Punctaporonins H–M: Caryophyllene-Type Sesquiterpenoids from the Sponge-Associated Fungus Hansfordia sinuosae




Abstract
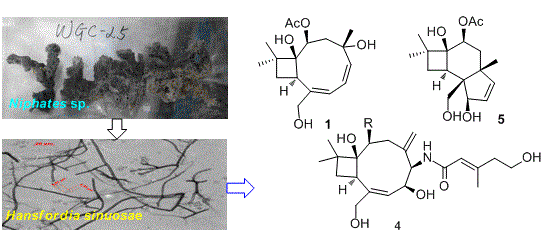
:
1. Introduction
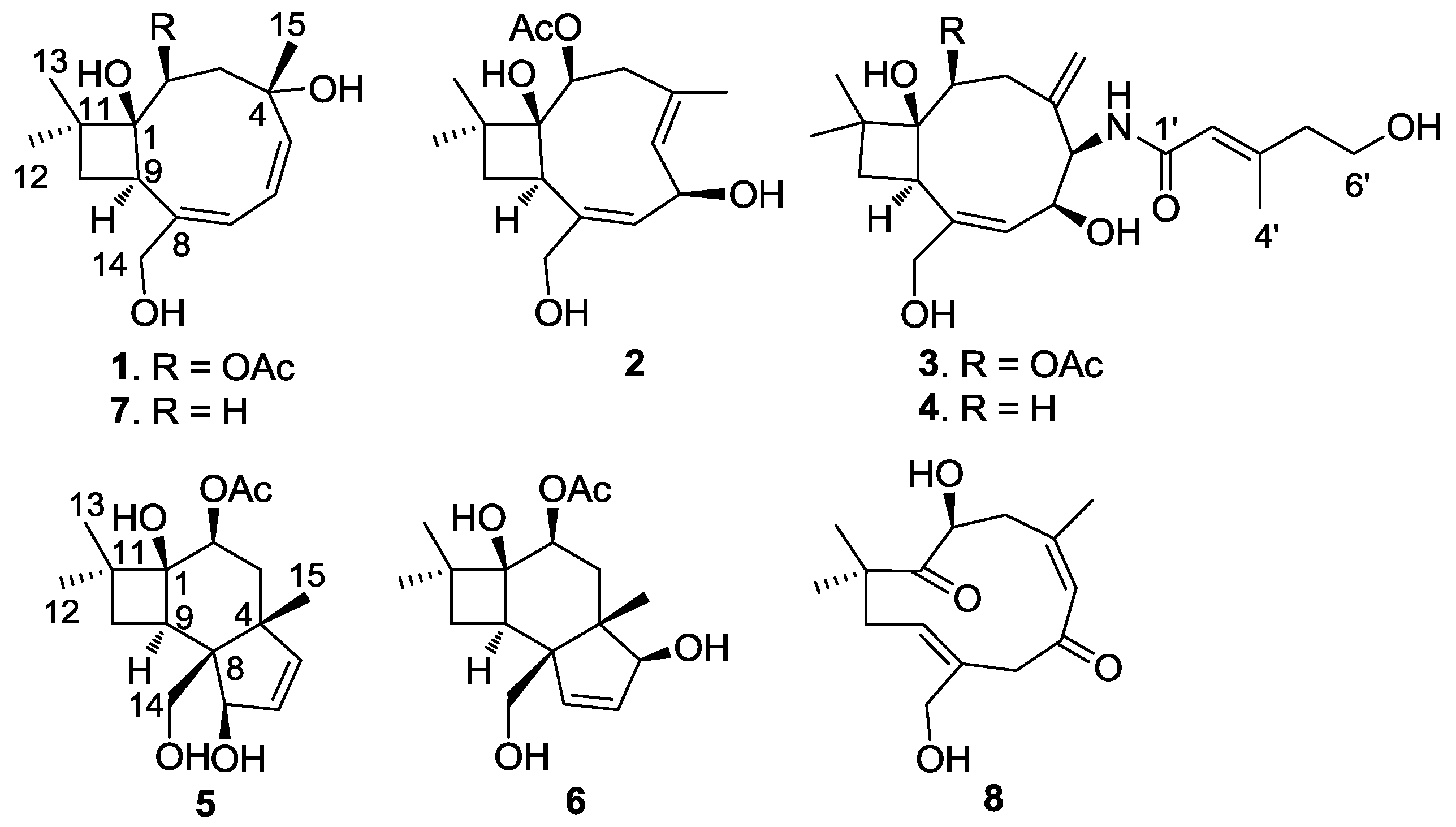
