
Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation?



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chitosan Characterization
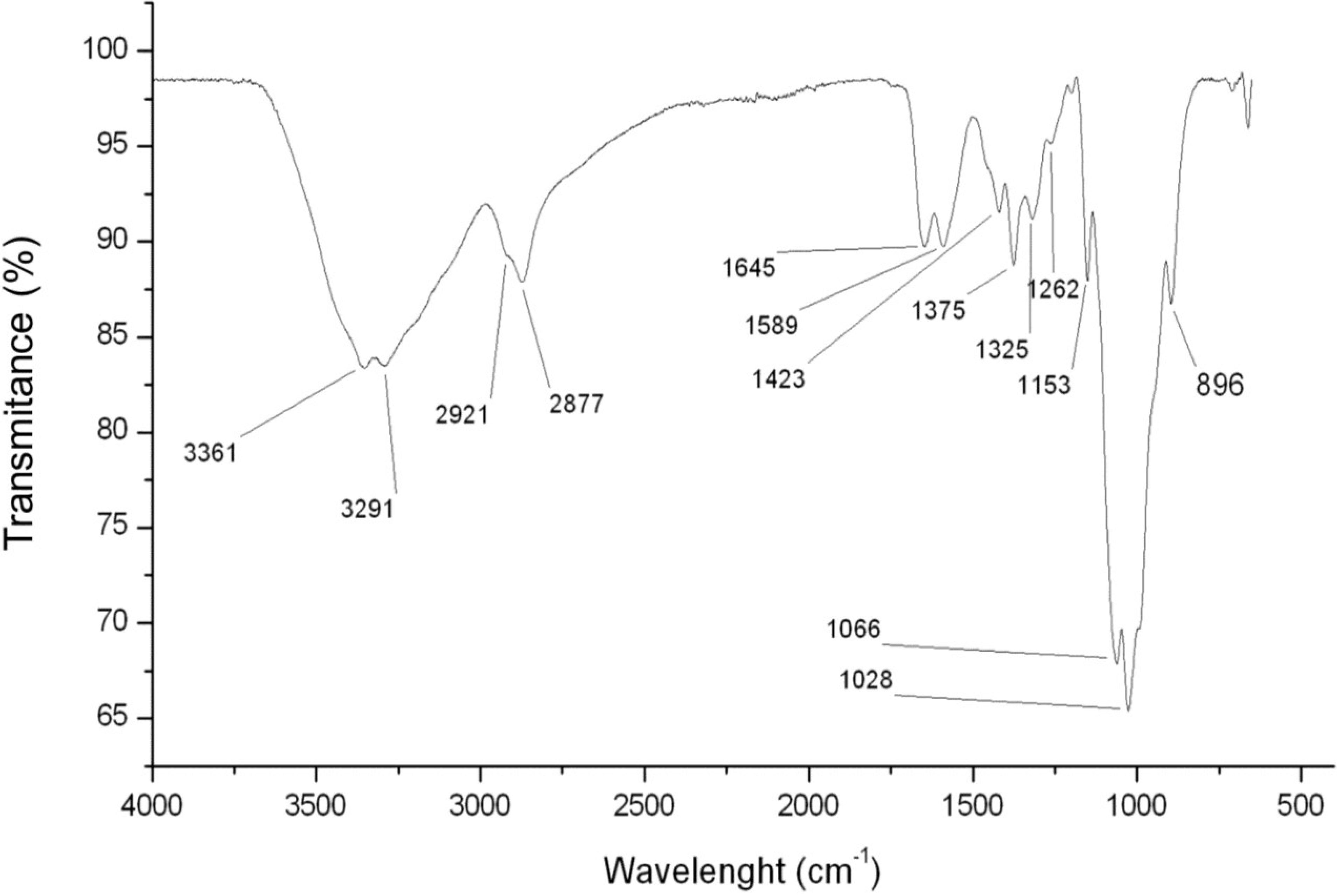
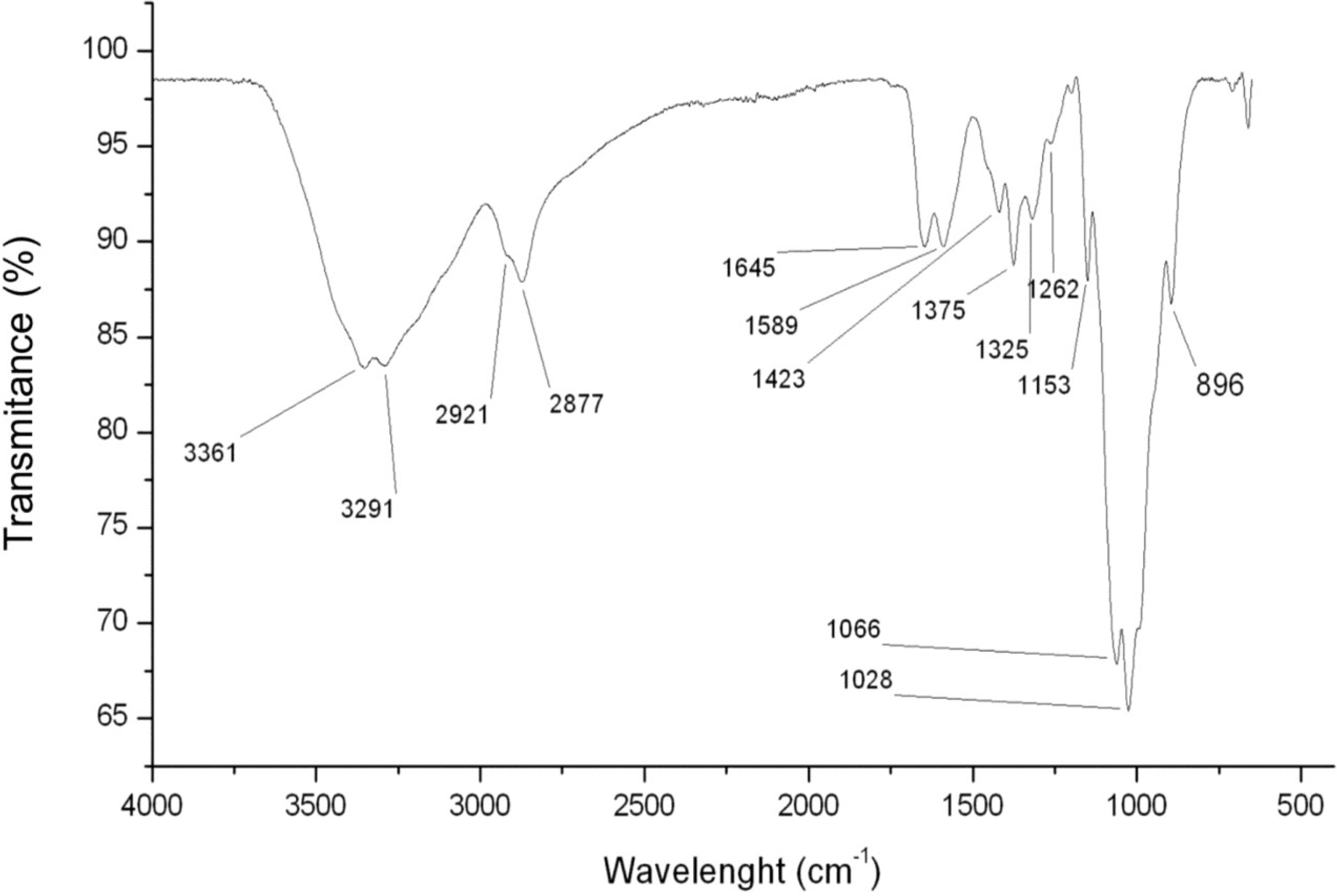
2.1.1. Chitosan FTIR Analyses
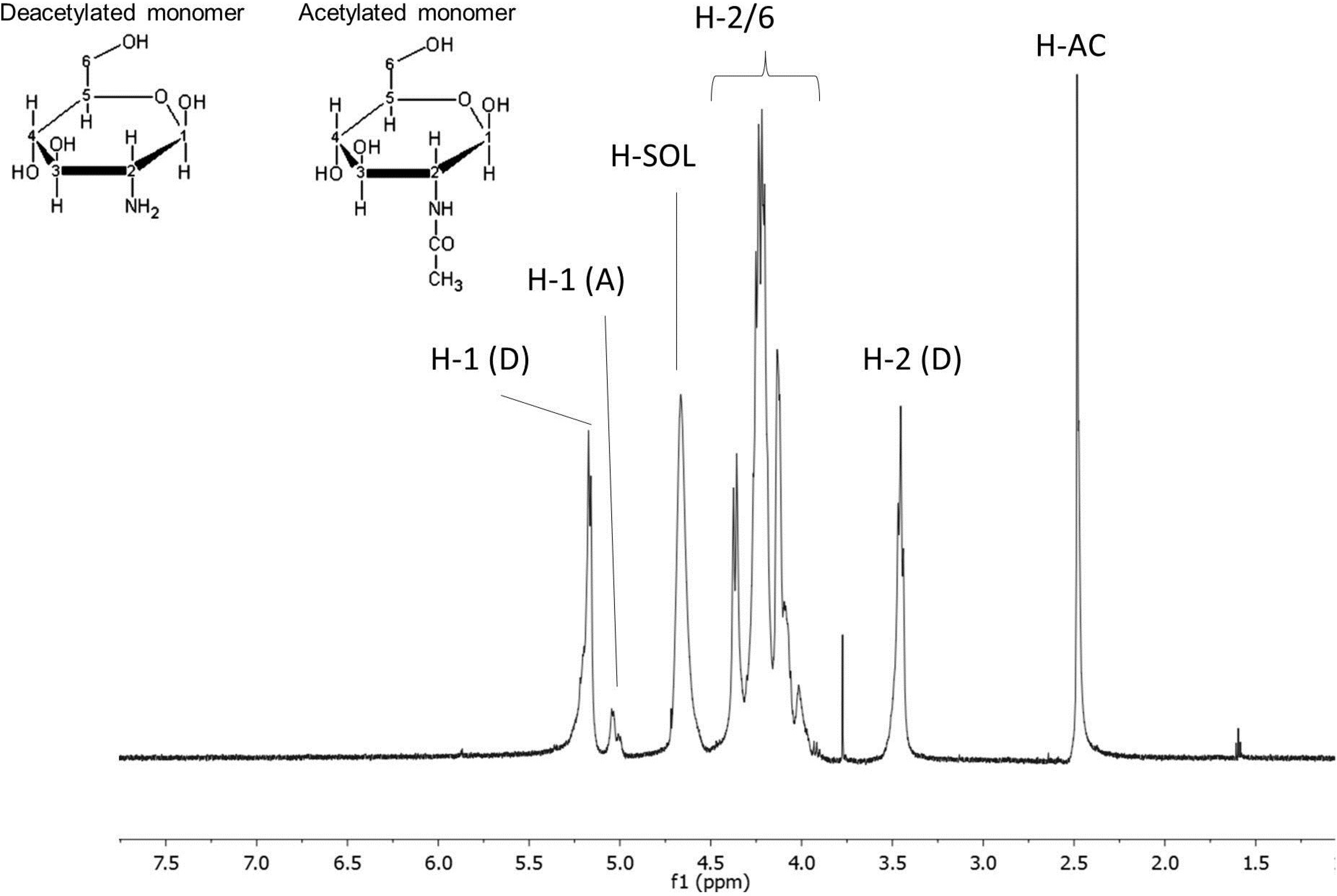
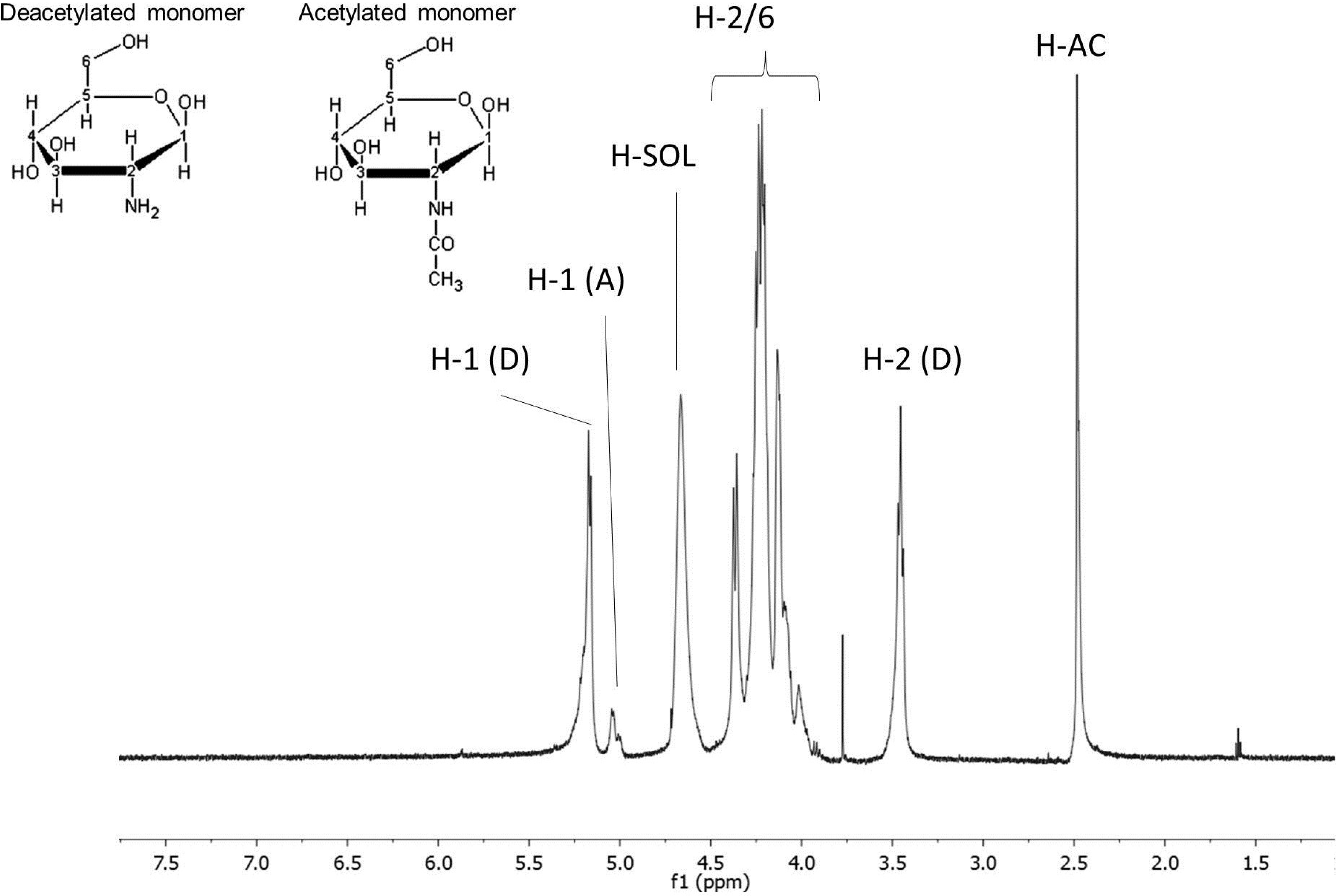
2.1.2. Chitosan 1H NMR Analyses
2.2. Antioxidant Activities
2.3. Crystal Formation
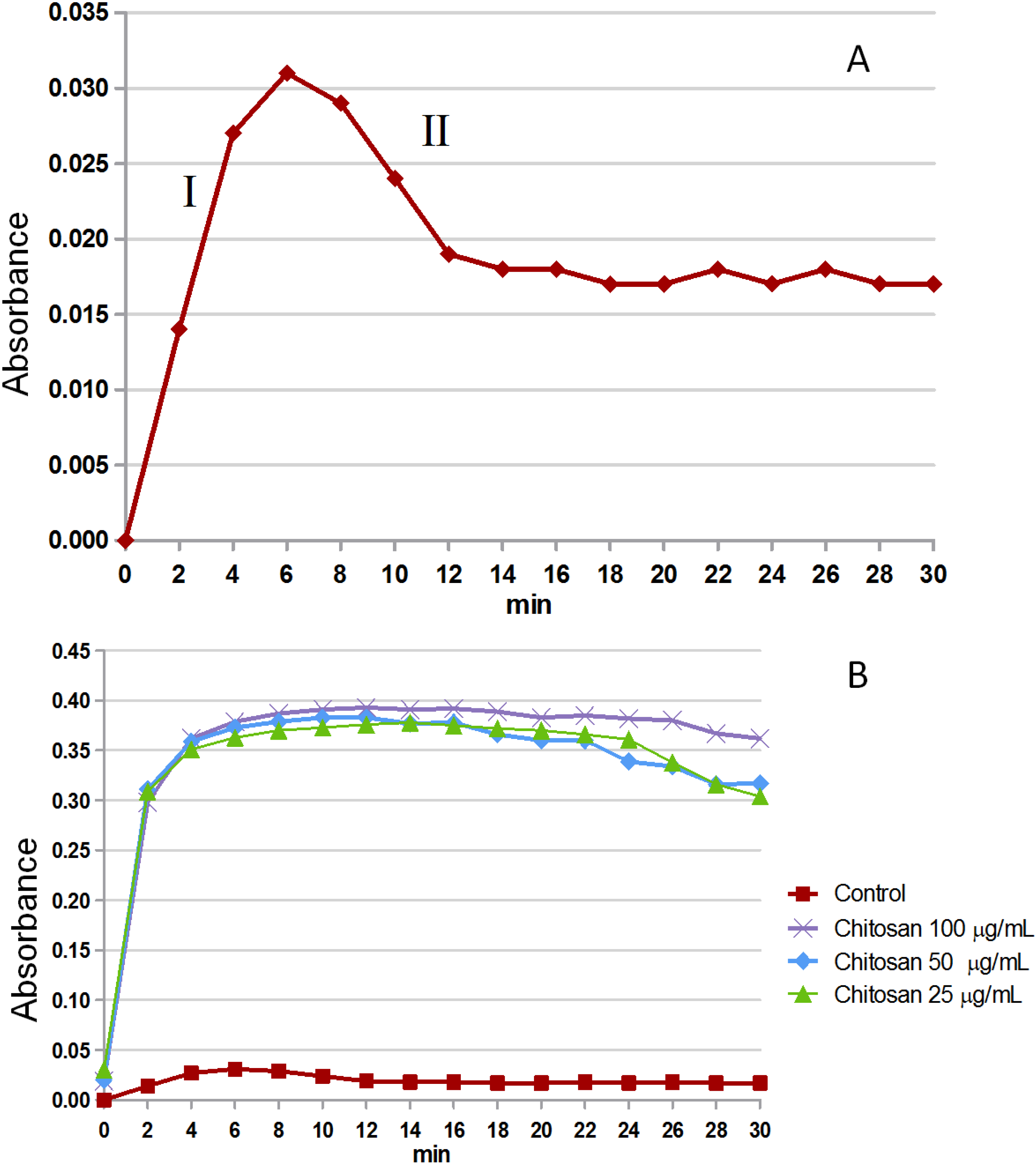
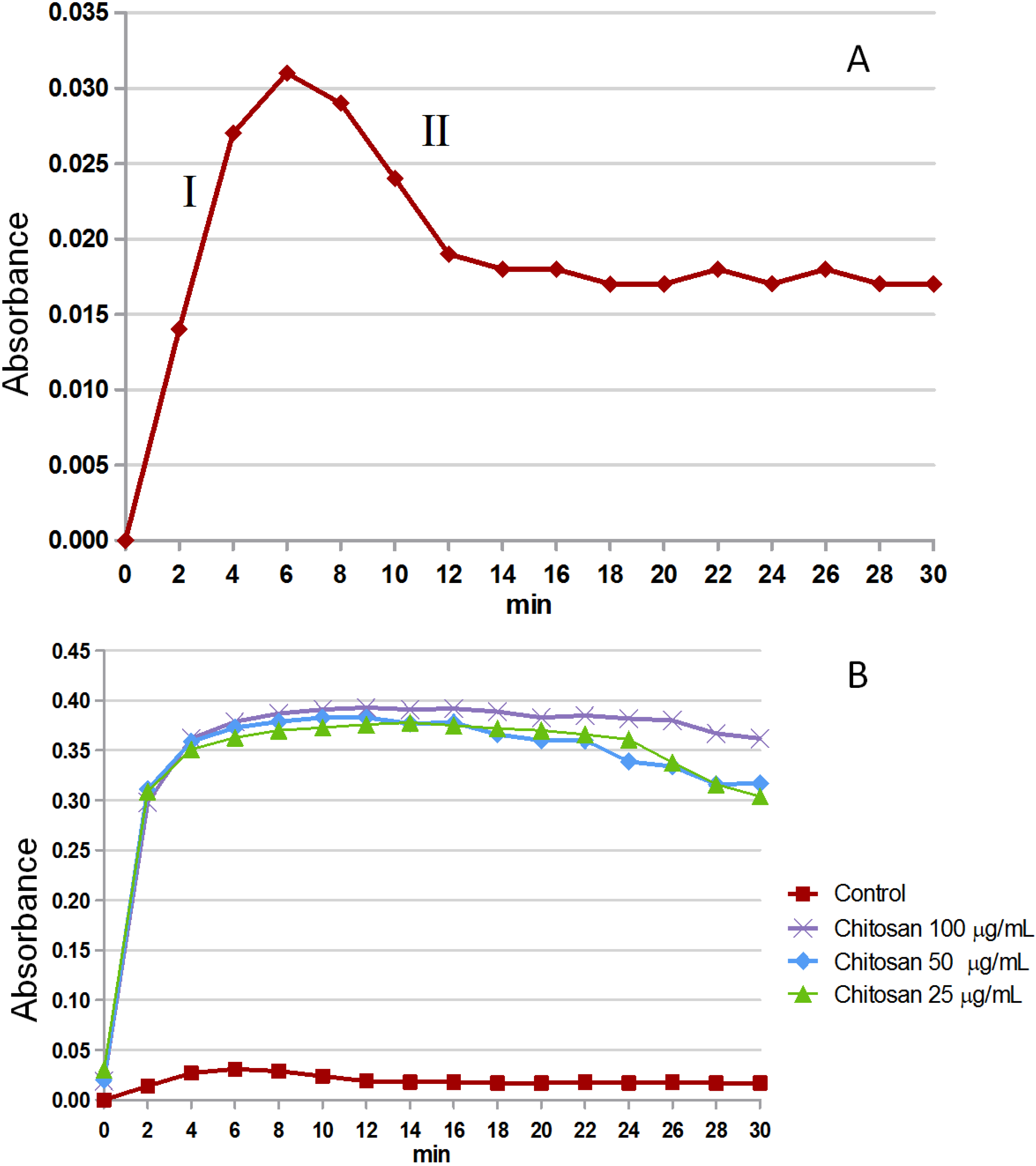
2.3.1. Crystal Formation in Vitro
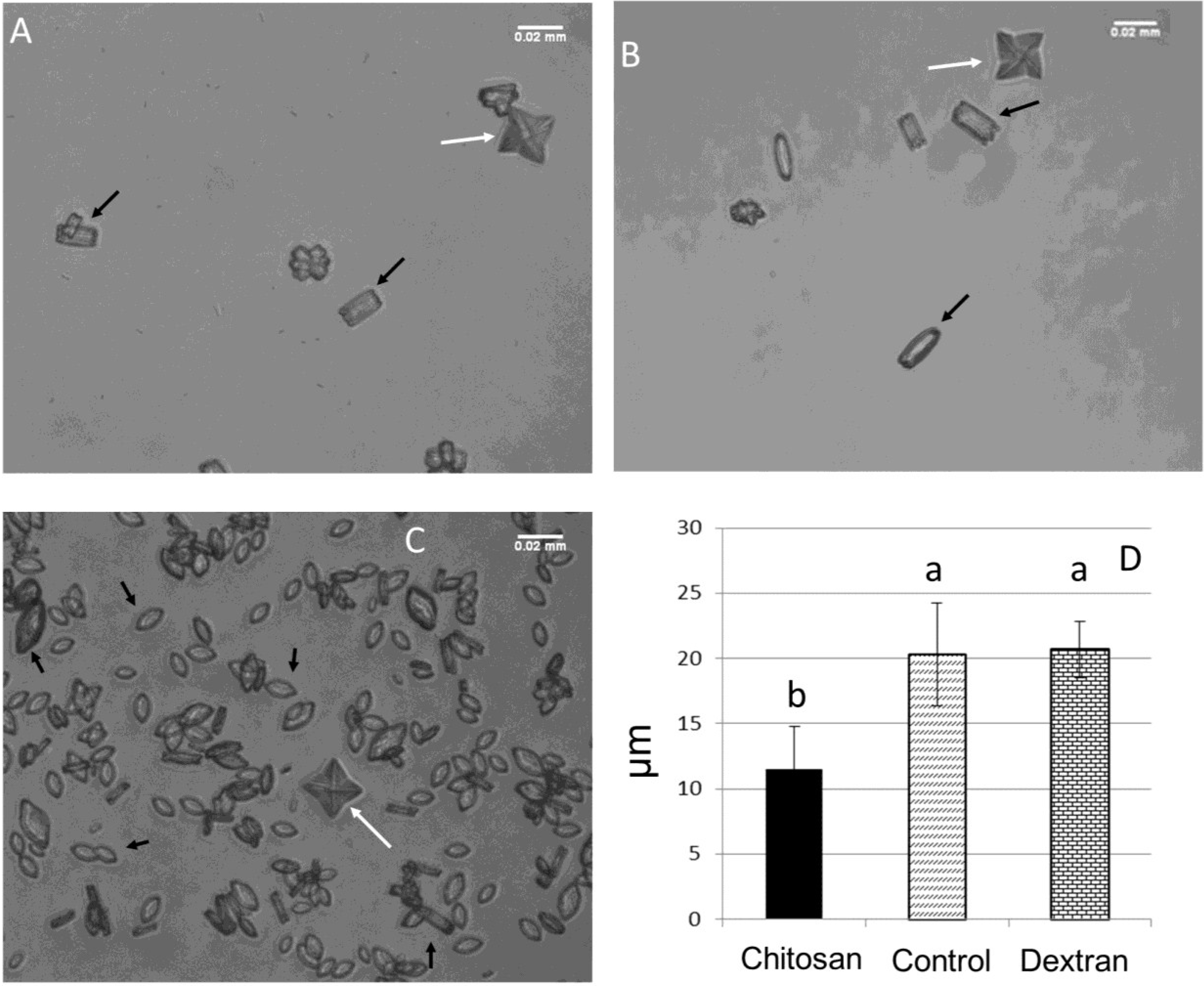
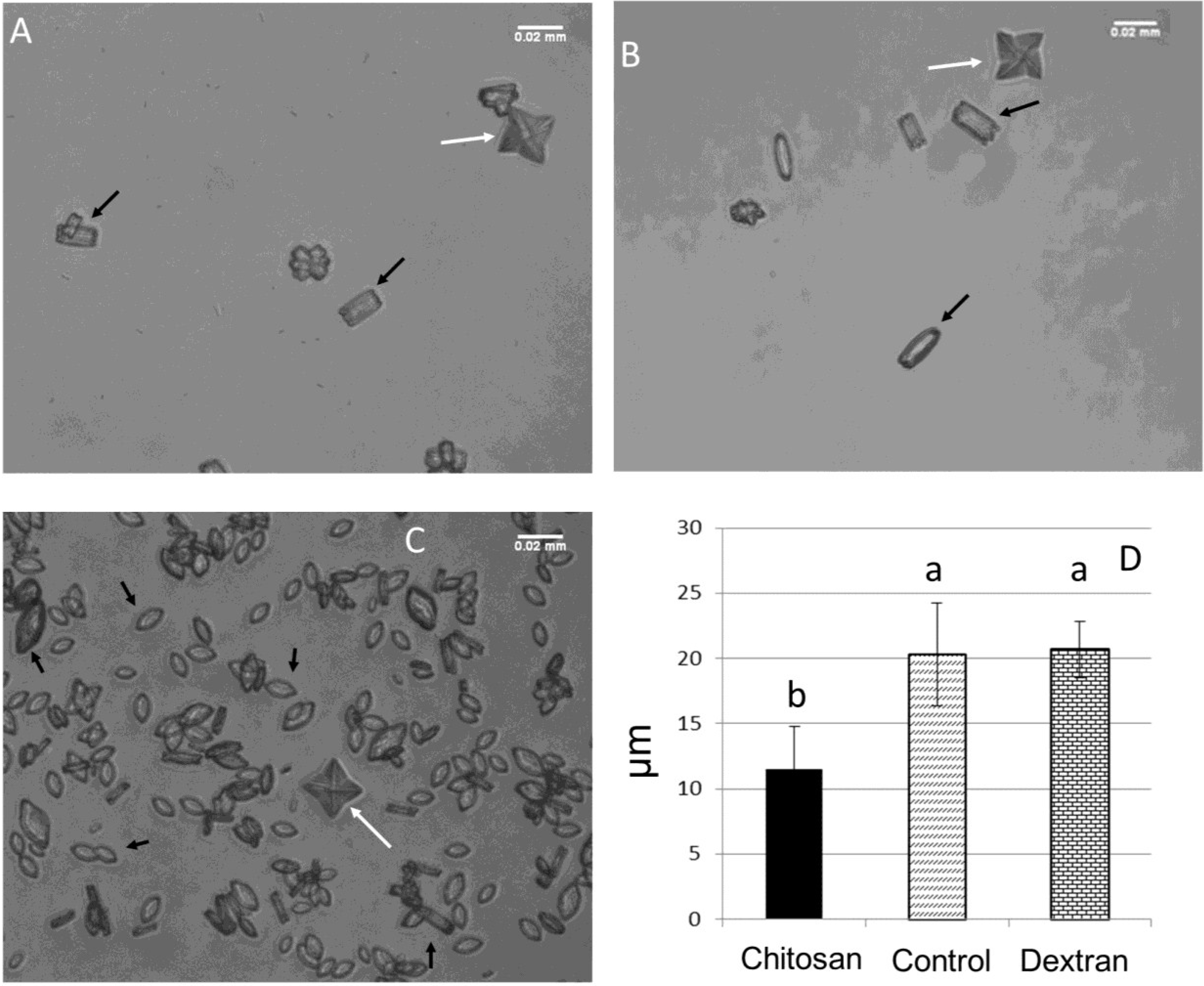
2.3.2. Crystal Morphology
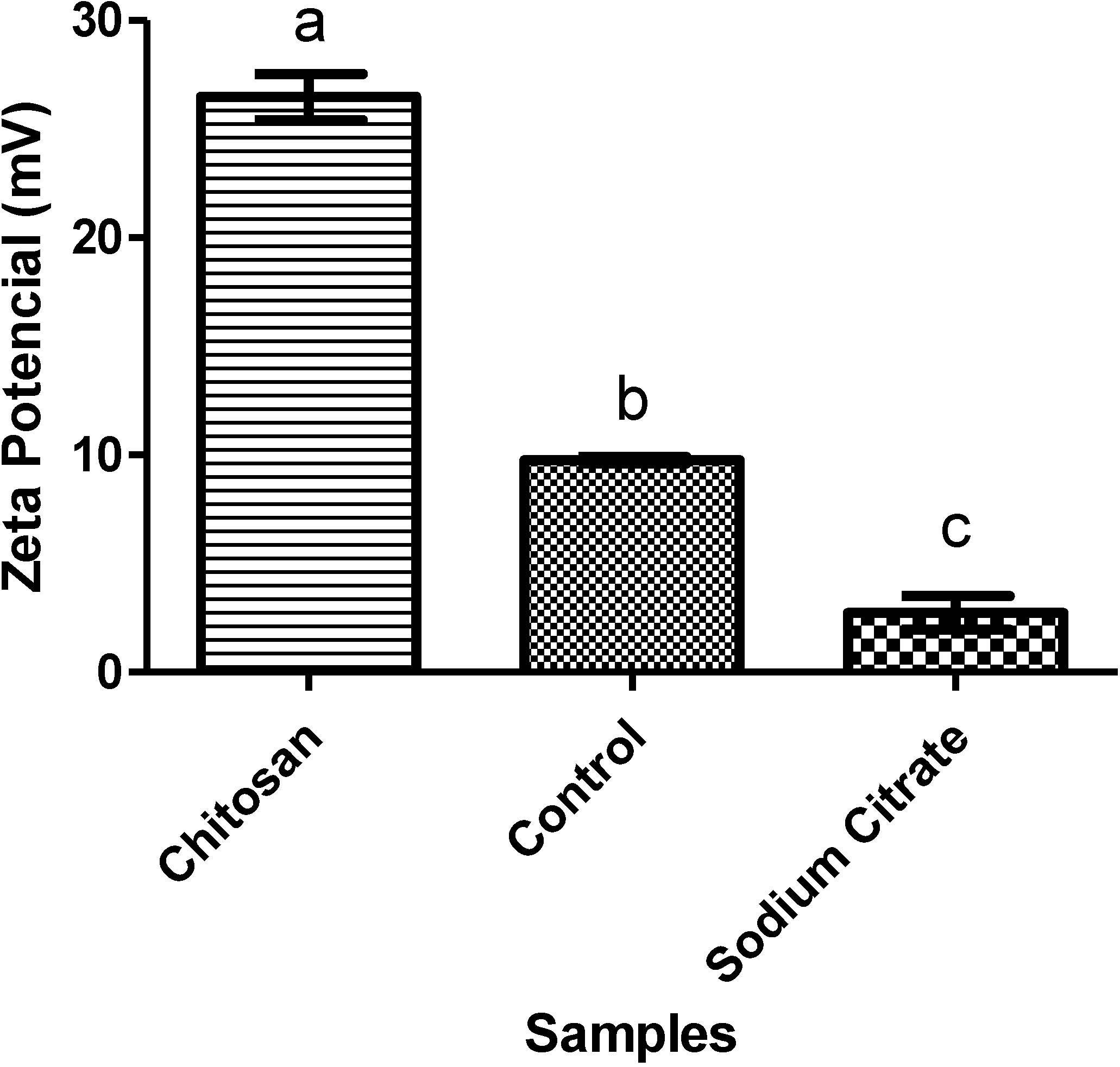
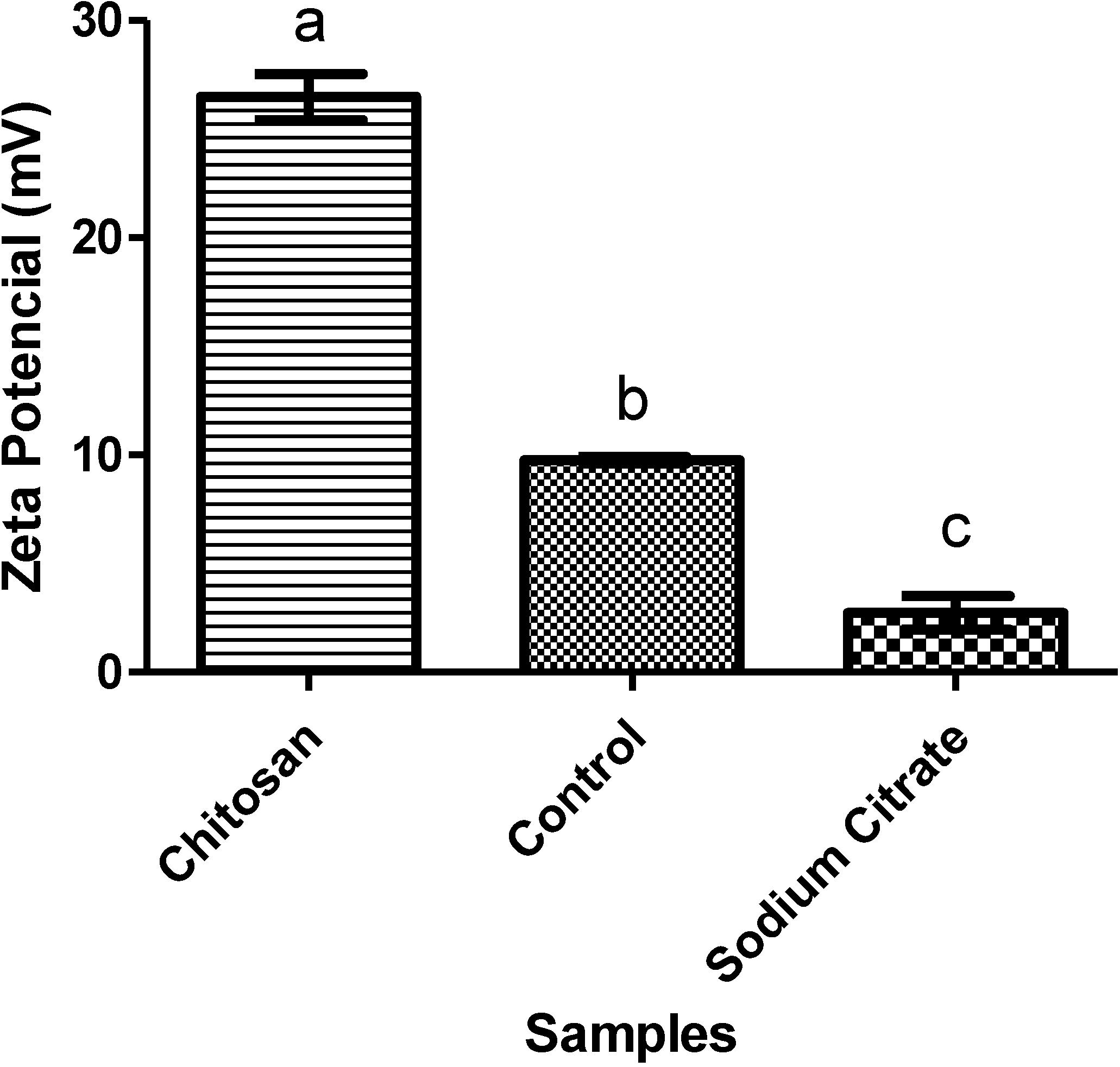
2.3.3. Zeta Potential (ζ)
3. Experimental
3.1. Chitosan
3.2. Fourier Transformed Infrared Spectroscopy (FTIR)
3.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
Determination of Deacetylation Degree
3.4. Determination of Chitosan Molecular Weight
3.5. Antioxidant Activity
3.5.1. Determination of Total Antioxidant Capacity
3.5.2. Reducing Power
3.5.3. Hydroxyl Radical Scavenging Activity Assay
3.5.4. Ferrous Chelating
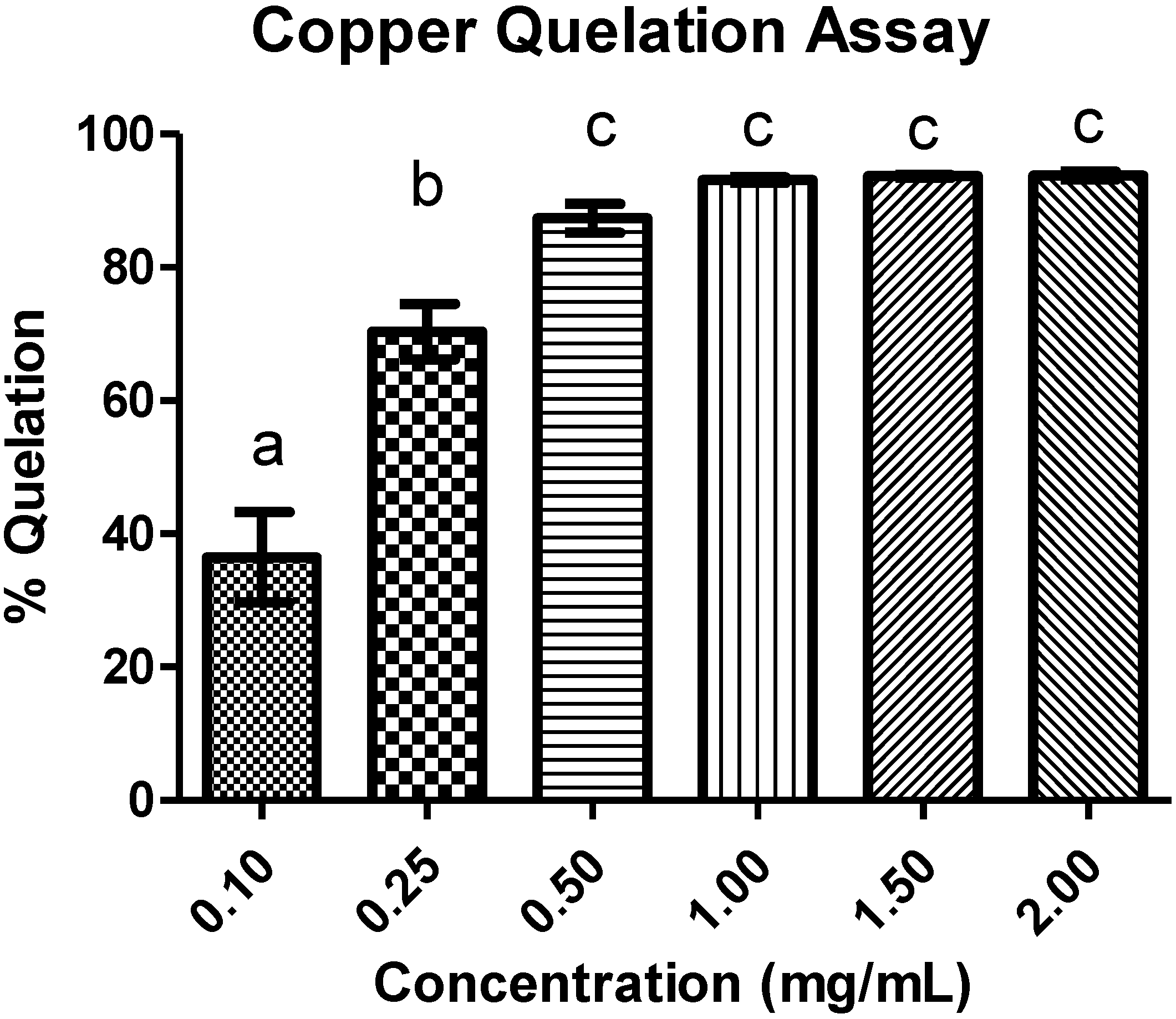
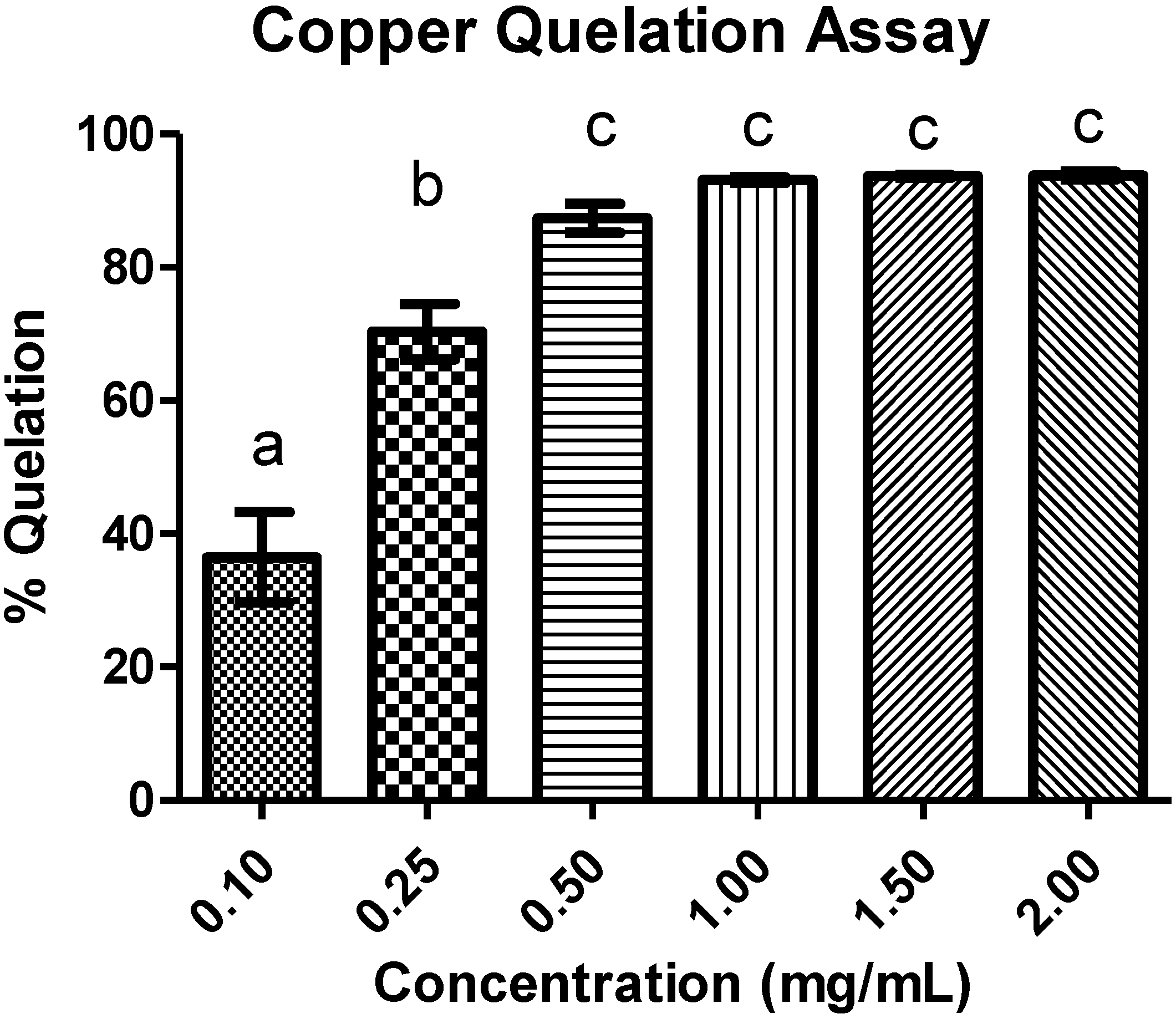
3.5.5. Copper Chelating
3.6. Calcium Oxalate Crystallization Assay
3.7. Image Analysis Crystal Morphology
3.8. Zeta Potential (ζ) Measurements
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hayes, M. Chitin, Chitosan and their Derivatives from Marine Rest Raw Materials: Potential Food and Pharmaceutical Applications. In Marine Bioactive Compounds; Springer: New York, NY, USA, 2012; pp. 115–128. [Google Scholar]
