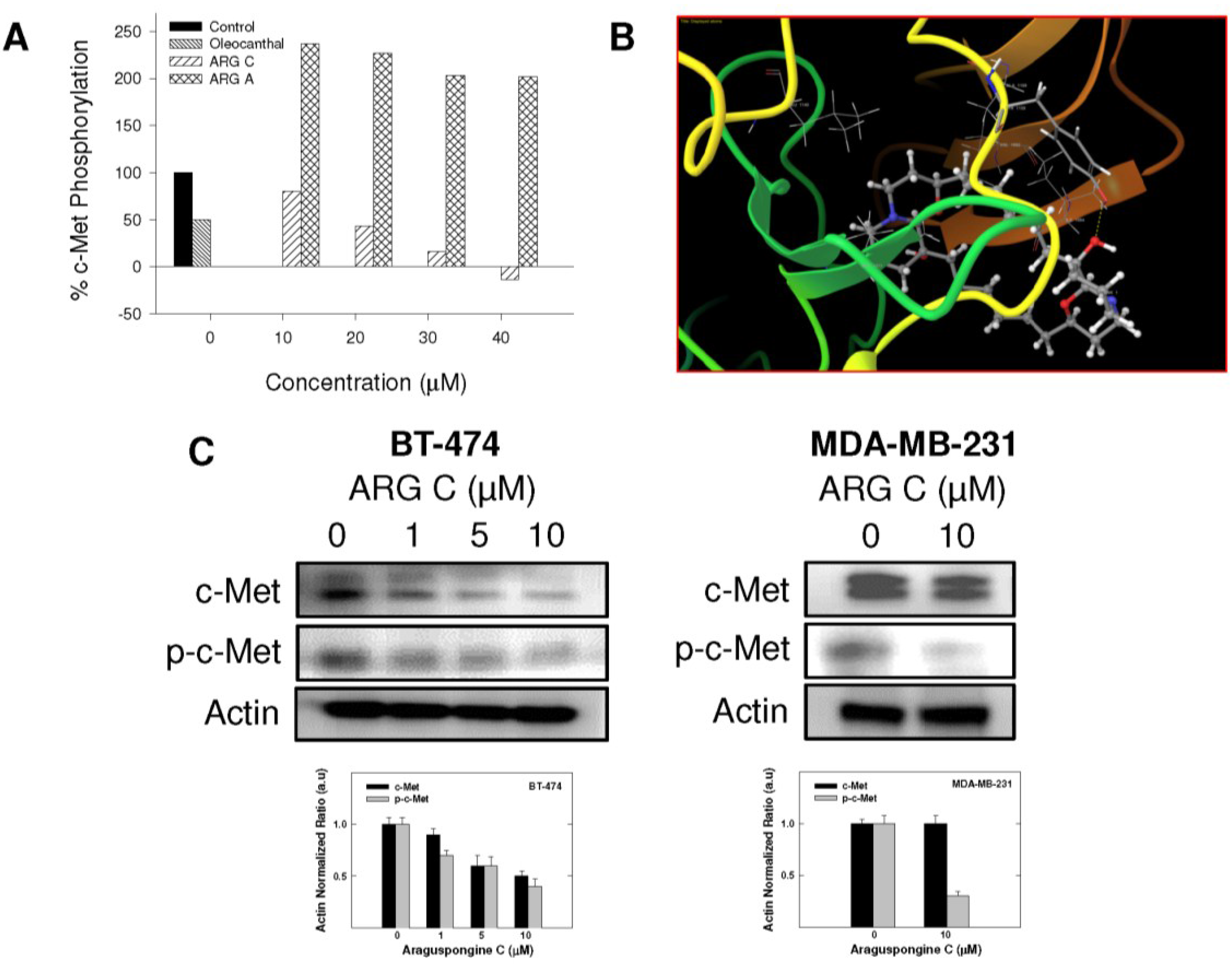
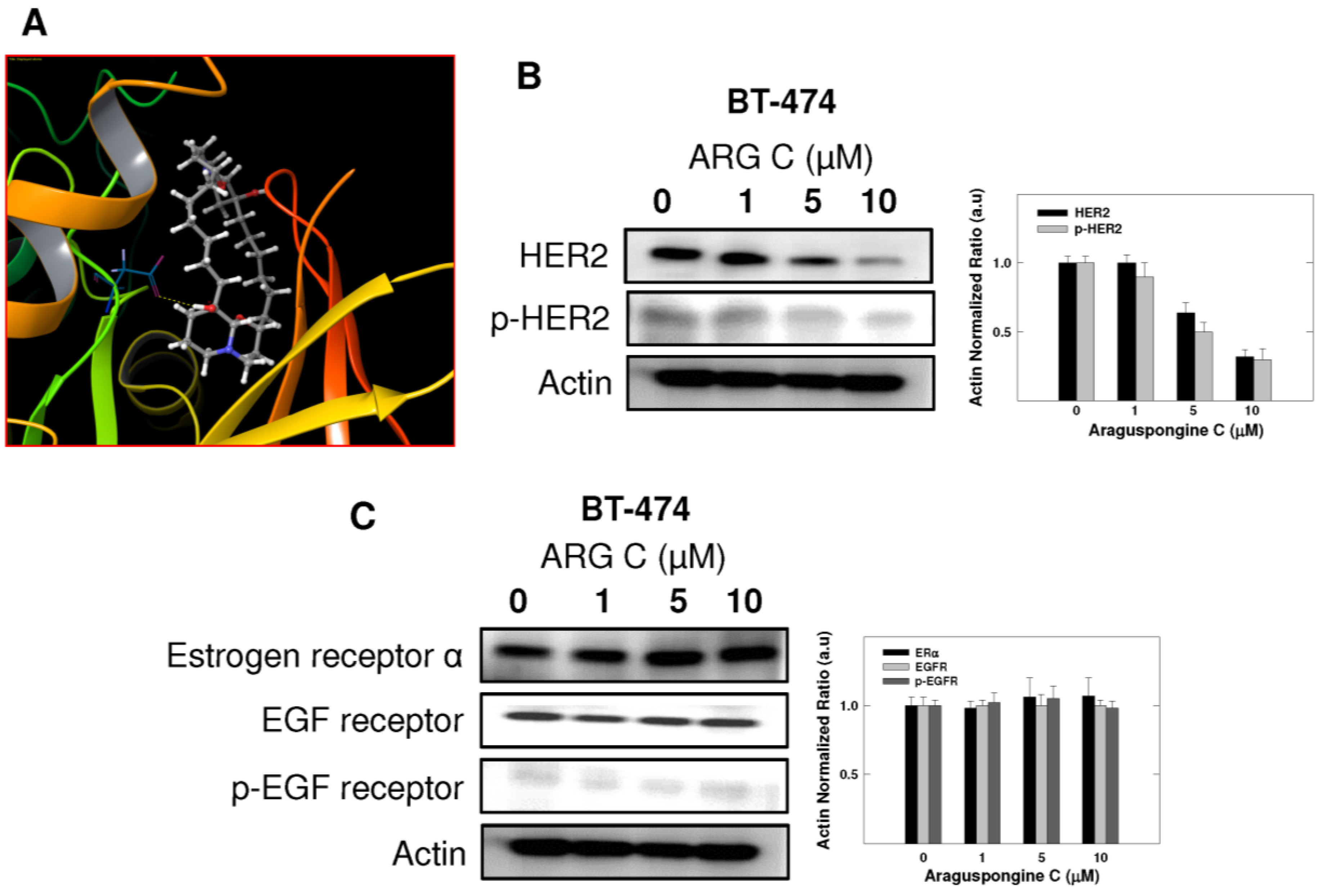
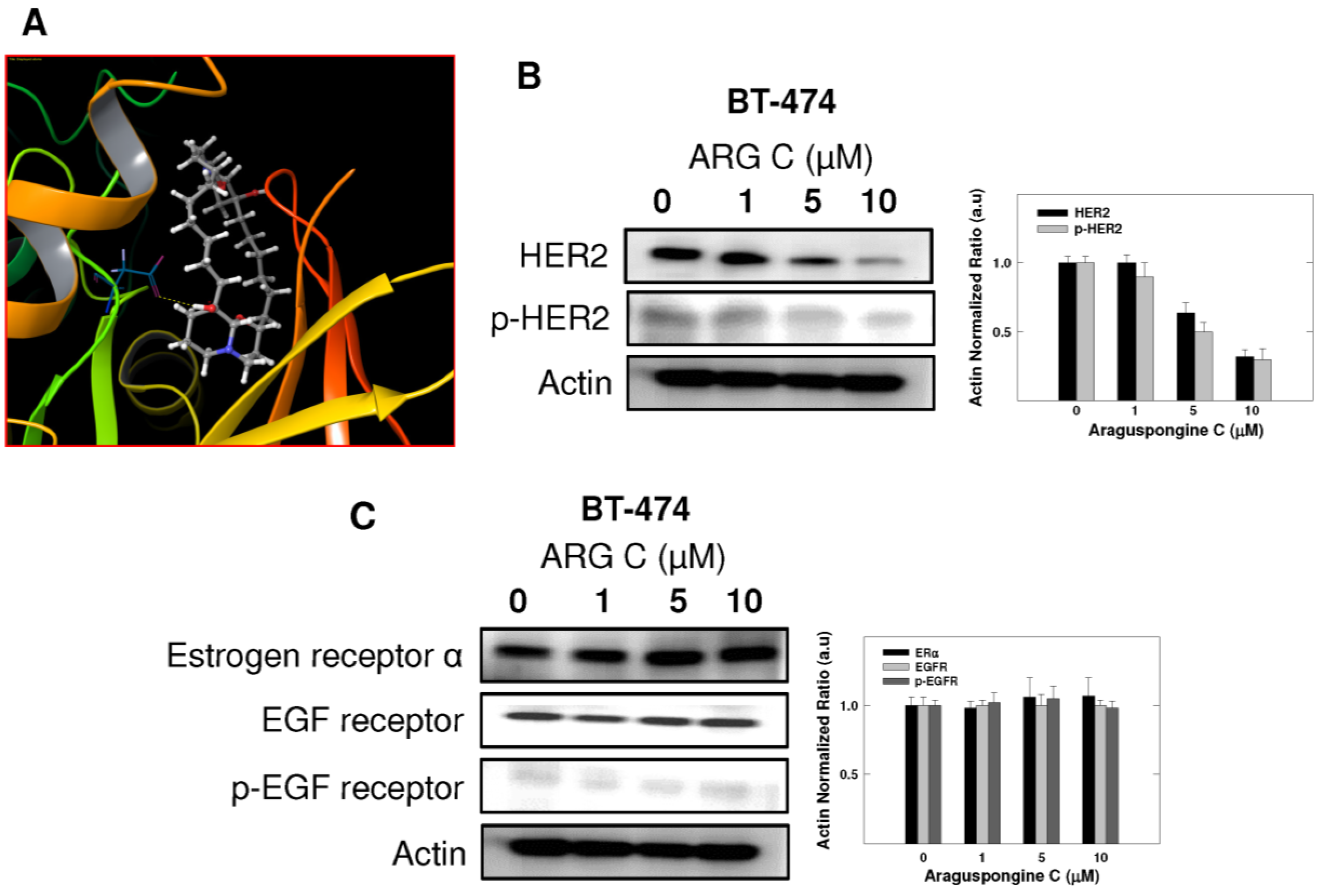
2.4. Effect of Araguspongine C on c-Met and HER2 Receptor Tyrosine Kinase Activation
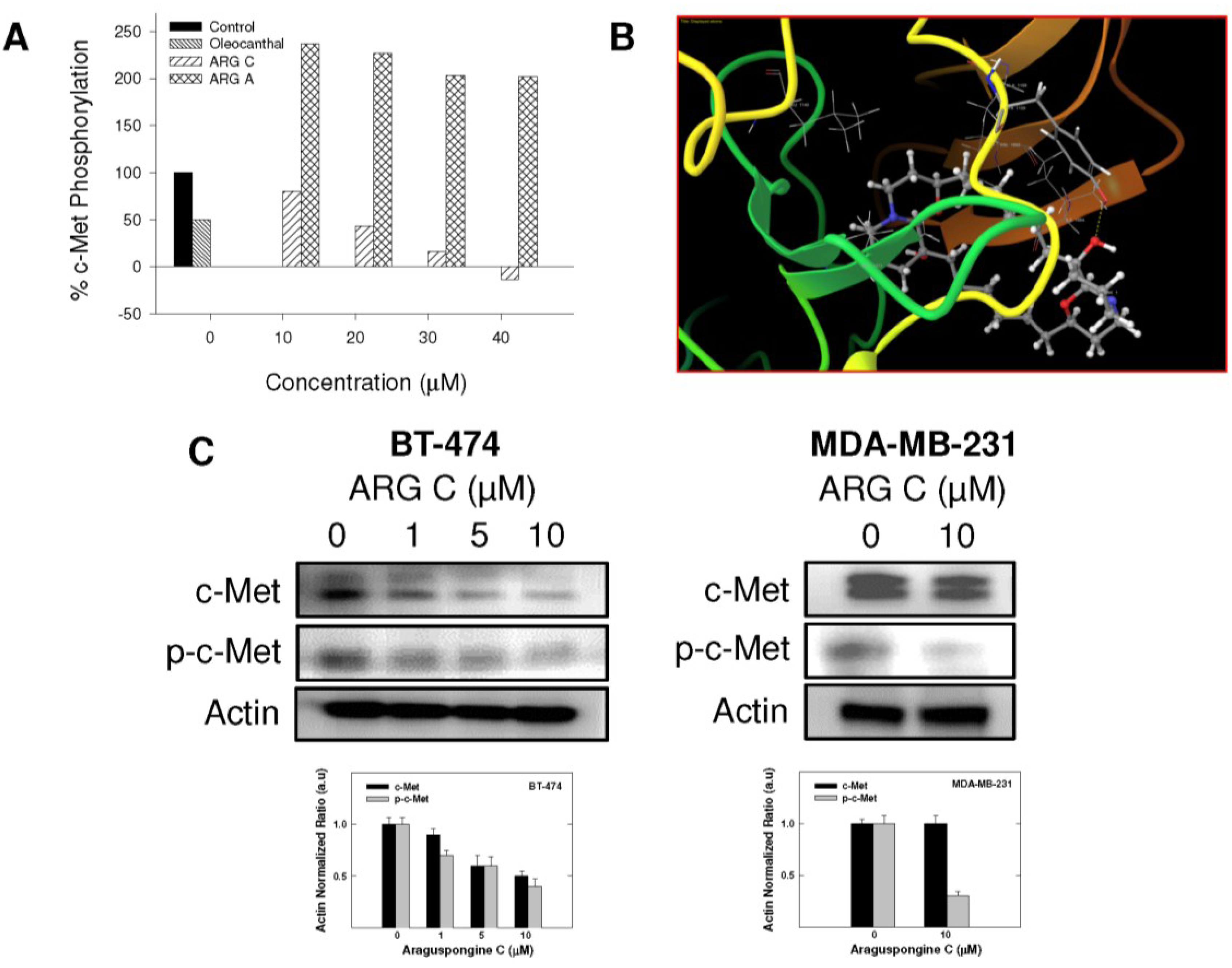
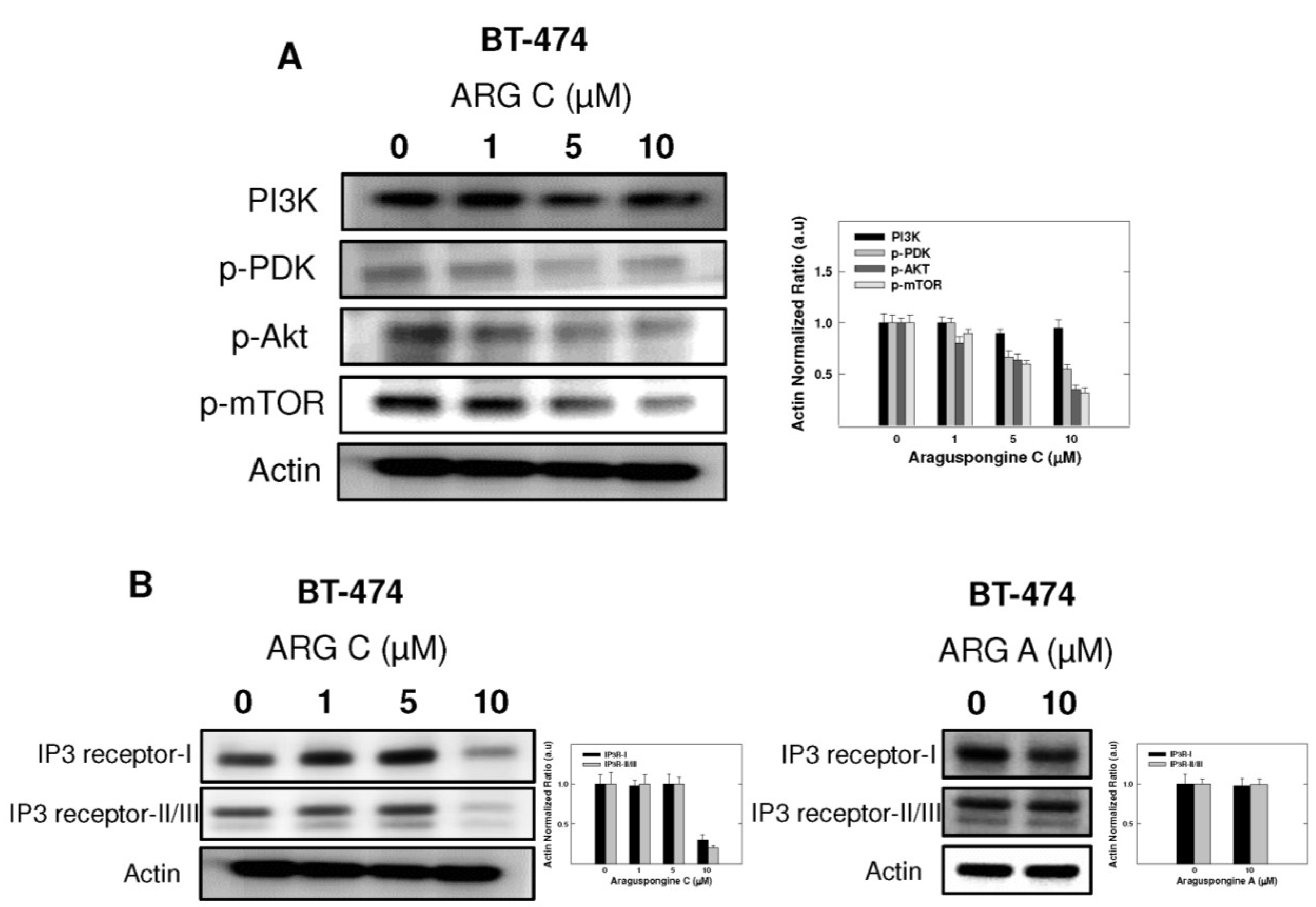
2.5. Effect of Araguspongine C on PI3K/Akt/mTOR Signaling Pathway and IP3 Receptor Levels

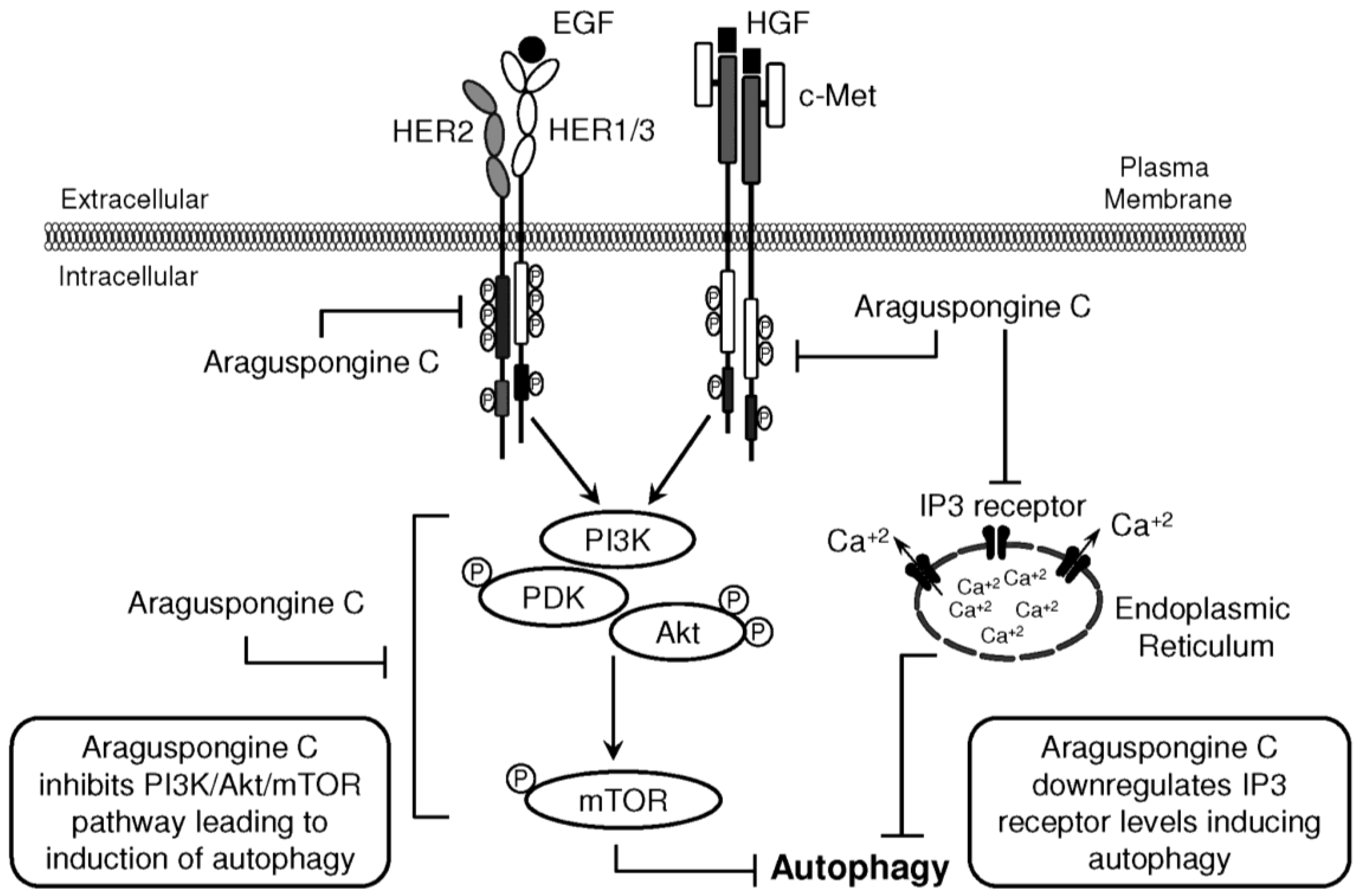
3. Discussion

4. Experimental Section
4.1. Chemicals, Reagents and Antibodies
4.2. Cell Lines and Culture Conditions
4.3. Measurement of Viable Cell Number
4.4. Cell Viability Assays
4.5. Soft Agar Assay
4.6. Western Blot Analysis
4.7. Apoptosis Analysis with Annexin V Staining by Flow Cytometry
4.8. Cyto-ID Staining Assay
4.9. Z-LYTE c-Met Kinase Assay
4.10. Molecular Modeling
4.10.1. Protein Structure Preparation
4.10.2. Ligand Structure Preparation
4.11. Molecular Docking
4.12. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| ER | estrogen receptor |
| EGFR | epidermal growth factor receptor |
| HER-2 | human epidermal growth factor receptor 2 |
| HGF | hepatocyte growth factor |
| PDB | protein data bank |
| H & E | hematoxylin and eosin |