2. Results and Discussion
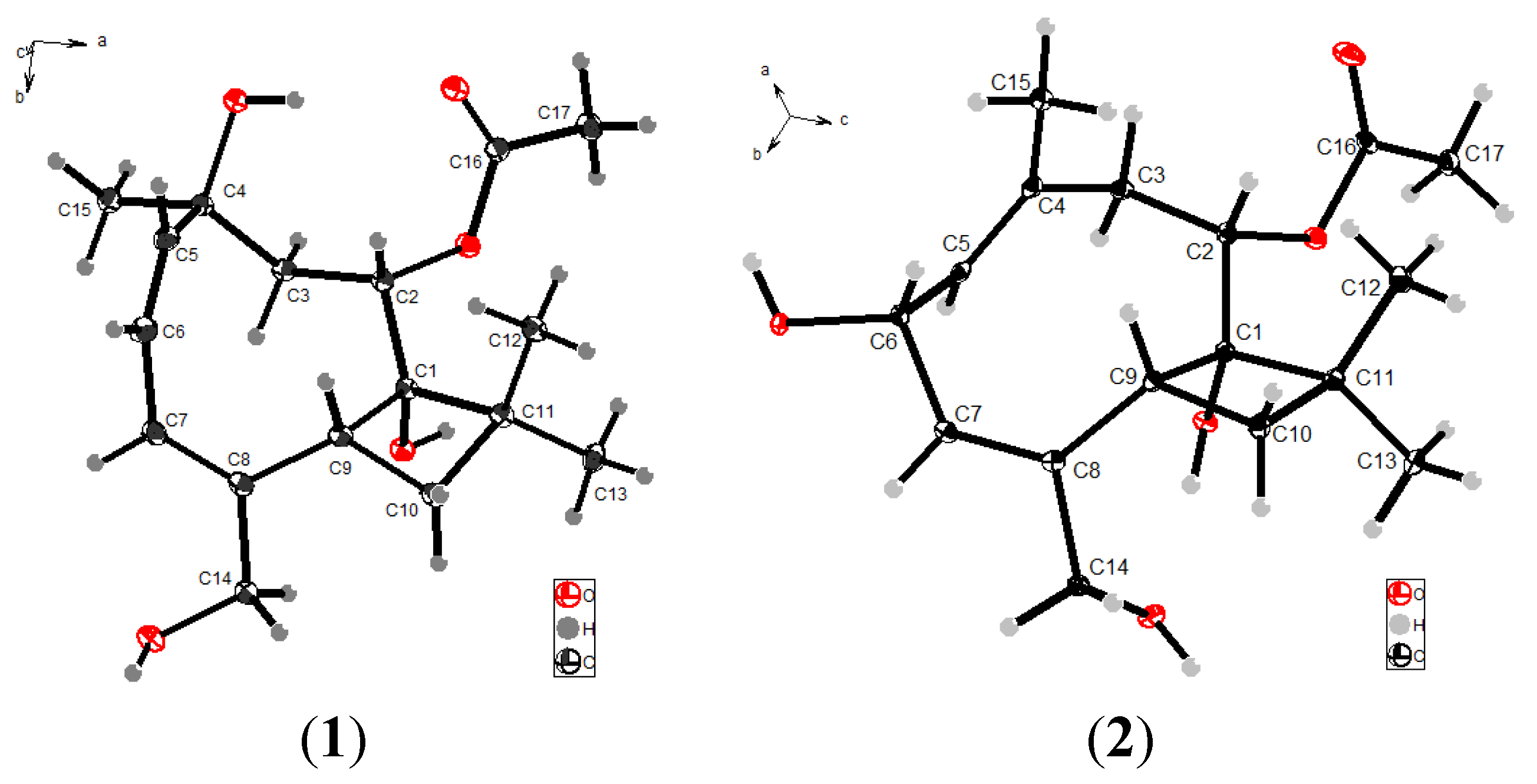
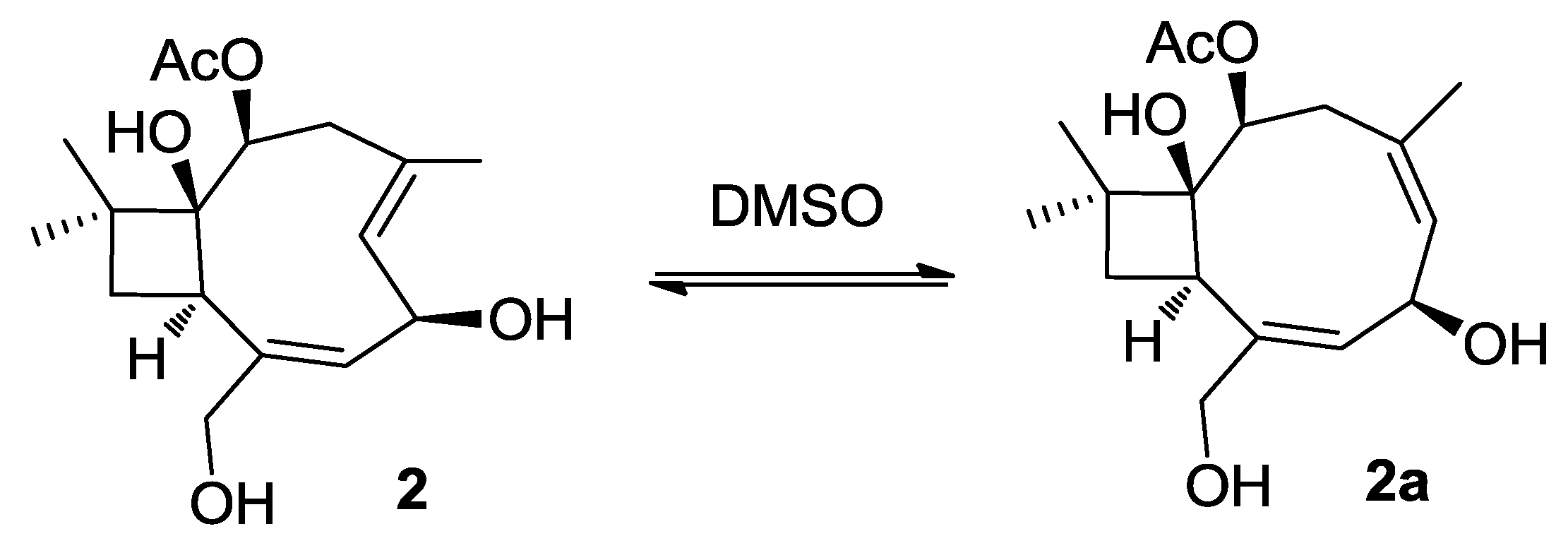