- Jayakumara, R.; Menon, D.; Manzoor, K.; Nair, S.V.; Tamura, H. Biomedical applications of chitin and chitosan based nanomaterials—A short review. Carbohydr. Polym. 2010, 82, 227–232. [Google Scholar] [CrossRef]
- Laurienzo, P. Marine Polysaccharides in Pharmaceutical Applications: An Overview. Mar. Drugs 2010, 8, 2435–2465. [Google Scholar] [CrossRef] [PubMed]
- Casettari, L.; Vllasaliu, D.; Lam, J.K.; Soliman, M.; Illum, L. Biomedical applications of amino acid-modified chitosans: A review. Biomaterials 2012, 33, 7565–7583. [Google Scholar] [CrossRef] [PubMed]
- Melo-Silveira, R.F.; Almeida-Lima, J.; Rocha, H.A.O.; Pomin, V.H. Marine Medicinal Glycomics, 1st ed.; Nova Science: New York, NY, USA, 2013; pp. 454–463. [Google Scholar]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug. Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Baldrick, P. The safety of chitosan as a pharmaceutical excipient. Regul. Toxicol. Pharmacol. 2010, 56, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fuentes, M.; Alonso, M.J. Chitosan-based drug nanocarriers: Where do we stand? J. Control. Release 2012, 161, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zeng, Z.W.; Xiao, R.Z.; Xie, T.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar]
