Abstract
Two new indole-diterpenoids (1 and 2) and a new isocoumarin (3), along with the known β-aflatrem (4), paspalinine (5), leporin B (6), α-cyclopiazonic acid (7), iso-α-cyclopiazonic acid (8), ditryptophenaline (9), aflatoxin B1 (10), 7-O-acetylkojic acid (11) and kojic acid (12), were isolated from the fermentation broth of the marine-derived fungus, Aspergillus flavus OUCMDZ-2205. The structures of Compounds 1–12 were elucidated by spectroscopic analyses, quantum ECD calculations and the chemical method. New Compound 1 exhibited antibacterial activity against Staphylococcus aureus with a MIC value of 20.5 μM. Both new Compounds 1 and 2 could arrest the A549 cell cycle in the S phase at a concentration of 10 μM. Compound 1 showed PKC-beta inhibition with an IC50 value of 15.6 μM. In addition, the absolute configurations of the known compounds, 4–6 and leporin A (6a), were also determined for the first time.
1. Introduction
Indole diterpenoids have attracted extensive attention for their diverse skeletons [1,2,3], as well as their different biological activities, such as cytotoxicity [4], antiinsectan activity [1] and tremorgenic activity [5]. Our previous study identified six new and five known indole diterpenoids with anti-H1N1 activity from an aciduric mangrove fungus, Penicillium camemberti OUCMDZ-1492 [6]. To enrich the chemodiversity of indole diterpenoids from marine-derived fungi, a fungal strain, Aspergillus flavus OUCMDZ-2205, was isolated from the marine prawn, Penaeus vannamei. Chemical study resulted in the isolation and identification of two new indole diterpenoids, (2R,4bR,6aS,12bS,12cS,14aS)-4b-deoxy-β-aflatrem (1) and (2R,4bS,6aS,12bS,12cR)-9-isopentenyl paxilline (2), and a new isocoumarin, (S)-(−)-6,8-di-O-methylcitreoisocoumarin (3), along with the known β-aflatrem (4) [7], paspalinine (5) [8], leporin B (6) [9], α-cyclopiazonic acid (7) [10], iso-α-cyclopiazonic acid (8) [11], ditryptophenaline (9) [12], aflatoxin B1 (10) [13], 7-O-acetylkojic acid (11) [14] and kojic acid (12) [15] (Figure 1, Supplementary Figure S2). Compound 1 exhibited antibacterial activity against Staphylococcus aureus with a MIC value of 20.5 μM. Both new Compounds 1 and 2 could arrest the A549 cell cycle in the S phase at a concentration of 10 μM. Additionally, Compound 1 showed PKC-beta inhibition with an IC50 value of 15.6 μM. Details of the isolation, structure determination and biological activities are presented here.
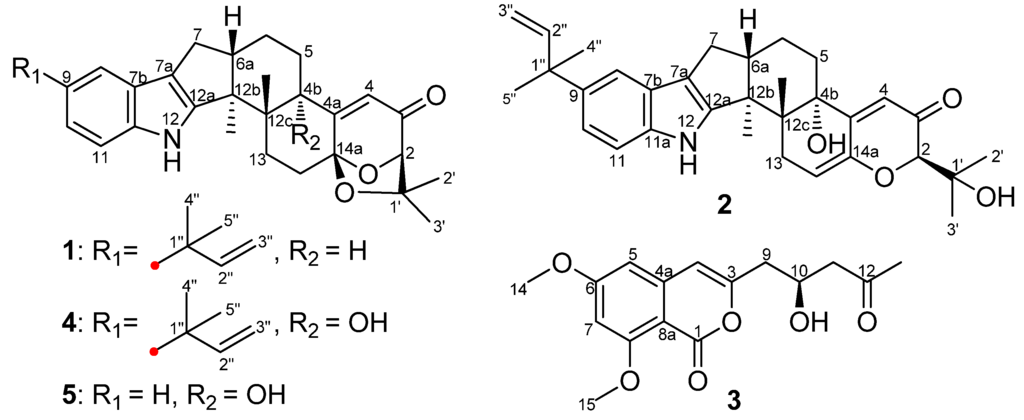
Figure 1.
Structures of Compounds 1–5 from Aspergillus flavus OUCMDZ-2205.
2. Results and Discussion
2.1. Structure Elucidation
The EtOAc extract of the fermentation broth of A. flavus OUCMDZ-2205 was separated by silica gel column, Sephadex LH-20 column and semi-preparative HPLC to give Compounds 1–12.
Compound 1 was obtained as a white amorphous powder. The molecular formula was determined to be C32H39NO3 on the basis of a HRESIMS peak at m/z 486.2996 [M + H]+ (calcd. 486.3008), indicating 14 degrees of unsaturation. The UV spectrum showed characteristic peaks of an indole chromophore at λmax (logε) 235 (4.11) and 245 (4.36) nm [1]. The IR absorption band at 1725 cm−1 suggested the presence of a carbonyl group. The 13C NMR spectrum was similar to that of β-aflatrem (4), except that a methine (δC/H 40.5/3.12) replaced an oxygenated quaternary carbon (δC 76.5) (Supplementary Table S1), indicating 1 as an isopentenylated indole diterpenoid. In addition, obvious shifts for C-12c, C-4a and C-5 were observed, suggesting 1 as the deoxy derivative of 4 at C-4b. The deduction was further confirmed by the key COSY correlations of H-4b/H-5/H-6 and the key HMBC correlations from H-4b (δH 3.12) to C-12b (δC 51.2), C-4 (δC 118.7) and Me-12c (δC 21.6) (Figure 2).