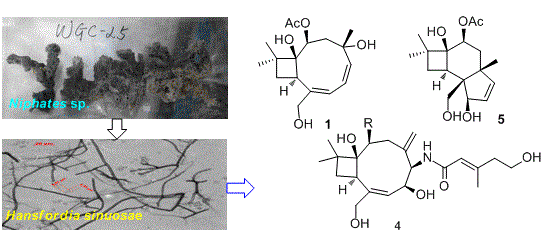
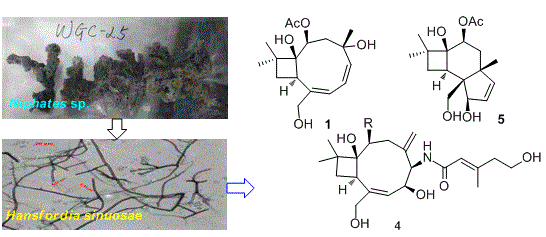
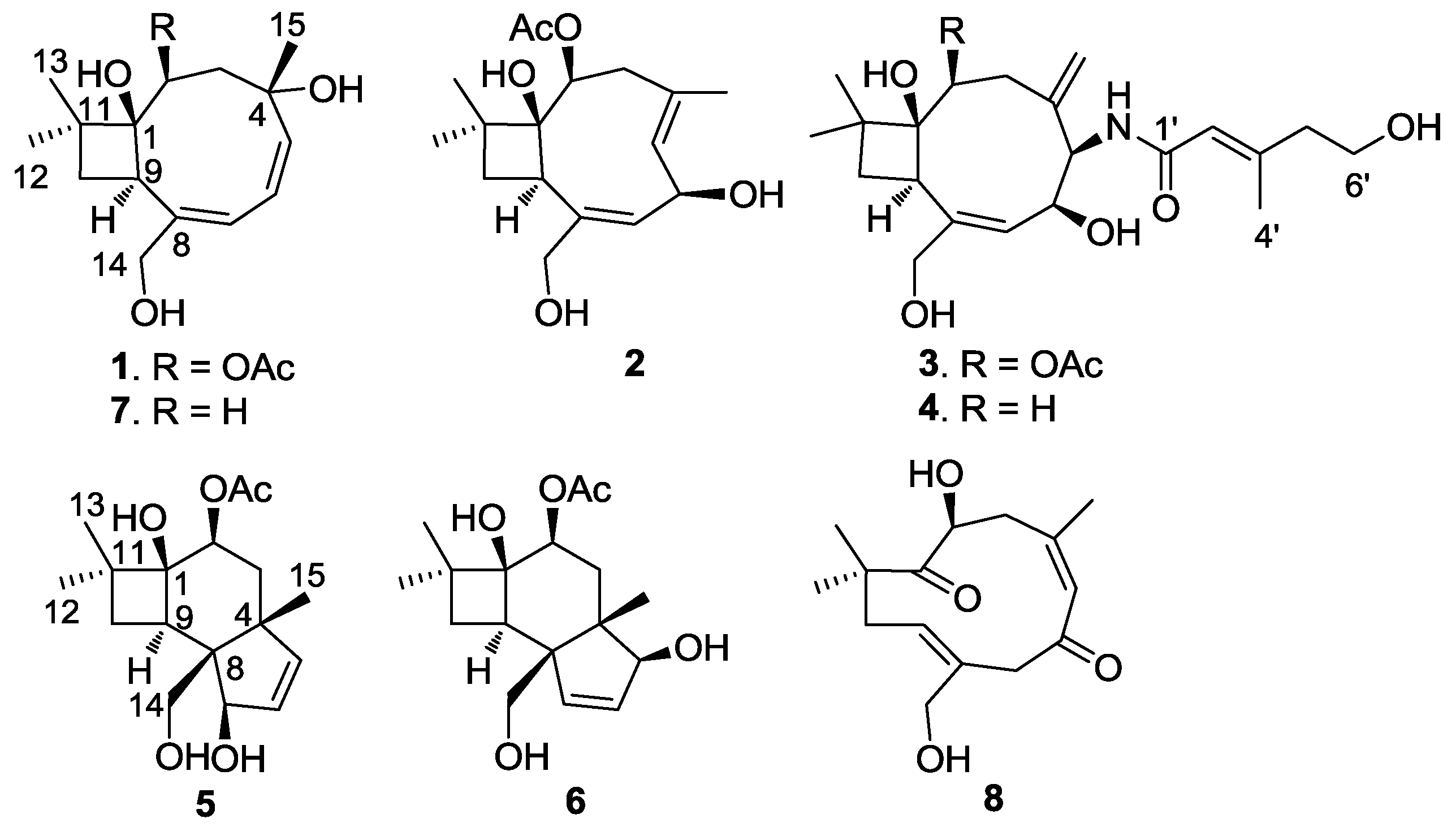
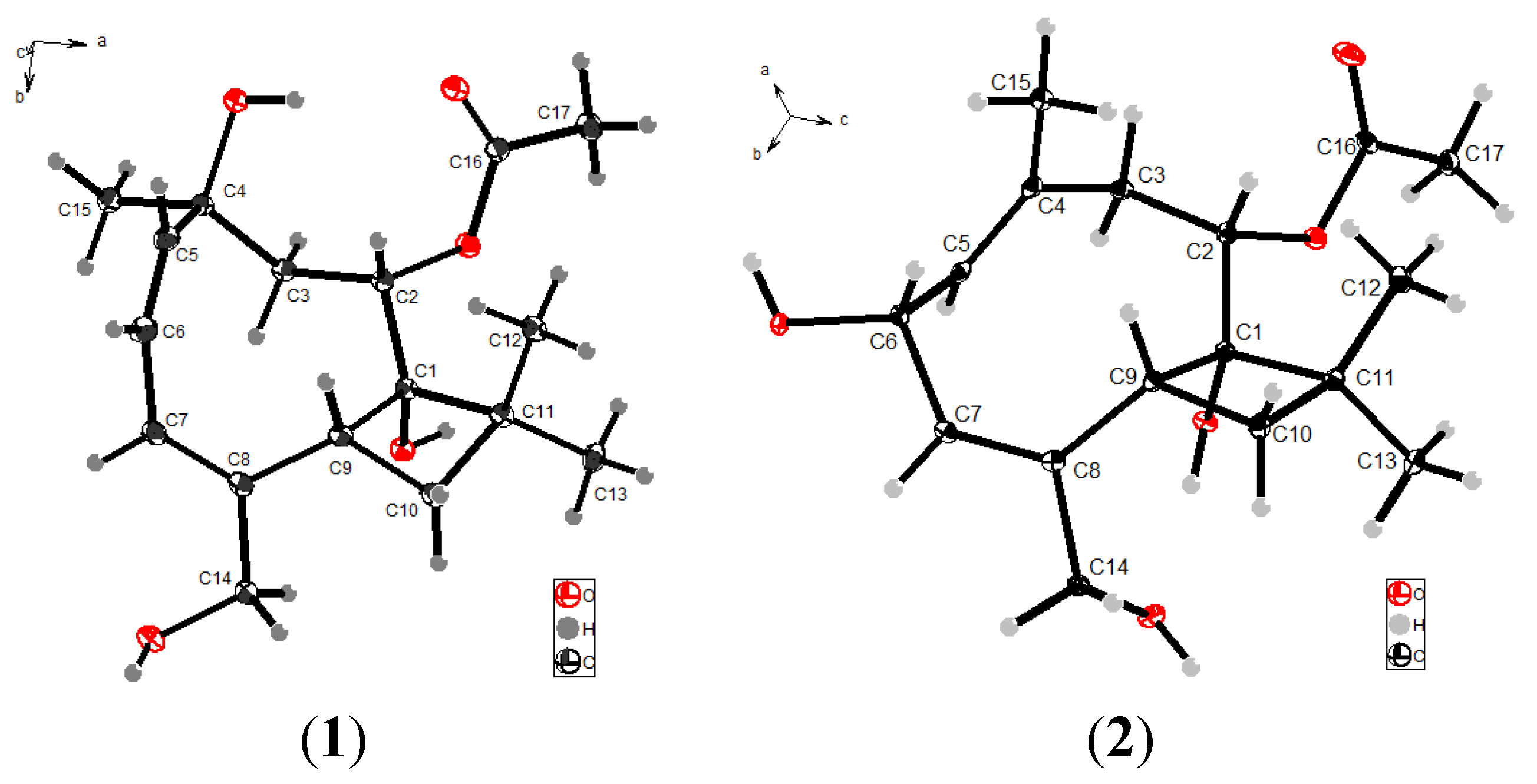
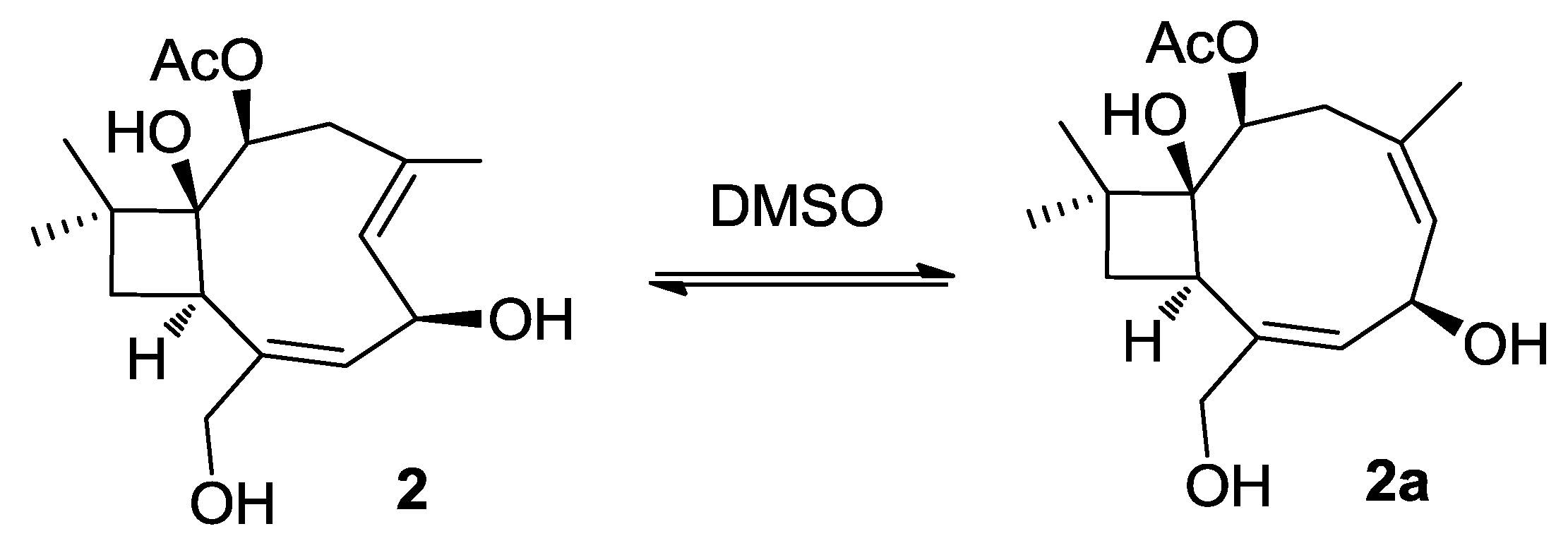