- Zhang, H.L.; Tao, Y.; Guo, J.; Hu, Y.M.; Su, Z.Q. Hypolipidemic effects of chitosan nanoparticles in hyperlipidemia rats induced by high fat diet. Int. Immunopharmacol. 2011, 11, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Yuan, Z.X.; Zhang, Z.R.; Zhu, D.; Sun, X.; Gong, T.; Liu, J.; Luan, C.T. Specific renal uptake of randomly 50% N-acetylated low molecular weight chitosan. Mol. Pharm. 2009, 6, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Nishimura, Y.; Watanabe, Y.; Takita, T.; Innami, S. Accelerating effect of chitosan Intake on Urinary calcium excretion by rats. Biosci. Biotech. Biochem. 1997, 61, 1206–1208. [Google Scholar] [CrossRef]
- Mahmood, A.; Silbergleit, A.; Olson, R.; Cotant, M. Urolithiasis: The influence of stone size on management. Nat. Clin. Pract. Urol. 2007, 4, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, B. Phisicochemical aspects of urolithiasis. Kidney Int. 1978, 13, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Miyazato, T.; Hatano, T. Oxalate and Urinary Stones. World J. Surg. 2000, 24, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Thamilselvan, S.; Hackett, R.L.; Khan, S.R. Lipid peroxidation in ethylene glycol induced hyperoxaluria and calcium oxalate nephrolithiasis. J. Urol. 1997, 157, 1059–1063. [Google Scholar]
- Selvam, R. Calcium oxalate stone disease: Role of lipid peroxidation and antioxidants. Urol. Res. 2002, 30, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Anbazhagan, M.; Hariprasad, C.; Samudram, P.; Latha, P.; Latha, M.; Selvam, R. Effect of oral supplementation of vitamin E in hyperoxaluric patients on urinary risk factors. J. Clin. Biochem. Nutr. 1999, 27, 37–47. [Google Scholar] [CrossRef]