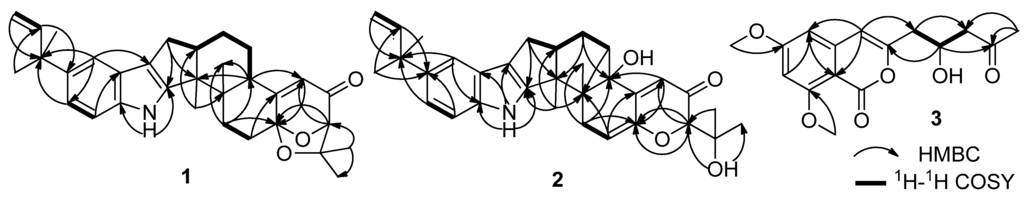
Figure 2.
Selected 1H-1H COSY and HMBC correlations for 1–3.
The relative configuration of 1 was assigned on the basis of the NOESY spectrum. The key NOESY correlations of Me-12b (δH 1.06) to H-13α (δH 2.40) and H-4b (δH 3.12) and of Me-12c (δH 1.03) to H-6a (δH 2.67) and H-13β (δH 1.92) (Figure 3) indicated that Me-12b, H-13α and H-4b are in the same orientation, while Me-12c, H-13β and H-6a are in the opposite orientation. Furthermore, the same relative configurations of C-14a and C-2 as those of Compounds 4/5 could be deduced from the good agreement of 13C NMR data of C-2/C-14/C-14a and C-1′/C-2′/C-3′ in 1 and 4/5 (Supplementary Table S1). This deduction was confirmed by the quantum chemical calculations of 13C NMR for 1 and (2S,14aR)-1 at the B3LYP/6-311++G(2d,p)//B3LYP/6-31G(d) level in Gaussian 03 (see Supplementary Information) ([16,17,18,19]). The magnetic shielding values were converted into chemical shifts after the corrections using the slope and intercept of the linear-square functions, and the relative errors of chemical shifts were computed by subtracting the calculated 13C NMR from the measured shifts. The maximum error of 1 is less than 13.0 ppm (C-4a), while the maximum error at C-2/C-3/C-1′ reached 23.0/23.1/‒20.5 ppm in (2S,14aR)-1, respectively (Supplementary Table S2). When the configuration changed from (2R,14aS) to (2S,14aR), the steric hindrance of Me-12c to C-1′ disappeared, while the steric hindrances of Me-12c to C-2 and Me-2′ to C-3 increased, resulting in the downfield shift of C-1′ and upfield shifts of C-2 and C-3 in (2S,14aR)-1. Thus, the structure of 1 is more reasonable than the fictitious (2S,14aR)-1, further indicating the same relative configurations of 1 and 4/5. In addition, our previous study revealed that the strongest negative CD Cotton effect at a short wavelength (λmax 210–250 nm) arose from the π–π* transitions of the indole nucleus, which could be used to determine the absolute configurations of C-4b, C-6a, C-12b and C-12c of the hexacyclic indole diterpenoids [6]. Thus, the strong negative Cotton effect at λmax 245 (Δε −8.0) nm (Figure 4) indicated the (4bR,6aS,12bS,12cS)-configuration of 1. The (2R,14aS)-configuration of 1 could be further supported by the electronic circular dichroism (ECD) calculations for 1 and (2S,14aR)-1 using time-dependent density functional theory (TDDFT) [19,20,21,22] at the B3LYP/6-31G(d) level in Gaussian 03. The result showed that the CD curve of 1 is consistent with the calculated ECD curve of 1, but opposite to that of (2S,14aR)-1 at a long wavelength (Figure 4). Thus, the structure of 1 was clearly elucidated as (2R,4bR,6aS,12bS,12cS,14aS)-4b-deoxy-β-aflatrem.
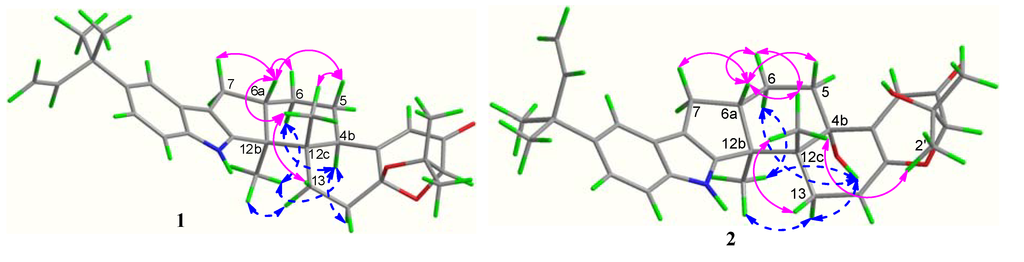
Figure 3.
Key NOESY correlations of 1 and 2.