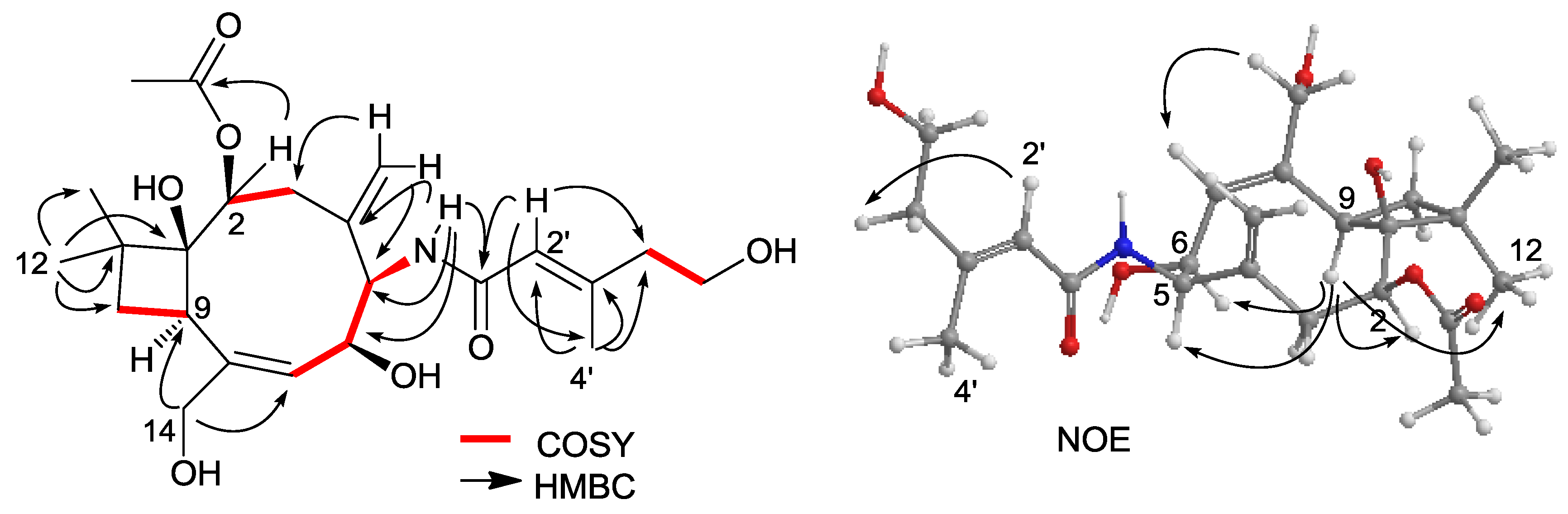
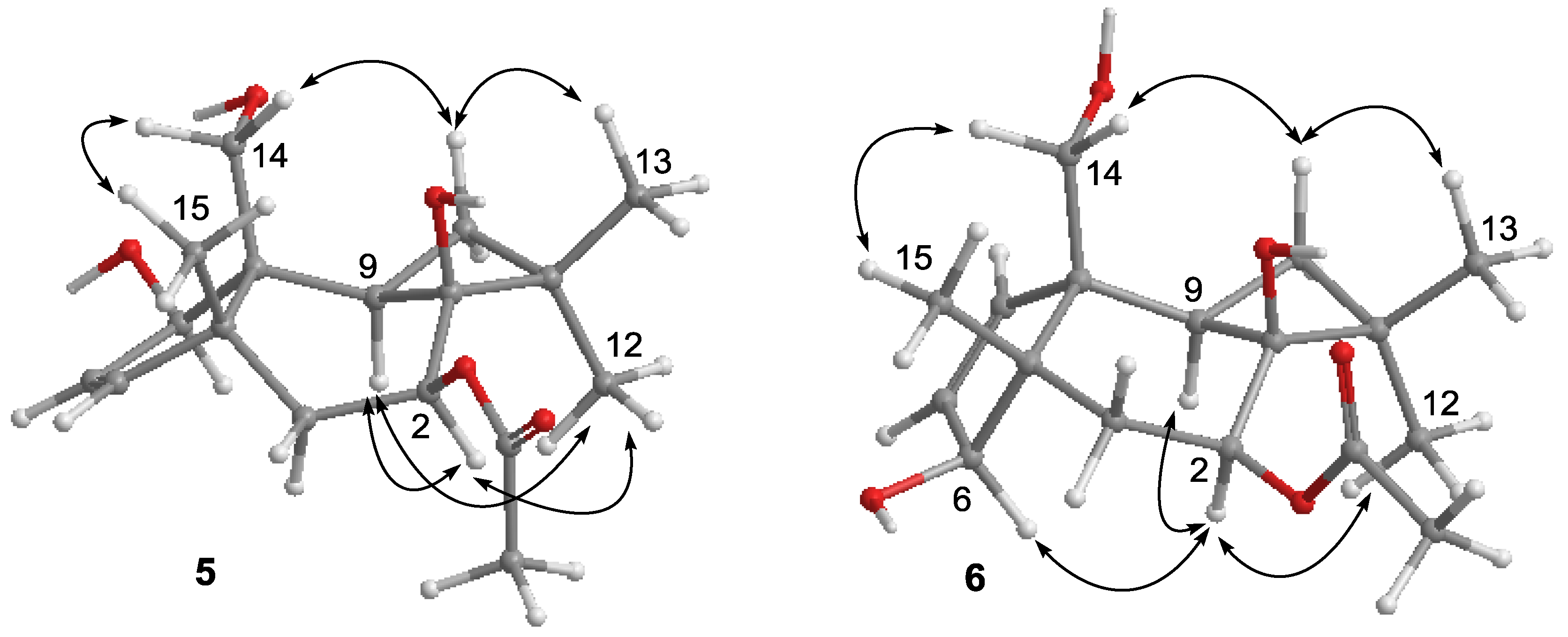
2.1. Structure Elucidation