- Davalos, M.; Konno, S.; Eshghi, M.; Choudhury, M. Oxidative renal cell injury induced by calcium oxalate crystal and renoprotection with antioxidants: A possible role of oxidative stress in nephrolithiasis. J. Endourol. 2010, 24, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Holoch, P.A.; Tracy, C.R. Antioxidants and self-reported history of kidney stones: The National Health and Nutrition Examination Survey. J. Endourol. 2011, 25, 1903–1908. [Google Scholar]
- Melo-Silveira, R.F.; Fidelis, G.P.; Costa, M.S.S.P.; Telles, C.B.S.; Dantas-Santos, N.; Elias, S.O.; Ribeiro, V.B.; Barth, A.L.; Macedo, A.J.; Leite, E.L.; et al. In Vitro Antioxidant, Anticoagulant and Antimicrobial Activity and in Inhibition of Cancer Cell Proliferation by Xylan Extracted from Corn Cobs. Int. J. Mol. Sci. 2012, 13, 409–426. [Google Scholar] [CrossRef]
- Wolkers, W.F.; Oliver, A.E.; Tablina, F.; Crowea, J.H. A Fourier-transform infrared spectroscopy study of sugar glasses. Carbohydr. Res. 2004, 339, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.R.F.; Dore, C.M.P.G.; Marques, C.T.; Nascimento, M.S.; Benevides, N.M.B.; Rocha, H.A.O.; Chavante, S.F.; Leite, E.L. Anticoagulant activity, paw edema and pleurisy induced carrageenan: Action of major types of commercial carrageenans. Carbohydr. Polym. 2010, 79, 26–33. [Google Scholar] [CrossRef]
- Lim, S.H.; Hudson, S.M. Synthesis and antimicrobial activity of a water-soluble chitosan derivative with a fiber-reactive group. Carbohydr. Res. 2004, 339, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Vino, A.B.; Ramasamy, P.; Shanmugam, V.; Shanmugam, A. Extraction, characterization and in vitro antioxidative potential of chitosan and sulfated chitosan from Cuttlebone of Sepia aculeata Orbigny, 1848. Asian. Pac. J. Trop. Biomed. 