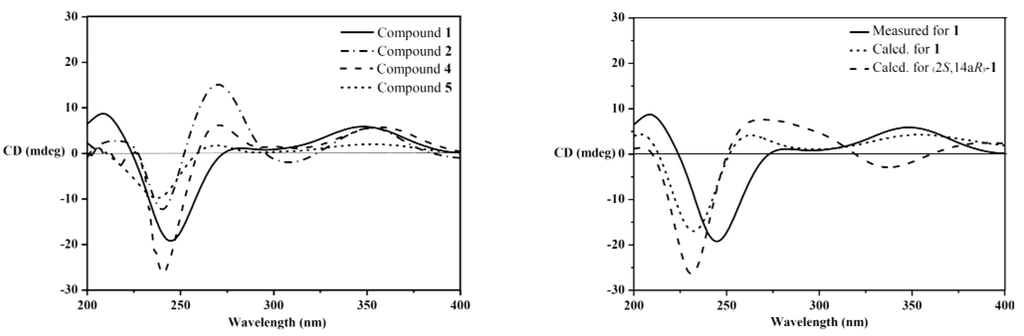
Figure 4.
CD spectra of 1, 2, 4 and 5 (left); measured CD and calculated ECD spectra for 1 and (2S,14aR)-1 (right).
Compound 2 was obtained as a yellowish amorphous powder. The molecular formula was C32H39NO4 on the basis of a HRESIMS peak at m/z 502.2939 [M + H]+ (calcd. 502.2957), indicating 14 degrees of unsaturation. The UV spectrum showed characteristic peaks of an indole diterpenoid nucleus at λmax (logε): 233 (4.15) and 248 (4.28) nm. The similarity of 1H and 13C NMR data between 2 and 4 suggested that Compound 2 is also a hexacyclic indole diterpenoid skeleton. The detailed comparison of NMR data between 2 and 4 revealed that a trisubstituted ethylene moiety (δC/H 111.4/5.64, 145.6) replaced the corresponding 1,1,1,2-tetrasubstituted ethane moiety (δC/H 28.4/2.72 & 1.87, 104.7); meanwhile, an additional exchangeable proton (δH 4.55) emerged in 2. This result was further confirmed by the key COSY correlations of H2-13 (δH 3.01, 2.40) to H-14 (δH 5.64) and the key HMBC correlations from H-14 (δH 5.64) to C-4a (δC 155.0) and C-12c (δC 43.3), from H-2 (δH 4.03) and H-4 (δH 5.81) to C-14a (δC 145.6) and from OH-1′ (δH 4.55) to C-2/1′/2′ (δC 86.4/73.4/27.6) (Figure 2). Thus, the planar structure of 2 was identified as 9-isopentenylpaxilline. The relative configuration of 2 was assigned on the basis of NOESY spectrum. The NOESY spectrum showed key correlations of Me-12b (δH 1.29) to H-13α (δH 3.01) and OH-4b (δH 4.91) and Me-12c (δH 1.00) to H-6a (δH 2.71) and H-13β (δH 2.40), H3-2′ (δH 1.25) and H3-3′ (δH 1.14) (Figure 3), indicating that Me-12b/H-13α/OH-4b are in the same orientation, while H-13β/H-6a/Me-12c/H3-2′ are all in the opposite orientation. The negative CD Cotton effect at λmax 240 (Δε −5.2) nm indicated the (4bS,6aS,12bS,12cR)-configuration [6]. Additionally, the (2R)-configuration was deduced from the cis-orientation of Me-12c and Me-2′, which was further supported by ECD calculation. The ECD calculation revealed that the CD curve of 2 at a long wavelength (λmax 327 nm) is consistent with the calculated ECD curve of 2, but opposite to its 2-epimer, (2S)-2 (Supplementary Figure S3). Therefore, Compound 2 was accurately identifiedas (2R,4bS,6aS,12bS,12cR)-9-isopentenylpaxilline.
Although the relative configurations of Compounds 4 [7] and 5 [8] have been published, the absolute configurations have not been firmly established yet. Thus, the same relative configurations of 4 and 5 were established by the agreement of 1H and 13C NMR data (Supplementary Table S1) with those reported [7,8]. The similar CD Cotton effects of 4 (λmax 240 (Δε −11.3) and 357 (Δε +2.6) nm) and 5 (λmax 238 (Δε −4.2) and 355 (Δε +0.9) nm) and 1 (λmax 245 (Δε −8.0), 347 (Δε +2.5) nm) (Figure 4) indicated that Compounds 4 and 5 shared the same absolute configurations, that is (2R,4bS,6aS,12bS,12cR,14aS).
The CD curves of Compounds 1, 4 and 5, along with our previously reported 2′-hydroxypaxilline, 9,10-diisopentenylpaxilline, paxilline, 21-isopentenylpaxilline and dehydroxypaxilline [6], revealed that all compounds with the (2R)-configuration showed a positive CD Cotton effect at λmax 300–350 nm arising from the n–π* transitions of α,β-unsaturated ketone. These data combined with the negative Cotton effects of the calculated ECD of (2S,14aR)-1 and (2S)-2 at λmax 300−350 nm revealed that the CD Cotton effects at a long wavelength (λmax 300−360 nm) could be used to determine the absolute configurations of C-2 in the α,β-unsaturated lactone moiety of hexacyclic indole diterpenoids. The positive effect means the (2R)-configuration, while the negative effect means (2S)-configuration.