| ATCC | American Type Culture Collection |
| RPMI | Roswell Park Memorial Institute |
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| RIPA | radioimmunoprecipitation assay |
| BCA | bicinchoninic acid assay |
| PVDF | polyvinylidene fluoride |
| BSA | bovine serum albumin |
| NIH | National Institutes of Health |
| HRP | horseradish peroxidase |
| MAPK | mitogen-activated protein kinase |
| PI3K | phosphatidylinositol 3-kinase |
| PLC-γ | phospholipase C-γ |
| STATs | transcription factors like the signal transducers and activators of transcription |
Conflicts of Interest
References
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Rho, O.; Kim, D.J.; Kiguchi, K.; Digiovanni, J. Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol. Carcinog. 2011, 50, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Salgia, R. MET pathway as a therapeutic target. J. Thorac. Oncol. 2009, 4, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Mendrola, J.M.; Shi, F.; Park, J.H.; Lemmon, M.A. Receptor tyrosine kinases with intracellular pseudokinase domains. Biochem. Soc. Trans. 2013, 41, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic autophagy in cancer therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Parys, J.B.; Decuypere, J.P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, W.; Sui, X.; Fang, Y.; Pan, H. Autophagy: A novel therapeutic target for hepatocarcinoma (Review). Oncol. Lett. 2014, 7, 1345–1351. [Google Scholar] [PubMed]
- Akl, M.R.; Foudah, A.I.; Ebrahim, H.Y.; Meyer, S.A.; El Sayed, K.A. The marine-derived sipholenol A-4-O-3′,4′-dichlorobenzoate inhibits breast cancer growth and motility in vitro and in vivo through the suppression of Brk and FAK signaling. Mar. Drugs 2014, 12, 2282–2304. [Google Scholar] [CrossRef] [PubMed]
- Orabi, K.Y.; El Sayed, K.A.; Hamann, M.T.; Dunbar, D.C.; Al-Said, M.S.; Higa, T.; Kelly, M. Araguspongines K and L, new bioactive bis-1-oxaquinolizidine N-oxide alkaloids from Red Sea specimens of Xestospongia exigua. J. Nat. Prod. 2002, 65, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.S.; MacMillan, J.B.; Olmstead, M.M.; Ta, T.A.; Pessah, I.N.; Molinski, T.F. (+)-7S-Hydroxyxestospongin A from the marine sponge Xestospongia sp. and absolute configuration of (+)-xestospongin D. J. Nat. Prod. 2002, 65, 249–254. [Google Scholar] [CrossRef]