2012, 2, S334–S341. [Google Scholar] [CrossRef]
- Song, C.; Yu, H.; Zhang, M.; Yang, Y.; Zhang, G. Physicochemical properties and antioxidant activity of chitosan from the blowfly Chrysomya megacephala larvae. Int. J. Biol. Macromol. 2013, 60, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.S.S.P.; Costa, L.S.; Cordeiro, S.L.; Almeida-Lima, J.; Dantas-Santos, N.; Magalhães, K.D.; Sabry, D.A.; Albuquerque, I.R.L.; Pereira, M.R.; Leite, E.L.; et al. Evaluating the possible anticoagulant and antioxidant effects of sulfated polysaccharides from the tropical green alga Caulerpa cupressoides var. flabellate. J. Appl. Phycol. 2012, 24, 1159–1167. [Google Scholar] [CrossRef]
- Czechowska-Biskup, R.; Jarosińska, D.; Rokita, B.; Ulański, P.; Rosiak, J.M. Determination of Degree of Deacetylation of Chitosan-Comparision of Methods. Prog. Chem. Appl. Chitin Deriv. 2012, XVII, 5–20. [Google Scholar]
- Lavertu, M.; Xia, Z.; Serreqi, A.; Berrada, M.; Rodrrigues, A.; Wang, D.; Buschmann, M.; Gupta, A. A validated H NMR method for the determination of the degree of deacetylation of chitosan. J. Pharm. Biomed. Anal. 2003, 32, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Wickens, A.P. Ageing and the free radical theory. Respir. Physiol. 