Compound 3 was obtained as a light yellow solid. The molecular formula was established as C16H18O6 by an HRESIMS peak at m/z 307.1180 [M + H]+ (calcd. 307.1176). The UV spectrum at λmax (logε) 236 (3.85) and 245 (4.18) nm indicated an isocoumarin nucleus [23]. Its 1H and 13C NMR data were similar to those of citreoisocoumarin with the exception of two additional methoxy groups at δC/H 56.3/3.87 and 56.5/3.86 [24,25], indicating 3 as the derivative of citreoisocoumarin. The key HMBC correlations from H-14 (δH 3.87) to C-6 (δC 165.6) and from H-15 (δH 3.86) to C-8 (δC 163.2) (Figure 2) supported that Compound 3 is the 6,8-di-O-methyl derivative of citreoisocoumarin. The S-configuration of 3 was deduced from the same specific rotation as that of (S)-(−)-citreoisocoumarin ([α]D −10) [24]. This deduction was further confirmed from the result of the Mosher’s method [26]. The distribution of Δδ values between (S)-and (R)-MTPA (α-methoxy-α-(trifluoromethyl)phenylacetic acid) esters (3a and 3b) clearly indicated the (S)-configuration (Figure 5 and Supplementary Information). Thus, Compound 3 was unambiguously determined as (S)-(−)-6,8-di-O-methyl citreoisocoumarin.
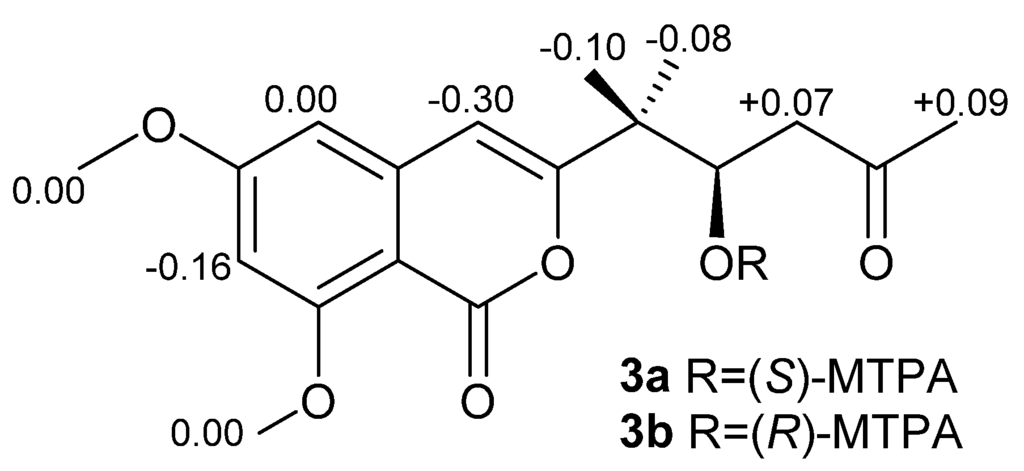
Figure 5.
Δδ (δS-δR) values of the two MTPA esters of 3.
The 1H and 13C NMR data and [α]D value of Compound 6 were in agreement with those reported for leporin B [9] (see Supplementary Information). Since there were no reports on the absolute configurations of leporin B (6) and its related leporin A (6a) in the literature, the CD spectrum was measured and the ECD was calculated. The results showed that the CD curve of 6 was in good agreement with the calculated ECD of the structure of 6, but opposite of the calculated ECD of ent-6 (Supplementary Figure S4). Thus, the absolute configuration of leporin B (6) was determined as (7S,8R,12R,13S). To determine the absolute configuration of leporin A (6a), Compound 6a was prepared from the methylation of 6 (see Supplementary Information). The synthetic-6a displayed the same 1H and 13C NMR data and a similar specific rotation as those reported (−36 vs. −22) [27]. Thus, the absolute configuration of leporin A (6a) is also (7S,8R,12R,13S).
Indole diterpenoids isolated from fungus can be traced back to 1980, and by now, 49 new indole diterpenoids have been identified from the fungal metabolites. According to the literature tracking, indole diterpenoids were produced by Aspergillus sp. [4], Penicillium sp. [6], Eupenicillium sp. [28], Dichotomomyces sp. [29], Claviceps sp. [30] and Nodulisporium sp. [31], respectively. This situation is common in the microbial community, that different microbes could produce the same type of natural products. We speculate that these different genera and species of fungi might have partial, similar biosynthetic gene clusters, which might be the reason why the same type of natural products can be isolated from different fungal metabolites.