- Nakagawa, M.; Endo, M.; Tanaka, N.; Gen-Pei, L. Structures of xestospongin A, B, C and D, novel vasodilativecompounds from marine sponge Xestospongia exigua. Tetrahed. Lett. 1984, 25, 3227–3230. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, O.B.; Doubek, D.L.; Tackett, L.; Schmidt, J.M.; Boyd, M.R.; Pettit, R.K.; Hooper, J.N.A. Isolation and X-ray crystal structure of racemic xestospongin D from the Singapore marine sponge Niphates sp. Bioorg. Med. Chem. Lett. 1996, 6, 1313–1318. [Google Scholar] [CrossRef]
- Pimentel, S.M.; Bojo, Z.P.; Roberto, A.V.; Lazaro, J.E.; Mangalindan, G.C.; Florentino, L.M.; Lim-Navarro, P.; Tasdemir, D.; Ireland, C.M.; Concepcion, G.P. Platelet aggregation inhibitors from Philippine marine invertebrate samples screened in a new microplate assay. Mar. Biotechnol. (NY). 2003, 5, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Jaimovich, E.; Mattei, C.; Liberona, J.L.; Cardenas, C.; Estrada, M.; Barbier, J.; Debitus, C.; Laurent, D.; Molgo, J. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108–15) cells. FEBS Lett. 2005, 579, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.A.; Feng, W.; Molinski, T.F.; Pessah, I.N. Hydroxylated xestospongins block inositol-1,4,5-trisphosphate-induced Ca2+ release and sensitize Ca2+-induced Ca2+ release mediated by ryanodine receptors. Mol. Pharmacol. 2006, 69, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009, 16, 1006–1017. [Google Scholar]