2001, 128, 379–391. [Google Scholar]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Filipe, H.M.; Centorrino, T.; Lepore, V.; Terranova, M.L.; Ciccolo, A.; Caputi, A.P.; Thiemermann, C. Beneficial effects of tempol, a membrane-permeable radical scavenger, on the multiple organ failure induced by zymosan in the rat. Crit. Care Med. 2001, 29, 102–111. [Google Scholar] [CrossRef]
- Costa, L.S.; Fidelis, G.P.; Cordeiro, S.L.; Oliveira, R.M.; de Sabry, D.A.; Camara, R.B.G.; Nobre, L.T.D.B.; da Costa, M.S.S.P.; de Lima, J.A.; de Farias, E.H.C.; et al. Biological activities of sulfated polysaccharides from tropical seaweeds. Biomed. Pharmacother. 2010, 64, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Prabu, K.; Natarajan, E. In Vitro Antimicrobial and Antioxidant Activity of Chitosan Isolated from Podophthalmus vigil. J. App. Pharm. Sci. 2012, 9, 075–082. [Google Scholar]
- Kuppusamy, S.K.; Karuppaiah, J. In Vitro evaluation of free radical scavenging activity of chitosan. Int. J. Pharm. Life Sci. 2013, 4, 2685–2690. [Google Scholar]
- Chien, P.-J.; Sheu, F.; Huang, W.-T.; Su, M.-S. Effect of molecular weight of chitosans on their antioxidative activities in apple juice. Food Chem. 2007, 155, 221–226. [Google Scholar]
- Xing, R.; Liu, S.; Guo, Z.; Yu, H.; Wang, P.; Lia, L.Z.; Lia, P. Relevance of molecular weight of chitosan and its derivatives and their antioxidant activities in vitro. Bioorg. Med. Chem. 2005, 13, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Melo, K.R.T.; Câmara, R.B.G.; Queiroz, M.F.; Vidal, A.A.J.; Lima, C.R.M.; Melo-Silveira, R.F.; Almeida-Lima, J.; Rocha, H.A.O. Evaluation of Sulfated Polysaccharides