2.2. Bioactivities the of Compounds Produced by Aspergillus flavus OUCMDZ-2205
Compounds 1–10 were assayed for their cytotoxic effects on the MCF-7 and A549 cell line by the MTT method [32] and the antibacterial activity against Enterobacter aerogenes, Bacillus subtilis, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Candida albicans using the agar dilution method [33], respectively. In addition, new Compounds 1 and 2 were also evaluated for their inhibitory effects on the A549 cell cycle [34] and the kinase, PKC-beta [35]. Because of a lack of quantity, only Compounds 4 and 6–12 were evaluated for their antiviral activities against the H1N1 virus by the CPE (cytopathic effect) inhibition assay [36]. The results showed that Compound 1 exhibited antibacterial activity against Staphylococcus aureus with a MIC value of 20.5 μM. The indole diterpenes (1, 2, 4 and 5) displayed weak cytotoxicity against MCF-7 and A549 cells with IC50 values of 18‒30 μM (Supplementary Table S3). Additionally, Compounds 1 and 2 could arrest the A549 cell cycle in the S phase at a concentration of 10 μM (Supplementary Figure S24), and 1 showed PKC-beta inhibition with an IC50 value of 15.6 μM (IC50 > 100 μM for 2). No anti-H1N1 activity (IC50 > 150 μM) was observed for Compounds 4 and 6–12, indicating that the further oxidation at C-14a decreases the anti-H1N1 activity of hexacyclic indole diterpenoids [6]. PKC beta mediates the signaling of inflammatory, mitogenic and angiogenic effects [37] and participates in the occurrence and development of colon cancer, breast cancer, lung cancer and neuroblastoma [38]. PKC-beta inhibitors are a new class of drugs that are effective in attenuating the vascular complications of diabetes [37] and had turned out to be a new ideal research target for anticancer drugs [38]. The weak cytotoxicity, the cell cycle arresting and the PKC-beta inhibition of 1 indicated that it could be a potential target compound for anticancer drug discovery.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured with a JASCO P-1020 digital polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were recorded on a Waters 2487 absorbance detector (Waters Corporation, Milford, MA, USA). CD spectra were measured on a JASCO J-715 and a J-815 spectropolarimeter (JASCO Corporation, Tokyo, Japan). IR spectra were taken on a Nicolet NEXUS 470 spectrophotometer (Nicolet Instrument Corporation, Madison, WI, USA) as KBr disks. 1H and 13C NMR, DEPT and 2D NMR spectra were recorded on a JEOL JNM-ECP 600 (JEOL Ltd, Tokyo, Japan) for Compounds 1−12 using TMS as the internal standard, and chemical shifts were recorded as δ values. ESIMS was measured on a Q-TOF ULTIMA GLOBAL GAA076 LC mass spectrometer (Micromass UK Ltd., Manchester, UK). Semi-preparative HPLC was performed using an ODS column (YMC-pack ODS-A, 10 × 250 mm, 5 μm, 4 mL/min). TLC and column chromatography (CC) were performed on plates precoated with silica gel GF254 (10−40 μm) and over silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China) and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden). Vacuum-liquid chromatography (VLC) was carried out over silica gel H (Qingdao Marine Chemical Factory, Qingdao, China). Sea salt was made by the evaporation of seawater collected in Laizhou Bay (Weifang Haisheng Chemical Factory, Weifang, China).
3.2. Fungal Material
The fungal strain, Aspergillus flavus OUCMDZ-2205, was isolated from the prawn, Penaeus vannamei, from the Lianyungang sea area, Jiangsu Province of China, in 2012. The prawn sample was successively washed with 75% EtOH and sterile seawater. Then, the sample was mashed with a mortar and diluted to 10−2 with sterile water. The 100 µL of diluents were evenly coated on potato dextrose agar medium (PDA; per liter containing potato extract, 200 g, glucose, 20 g, agar ,15 g, and seawater, 1 L) and cultivated at 28 °C for 7 days. The colony was collected and purified. It was identified according to its morphological characteristics and 18S rRNA gene sequence (GenBank Accession No. KC120773). A voucher specimen was deposited in Zhu’s laboratory at −80 °C. The working strain was prepared on PDA slants and stored at 4 °C.
3.3. Fermentation and Extraction
The fungus, A. flavus OUCMDZ-2205, was cultured in 1 L-conical flask containing 300 mL fermentation medium that was composed of glucose (10 g/L), maltose (20 g/L), mannitol (20 g/L), monosodium glutamate (10 g/L), KH2PO4 (0.5 g/L), MgSO4·7H2O (0.3 g/L), yeast extract (3 g/L), sea salt (33 g/L) and tap water (1 L, pH 7.0) and was grown under static conditions for 32 days at 25 °C. The fermented whole broth (25 L) was filtered through cheesecloth to separate the filtrate from the mycelia. Then, the filtrate was extracted three times with an equivalent volume of EtOAc to give an EtOAc solution. The mycelia were extracted three times with 80% acetone. The acetone solution was concentrated under reduced pressure to afford an aqueous solution that was extracted three times with an equivalent volume of EtOAc to give another EtOAc solution. Both EtOAc solutions were combined and concentrated under reduced pressure to give EtOAc extract (70.2 g).