- Tanaka, J.; Higa, T.; Garcia, G.D.; Ruffles, G.K. bis-1-Oxaquinolizidine Alkaloids from a Marine Sponge with Antitumor Activity. PCT International Application WO 9704783 CAN 126, 207511, 13 February 1997. [Google Scholar]
- Akl, M.R.; Ayoub, N.M.; Mohyeldin, M.M.; Busnena, B.A.; Foudah, A.I.; Liu, Y.Y.; El Sayed, K.A. Olive phenolics as c-Met inhibitors: (−)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models. PLoS One 2014, 9, e97622. [Google Scholar] [CrossRef] [PubMed]
- Elnagar, A.Y.; Sylvester, P.W.; El Sayed, K.A. (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Med. 2011, 77, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.H.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J. Pharm. Pharmacol. 2013, 65, 1622–1642. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Miki, H.; Kamiguchi, H.; Tanaka, T.; Habuka, N.; Sogabe, S.; Yano, J.; Aertgeerts, K.; Kamiyama, K. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Vassilakopoulou, M.; Togun, T.; Dafni, U.; Cheng, H.; Bordeaux, J.; Neumeister, V.M.; Bobos, M.; Pentheroudakis, G.; Skarlos, D.V.; Pectasides, D.; et al. In situ quantitative measurement of HER2mRNA predicts benefit from trastuzumab-containing chemotherapy in a cohort of metastatic breast cancer patients. PLoS One 2014, 9, e99131. [Google Scholar]
- Graveel, C.R.; DeGroot, J.D.; Su, Y.; Koeman, J.; Dykema, K.; Leung, S.; Snider, J.; Davies, S.R.; Swiatek, P.J.; Cottingham, S.; et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12909–12914. [Google Scholar]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar]
- Szatkowski, C.; Parys, J.B.; Ouadid-Ahidouch, H.; Matifat, F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol. Cancer 2010, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar] [PubMed]
- Jain, K.; Paranandi, K.S.; Sridharan, S.; Basu, A. Autophagy in breast cancer and its implications for therapy. Am. J. Cancer Res. 2013, 3, 251–265. [Google Scholar] [PubMed]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chi, C.W.; Su, W.C.; Huang, H.L. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget 2014, 5, 4845–4854. [Google Scholar] [PubMed]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Patane, S.; Porras, A.; Dono, R.; Maina, F. Met acts on Mdm2 via mTOR to signal cell survival during development. Development 2007, 134, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Lu, C.