from the Brown Seaweed Dictyopteris Justii as Antioxidant Agents and as Inhibitors of the Formation of Calcium Oxalate Crystals. Molecules 2013, 18, 14543–14563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Wu, W.H.; Wang, J.; Lan, M.B. Antioxidant properties of polysaccharide from the brown seaweed Sargassum graminifolium (Turn.), and its effects on calcium oxalate crystallization. Mar. Drugs 2012, 10, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kulaksizoglu, S.; Sofikerim, M.; Cevik, C. In vitro effect of lemon and orange juices on calcium oxalate crystallization. Int. Urol. Nephrol. 2008, 40, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Green, W.; Ratan, H. Molecular mechanisms of urolithiasis. Urology 2013, 81, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Lieske, J.C.; Deganello, S.; Toback, F.G. Cell-crystal interactions and kidney stone formation. Nephron 1999, 81, 8–17. [Google Scholar]
- Yuen, J.W.; Gohel, M.D.; Poon, N.W.; Shum, D.K.; Tam, P.C.; Au, D.W. The initial and subsequent inflammatory events during calcium oxalate lithiasis. Clin. Chim. Acta 2010, 411, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- BanÞeld, J.F.; Welch, S.A.; Zhang, H.; Ebert, T.T.; Penn, R.L. Aggregation-Based Crystal Growth and Microstructure Development in Natural Iron Oxyhydroxide Biomineralization Products. Science 2000, 289, 751–754. [Google Scholar]
- Escobar, C.; Neira-Carrillo, A.; Fernández, M.S.; Arias, J.L. Biomineralization: From Paleontology to Materials Science; Arias, J.L., Fernández, M.S., Eds.; Editorial Universitaria: Santiago, Chile, 2007; pp. 343–358. [Google Scholar]