3.4. Purification and Identification
The EtOAc extract (70.2 g) was subjected to a silica gel VLC column, eluting with a stepwise gradient of petroleum ether–CH2Cl2 (1:1 and 0:1) and then of CH2Cl2–MeOH (100%–0%) to yield eight primary fractions (Fractions 1–8). Fraction 2 (3.8 g) eluted with petroleum CH2Cl2–MeOH (100:1) was further purified by Sephadex LH-20 (1:1 CH2Cl2–MeOH) to afford Fraction 2.1 and Fraction 2.2, which were purified by semi-preparative HPLC eluting with 90% MeOH–H2O to yield Compounds 1(2.0 mg, tR6.0 min) and 2 (2.3 mg, tR8.0 min), respectively. Fraction 3 (7.7 g) eluted with CH2Cl2–MeOH (90:1) was subjected to RP-18 silica column eluting with a stepwise gradient of 40%–100% MeOH/H2O to give four subfractions (Fractions 3.1–3.4). Fraction 3.1 (2.4 g) was further purified by Sephadex LH-20 eluting with CH2Cl2–MeOH (1:1) to give Fractions 3.1.1–3.1.3. Fraction 3.1.3 (180 mg) was purified by semi-preparative HPLC eluting with 85% MeOH/H2O to yield Compounds 5 (1.3 mg, tR 16.5 min) and 3 (3.0 mg, tR 10.0 min). Fraction 3.3 (800 mg) was subjected to a Sephadex LH-20 column eluting with CH2Cl2–MeOH (1:1) and further purified by semi-preparative HPLC (85% MeOH/H2O) to yield 6 (6.4 mg, tR 17.8 min). Similarly, Fraction 3.4 (1.2 g) was subjected to a Sephadex LH-20 column (1:1 CH2Cl2–MeOH) and then was further purified by semi-preparative HPLC (80% MeOH/H2O) to yield Compounds 7 (3.3 mg, tR 7.3 min) and 12 (4.1 mg, tR 10.6 min). Fraction 4 (6.1 g) eluted with CH2Cl2–MeOH (80:1) was further subjected to RP-18 silica column eluting with a stepwise gradient of 30%–100% MeOH/H2O to afford four subfractions (Fractions 4.1–4.4). Fraction 4.1 (2.0 g) was further isolated by Sephadex LH-20 eluting with MeOH to provide Fraction 4.1.1 and Fraction 4.1.2. Compounds 10 (5 mg, tR 7.3 min) and 11 (6.4 mg, tR 3.9 min) were purified from Fraction 4.1.1 and Fraction 4.1.2 by semi-preparative HPLC eluting with 75% MeOH/H2O, respectively. Furthermore, Fraction 4.3 (130 mg) was further purified by Sephadex LH-20 with MeOH and then was purified by semi-preparative HPLC (70% MeOH/H2O) to yield 8 (3.2 mg, tR 10.0 min). Fraction 4.4 (1.6 g) was subjected to Sephadex LH-20 column (MeOH) to give Fraction 4.4.1 and Fraction 4.4.2. Fraction 4.4.1 (96 mg) and Fraction 4.4.2 (85 mg) were further purified by semi-preparative HPLC eluting with 70% MeOH/H2O and 80% MeOH/H2O to afford Compounds 9 (6.1 mg, tR 6.7 min) and 4 (4.8 mg, tR 16.3 min), respectively.
(2R,4bR,6aS,12bS,12cS,14aS)-4b-Deoxy-β-aflatrem (1): white amorphous powder; [α]D20 +62 (c 2.2, CHCl3); UV (MeOH) λmax (logε) 235 (4.11), 245 (4.36) nm; CD (c 0.1, MeOH) λmax (Δε) 209 (+3.8), 245 (−8.0), 280 (+0.5), 347 (+2.5) nm; IR (KBr) νmax 3396, 3334, 2922, 2851, 1664, 1606, 1446 cm−1; 1H and 13C NMR data; see Supplementary Table S1; HRESIMS m/z 486.2996 [M + H]+ (calcd. for C32H40NO3, 486.3008).
(2R,4bS,6aS,12bS,12cR)-9-Isopentenylpaxilline D (2): light yellow amorphous powder; [α]D20 +17 (c 0.4, CHCl3); UV (MeOH) λmax (logε) 233 (4.15), 248 (4.28) nm; CD (c 0.3, MeOH) λmax (Δε): 240 (−5.2), 270 (+6.5), 307 (−0.9), 350 (+2.5) nm; IR (KBr) νmax 3396, 3334, 2922, 2851, 1664, 1606, 1446; 1H and 13C NMR data; see Supplementary Table S1; HRESIMS m/z 502.2939 [M + H]+ (calcd. for C32H40NO4, 502.2957).
(S)-(−)-6,8-Di-O-Methylcitreoisocoumarin (3): light yellow solid; [α]D20 −10 (c 2.0, CHCl3); UV (MeOH) λmax (logε) 209 (3.38), 236 (3.85), 245 (4.18); 1H NMR (600 MHz, DMSO-d6) δ 6.38 (s, 1H, H-4), 6.61 (d, 1H, J = 2.2 Hz, H-5), 6.57 (d, 1H, J = 2.3 Hz, H-7), 2.48 (dd, 1H, J = 14.3, 7.9 Hz, H-9a), 2.54 (dd, 1H, J = 14.5, 5.1 Hz, H-9b), 4.27 (m, 1H, H-10), 2.57 (d, 2H, J = 6.2 Hz, H-11), 2.11 (s, 3H, H-13), 3.87 (s, 3H, H-14), 3.86 (s, 3H, H-15), 5.06 (s, 1H, HO-10); 13C NMR (150 MHz, DMSO-d6) δ 158.5 (C, C-1), 156.1 (C, C-3), 105.2 (CH, C-4), 142.3 (C, C-4a), 100.8 (CH, C-5), 165.6 (C, C-6), 98.8 (CH, C-7), 163.2 (C, C-8), 102.4 (C, C-8a), 41.4 (CH2, C-9), 65.0 (CH, C-10), 50.8 (CH2, C-11), 207.9 (C, C-12), 31.0 (CH3, C-13), 56.3 (CH3, C-14), 56.5 (CH3, C-15); HRESIMS m/z 307.1180 [M + H]+ (calcd. for C16H19O6, 307.1176).