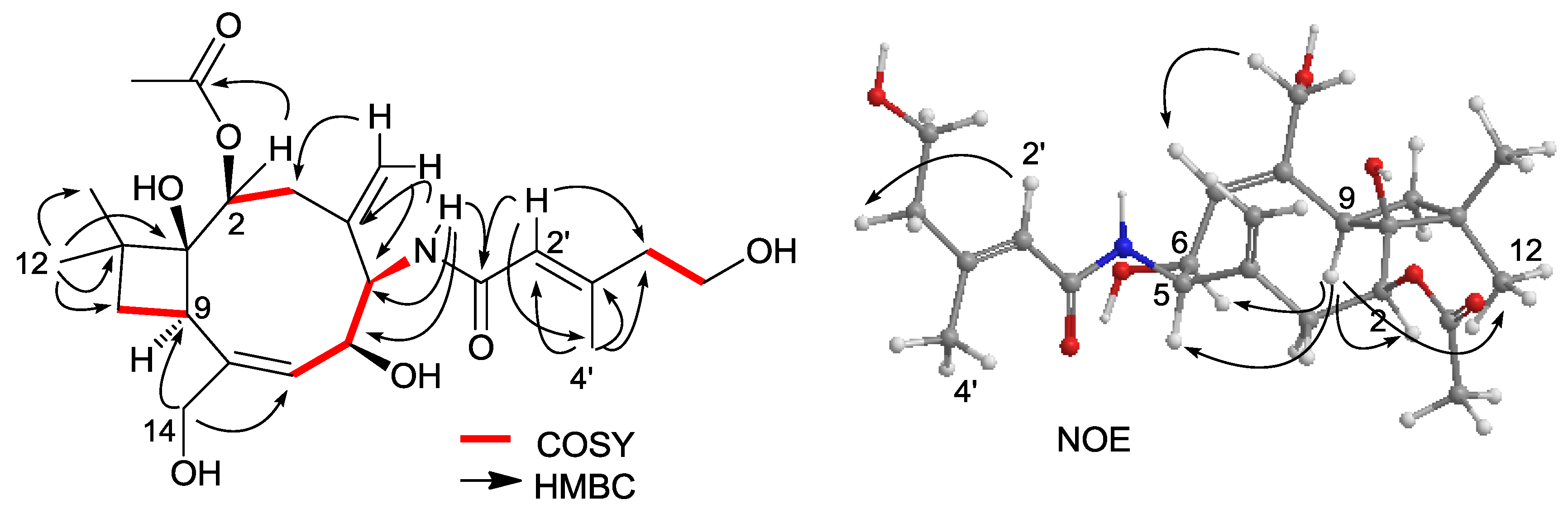
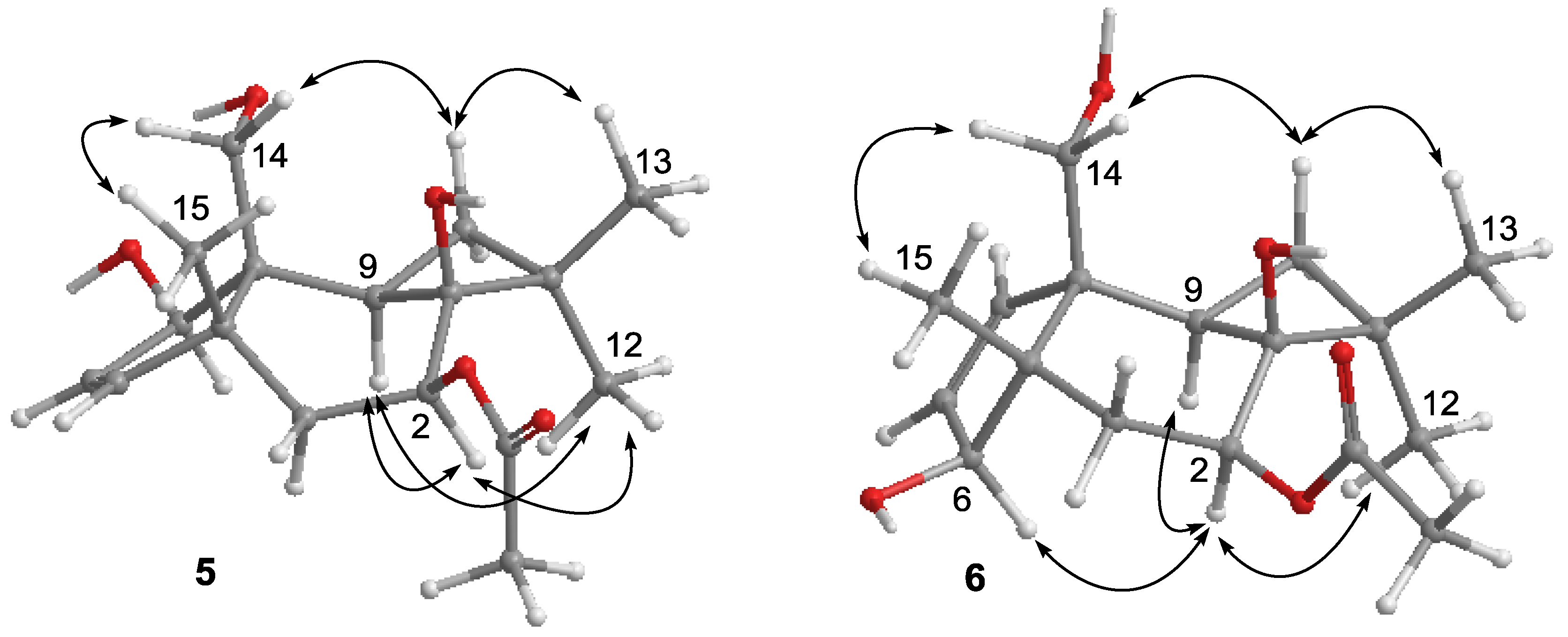