- Gades, M.D.; Stern, J.S. Chitosan supplementation and fat absorption in men and women. J. Am. Diet. Assoc. 2005, 105, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Giri, T.K.; Thakur, A.; Alexander, A.; Ajazuddin; Badwaik, H.; Tripathi, D.K. Modified chitosan hydrogels as drug delivery and tissue engineering systems: Present status and applications. Acta Pharm. Sin. B 2012, 2, 439–449. [Google Scholar] [CrossRef]
- Anton, A. Colorimetric Estimation of Aluminum with Pyrocatechol Violet. Anal. Chem. 1960, 32, 725–726. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes Queiroz, M.; Melo, K.R.T.; Sabry, D.A.; Sassaki, G.L.; Rocha, H.A.O. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Mar. Drugs 2015, 13, 141-158. https://doi.org/10.3390/md13010141
Fernandes Queiroz M, Melo KRT, Sabry DA, Sassaki GL, Rocha HAO. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Marine Drugs. 2015; 13(1):141-158. https://doi.org/10.3390/md13010141
Chicago/Turabian StyleFernandes Queiroz, Moacir, Karoline Rachel Teodosio Melo, Diego Araujo Sabry, Guilherme Lanzi Sassaki, and Hugo Alexandre Oliveira Rocha. 2015. "Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation?" Marine Drugs 13, no. 1: 141-158. https://doi.org/10.3390/md13010141
APA StyleFernandes Queiroz, M., Melo, K. R. T., Sabry, D. A., Sassaki, G. L., & Rocha, H. A. O. (2015). Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Marine Drugs, 13(1), 141-158. https://doi.org/10.3390/md13010141