3.5. Supplementary Information Available
Following information can be found in supplementary file: 18S rRNA gene sequences of A. flavus OUCMDZ-2205; bioassay protocols used; the physicochemical data of the known compounds, 4–12 and 6a; chemical transformation of leporin B (6) into leporin A (6a); X-ray crystal data for 9 and 12; theory and calculation details; 13C NMR quantum chemical calculations of 1 and (2S,14aR)-1; crystal structures of 9 and 12; measured CD and calculated ECD spectra of 2 and 6; the NMR spectra of Compounds 1–3; the cytotoxicities of Compounds 1–10; and the preparation of MTPA esters 3a and 3b and their 1H NMR data.
4. Conclusions
Three new compounds, (2R,4bR,6aS,12bS,12cS,14aS)-4b-deoxy-β-aflatrem (1), (2R,4bS,6aS,12bS, 12cR)-9-isopentenylpaxilline D (2) and (S)-(−)-6,8-Di-O-Methylcitreoisocoumarin (3), have been identified from the fermentation broth of the marine-derived A. flavus OUCMDZ-2205. Compound 1 exhibited anti-S. aureus and PKC-beta inhibition with MIC and IC50 values of 20.5 and 15.6 μM, respectively. Both 1 and 2 could arrest the A549 cell cycle at the S phase at a concentration of 10 μM. In addition, the absolute configurations of the known compounds 4–6 and leporin A (6a) were also determined for the first time.
Supplementary Files
Acknowledgments
This work was financially supported by grants from the NSFC (Nos. 21172204 and 41376148), the 973 Program of China (No. 2010CB833804), the 863 Program of China (Nos. 2013AA092901 and 2012AA092104) and the Special Fund for Marine Scientific Research in the Public Interest of China (No. 2010418022-3). The cell cycle and the kinase assays were performed by Shanghai Biofort Biotechnology CO., Ltd, Shanghai, China.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Li, C.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Thiersinines A and B: Novel antiinsectan indole diterpenoids from a new fungicolous Penicillium species (NRRL 28147). Org. Lett. 2002, 4, 3095–3098. [Google Scholar] [CrossRef]
- Gatenby, W.A.; Munday-Finch, S.C.; Wilkins, A.L.; Miles, C.O. Terpendole M, a novel indole-diterpenoid isolated from Lolium perenne infected with the endophytic fungus Neotyphodium lolii. J. Agric. Food Chem. 1999, 47, 1092–1097. [Google Scholar] [CrossRef]
- Li, C.; Gloer, J.B.; Wicklow, D.T. Thiersindoles A–C: New indole diterpenoids from Penicillium thiersii. J. Nat. Prod. 2003, 66, 1232–1235. [Google Scholar] [CrossRef]
- Laakso, J.A.; Gloer, J.B. Radarins A–D: New antiinsectan and cytotoxic indole diterpenoids from the sclerotia of Aspergillus sulphureus. J. Org. Chem. 1992, 57, 138–141. [Google Scholar]
- Gallagher, R.T.; Hawkes, A.D.; Steyn, P.S.; Vleggaar, R. Tremorgenic neurotoxins from perennial ryegrass causing ryegrass staggers disorder of livestock: Structure elucidation of lolitrem B. J. Chem. Soc. Chem. Commun. 1984, 9, 614–616. [Google Scholar]
- Fan, Y.Q.; Wang, Y.; Liu, P.P.; Fu, P.; Zhu, T.H.; Wang, W.; Zhu, W.M. Indole-diterpenoids with anti-H1N1 activity from the aciduric fungus Penicillium camemberti OUCMDZ-1492. J. Nat. Prod. 2013, 76, 1328–1336. [Google Scholar] [CrossRef]
- Mark, R.; Tepaske, J.B.; Gloer, D.T. Aflavarin and β-Aflatrem: New anti-insectan metabolites from the sclerotia of Aspergillus flavus. J. Nat. Prod. 1992, 55, 1080–1086. [Google Scholar] [CrossRef]
- Rex, T.G.; Janet, F.; Jon, C. Paspalinine, a tremorgenic metabolite from Claviceps paspali stevens et hall. Tetrahedron Lett. 1980, 21, 235–238. [Google Scholar]
- Zhang, C.W.; Jin, L.; Mondie, B.; Mitchell, S.S.; Castelhano, A.L.; Cai, W.Z.; Bergenhem, N. Leporin B: A novel hexokinase II gene inducing agent from an unidentified fungus. Bioorg. Med. Chem. Lett. 2003, 13, 1433–1435. [Google Scholar] [CrossRef]
- Holzapfel, C.W. The isolation and structure of cyclopiazonic acid, a toxic metabolite of penicillium cyclopium westling. Tetrahedron 1968, 24, 2101–2119. [Google Scholar] [CrossRef]
- Lin, A.Q.; Du, L.; Fang, Y.C.; Wang, F.Z.; Zhu, T.J.; Gu, Q.Q.; Zhu, W.M. iso-α-Cyclopiazonic acid, a new natural product isolated from the marine-derived fungus Aspergillus flavus CF-3. Chem. Nat. Compd. 2009, 45, 677–680. [Google Scholar] [CrossRef]
- Springer, J.P.; Bűchi, G.; Kobbe, B.; Demain, A.L.; Clardy, J. The structure of ditryptophenaline—A new metabolite of Aspergillus flavus. Tetrahedron Lett. 1977, 18, 2403–2406. [Google Scholar] [CrossRef]