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. Interaction between Her2 and Beclin-1 proteins underlies a new mechanism of reciprocal regulation. J. Biol. Chem. 2013, 288, 20315–20325. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell. 2011, 42, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- De Smet, P.; Parys, J.B.; Callewaert, G.; Weidema, A.F.; Hill, E.; de Smedt, H.; Erneux, C.; Sorrentino, V.; Missiaen, L. Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca2+ pumps. Cell Calcium. 1999, 26, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Boezio, A.A.; Berry, L.; Albrecht, B.K.; Bauer, D.; Bellon, S.F.; Bode, C.; Chen, A.; Choquette, D.; Dussault, I.; Fang, M.; et al. Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6307–6312. [Google Scholar]
- Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 1 September 2014).
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comp. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akl, M.R.; Ayoub, N.M.; Ebrahim, H.Y.; Mohyeldin, M.M.; Orabi, K.Y.; Foudah, A.I.; Sayed, K.A.E. Araguspongine C Induces Autophagic Death in Breast Cancer Cells through Suppression of c-Met and HER2 Receptor Tyrosine Kinase Signaling. Mar. Drugs 2015, 13, 288-311. https://doi.org/10.3390/md13010288
Akl MR, Ayoub NM, Ebrahim HY, Mohyeldin MM, Orabi KY, Foudah AI, Sayed KAE. Araguspongine C Induces Autophagic Death in Breast Cancer Cells through Suppression of c-Met and HER2 Receptor Tyrosine Kinase Signaling. Marine Drugs. 2015; 13(1):288-311. https://doi.org/10.3390/md13010288
Chicago/Turabian StyleAkl, Mohamed R., Nehad M. Ayoub, Hassan Y. Ebrahim, Mohamed M. Mohyeldin, Khaled Y. Orabi, Ahmed I. Foudah, and Khalid A. El Sayed. 2015. "Araguspongine C Induces Autophagic Death in Breast Cancer Cells through Suppression of c-Met and HER2 Receptor Tyrosine Kinase Signaling" Marine Drugs 13, no. 1: 288-311. https://doi.org/10.3390/md13010288
APA StyleAkl, M. R., Ayoub, N. M., Ebrahim, H. Y., Mohyeldin, M. M., Orabi, K. Y., Foudah, A. I., & Sayed, K. A. E. (2015). Araguspongine C Induces Autophagic Death in Breast Cancer Cells through Suppression of c-Met and HER2 Receptor Tyrosine Kinase Signaling. Marine Drugs, 13(1), 288-311. https://doi.org/10.3390/md13010288