- Cox, R.H.; Cole, R.J. Carbon-13 nuclear magnetic resonance studies of fungal metabolites, aflatoxins, and sterigmatocystins. J. Org. Chem. 1977, 42, 112–114. [Google Scholar] [CrossRef]
- Sugijanto, N.E.; Diesel, A.; Ebel, R.; Indrayanto, G.; Zaini, N.C. Chemical constituents of the endophytic fungus Lecythophora sp. isolated from Alyxia reinwardtii. Nat. Prod. Commun. 2009, 4, 1485–1488. [Google Scholar]
- Basappa, S.C.; Sreenivasamurthy, V.; Parpia, H. Aflatoxin and kojic acid production by resting cells of Aspergillus flavus Link. J. Gen. Microbiol. 1970, 61, 81–86. [Google Scholar] [CrossRef]
- Zheng, J.K.; Zhu, H.J.; Hong, K.; Wang, Y.; Liu, P.P.; Wang, X.; Peng, X. P.; Zhu, W.M. Novel cyclic hexapeptides from marine-derived fungus, Aspergillus sclerotiorum PT06-1. Org. Lett. 2009, 11, 5262–5265. [Google Scholar] [CrossRef]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, J.A.; Montgomery, T., Jr.; Vreven, K.N.; Kudin, J.C.; Burant, J M.; et al. Pople, Gaussian 03, Revision E. 01, Suite of Programs for ab Initio Calculation; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comput. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Dobson, J.F.; Petersilka, M. Density functional theory of time-dependent phenomena. In Density Functional Theory II; Nalewajski, R.F., Ed.; Springer-Verlag: Berlin, Germany, 1996; Volume 181, p. 81. [Google Scholar]
- Casida, M.E. Recent Advances in Density Functional Methods, Part I; Chong, D.P., Ed.; World Scientific: Singapore, Singapore, 1995; pp. 155–192. [Google Scholar]
- Gross, E.K.U.; Kohn, W. Time-dependent density functional theory. In Density Functional Theory of Many-Fermion Systems; Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1990; Volume 21, pp. 255–291. [Google Scholar]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Ellestad, G.A.; Lovell, F.M.; Perkinson, N.A.; Hargreaves, R.T.; McGahren, W.J. New zearalenone related macrolides and isocoumarins from an unidentified fungus. J. Org. Chem. 1978, 43, 2339–2343. [Google Scholar]
- Lai, S.; Shizuri, Y.; Yamamura, S.; Kawai, K.; Furukawa, H. Three new phenolic metabolites from Penicillium species. Heterocycles 1991, 32, 297–305. [Google Scholar] [CrossRef]
- Watanabe, A.; Ono, Y.; Fujii, I.; Sankawa, U.; Mayorga, M.E.; Timberlake, W.E.; Yutaka Ebizuka, Y. Aspergillus fumigatus alb1 encodes naphthopyrone synthase when expressed in Aspergillus oryzae. Tetrahydron Lett. 1998, 39, 7733–7736. [Google Scholar]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Tepaske, M.R.; Gloer, J.B. Leporin A: An antiinsectan N-alkoxypyridone from the sclerotia of Aspergillus leporis. Tetrahedron Lett. 1991, 32, 5687–5690. [Google Scholar] [CrossRef]
- Nakadate, S.; Nozawa, K.; Yaguchi, T. Two new eujindoles from Eupenicillium javanicum. Heterocycles 2011, 83, 1867–1871. [Google Scholar] [CrossRef]
- Ogata, M.; Ueda, J.; Hoshi, M.; Hashimoto, J.; Nakashima, T.; Anzai, K.; Takagi, M.; Shin-ya, K. A novel indole-diterpenoid, JBIR-03 with anti-MRSA activity from Dichotomomyces cejpii var. cejpii NBRC 103559. J. Antibiot. 2007, 60, 645–648. [Google Scholar] [CrossRef]
- Clardy, J.; Springer, J.P. Paspaline and paspalicine two indole mevalonate metabolites from Claviceps paspali. Tetrahedron Lett. 1980, 21, 231–234. [Google Scholar] [CrossRef]
- Singh, S.B.; Ondeyka, J.G.; Jayasuriya, H.; Zink, D.L.; Ha, S.N.; Dahl, R.A.; Greene, J.; Kim, J.A.; Smith, M.M.; Shoop, W.; et al. Nodulisporic acids D-F: Structure, biological activities, and biogenetic relationships. J. Nat. Prod. 2004, 67, 1496–1506. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Zaika, L.L. Spices and herbs: Their antimicrobial activity and its determination. J. Food Saf. 1988, 9, 97–118. [Google Scholar] [CrossRef]
- Li, J.S.; Kleeff, J.; Guo, J.C.; Fischer, L.; Giese, N.; Büchler, M.W.; Friess, H. Effects of STI571 (gleevec) on pancreatic cancer cell growth. Mol. Cancer 2003, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Somberg, R.; Pferdehirt, B.; Kupcho, K. A universal kinase aassay for a world of kinases. Promega Notes 2003, 83, 14–17. [Google Scholar]
- Grassauer, A.; Weinmuellner, R.; Meier, C.; Pretsch, A.; Prieschl-Grassauer, E.; Unger, H. Iota-Carrageenan is a potent inhibitor of rhinovirus infection. Virol. J. 2008, 5, 107. [Google Scholar] [CrossRef]
- Mehta, N.N.; Sheetz, M.; Price, K. Selective PKC beta inhibition with ruboxistaurin and endothelial function in type-2 diabetes mellitus. Cardiovasc. Drugs Ther. 2009, 23, 17–24. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).