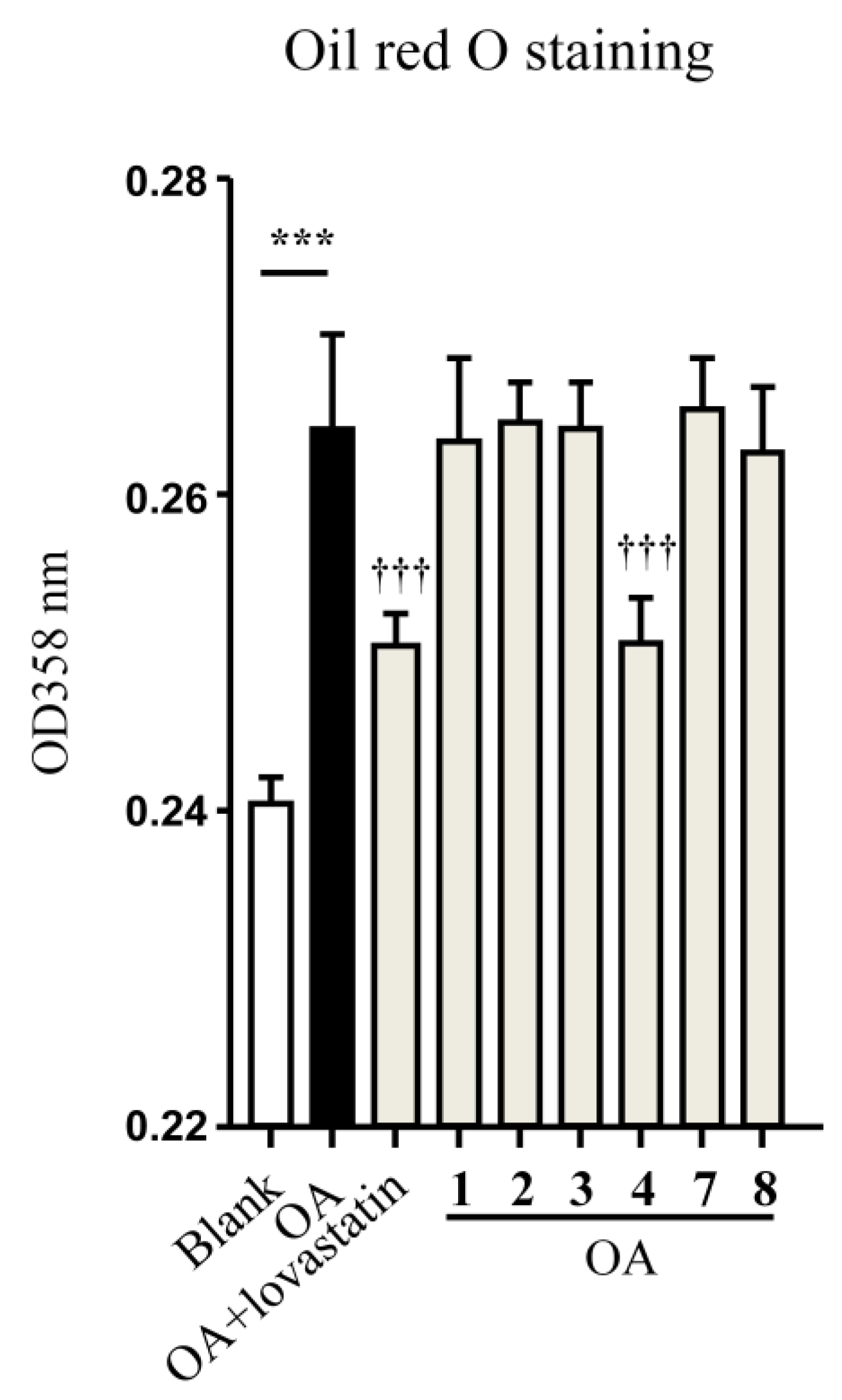
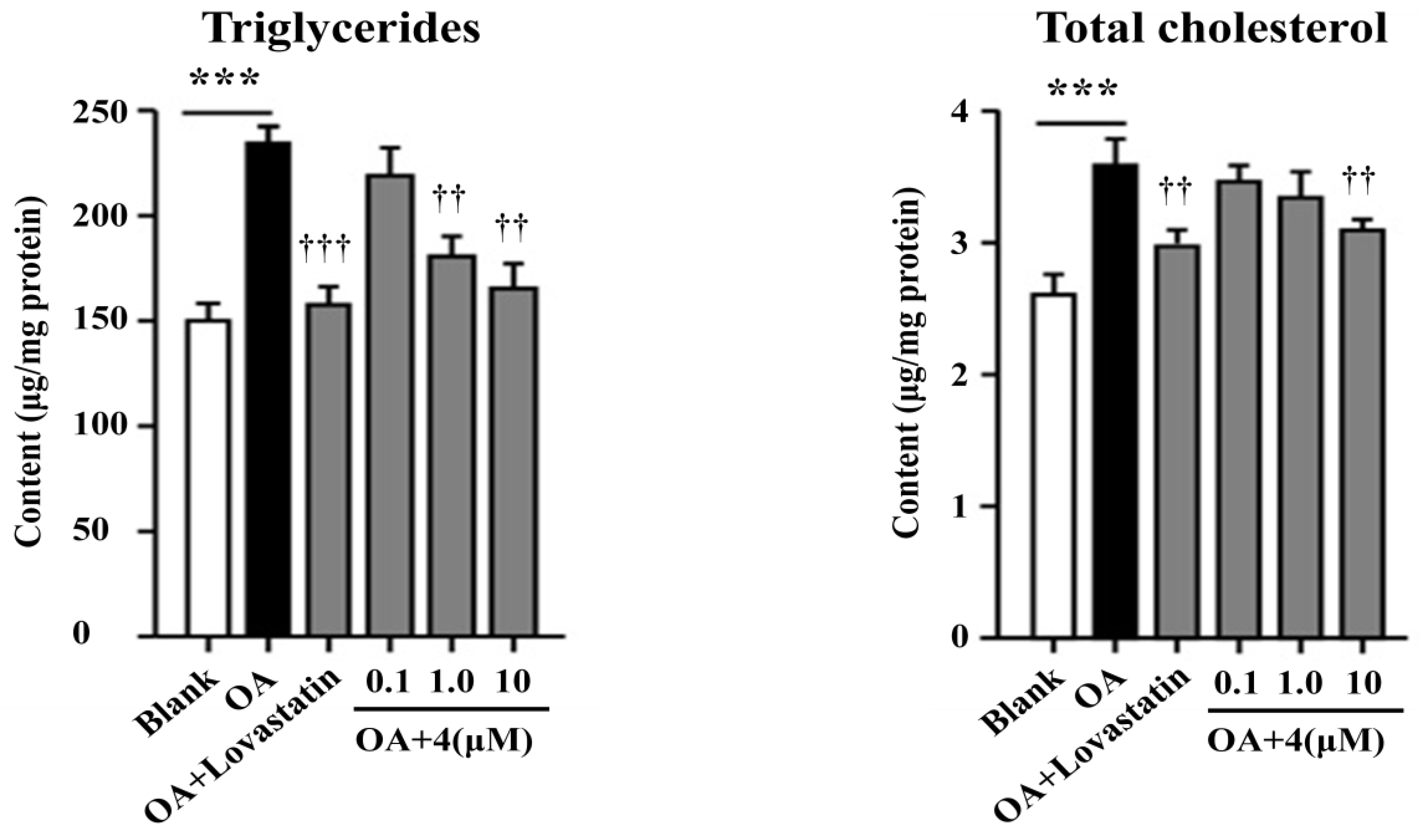
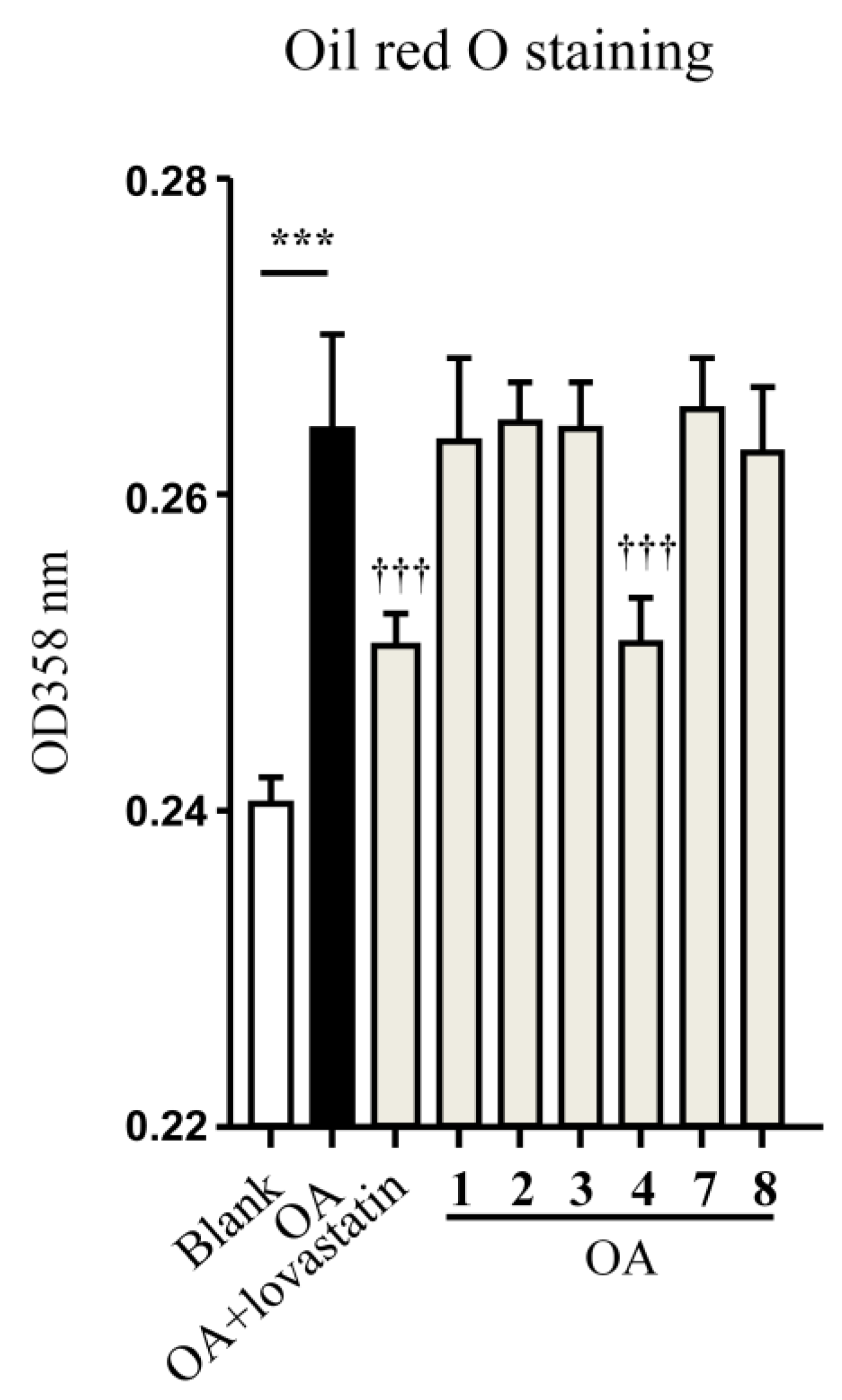
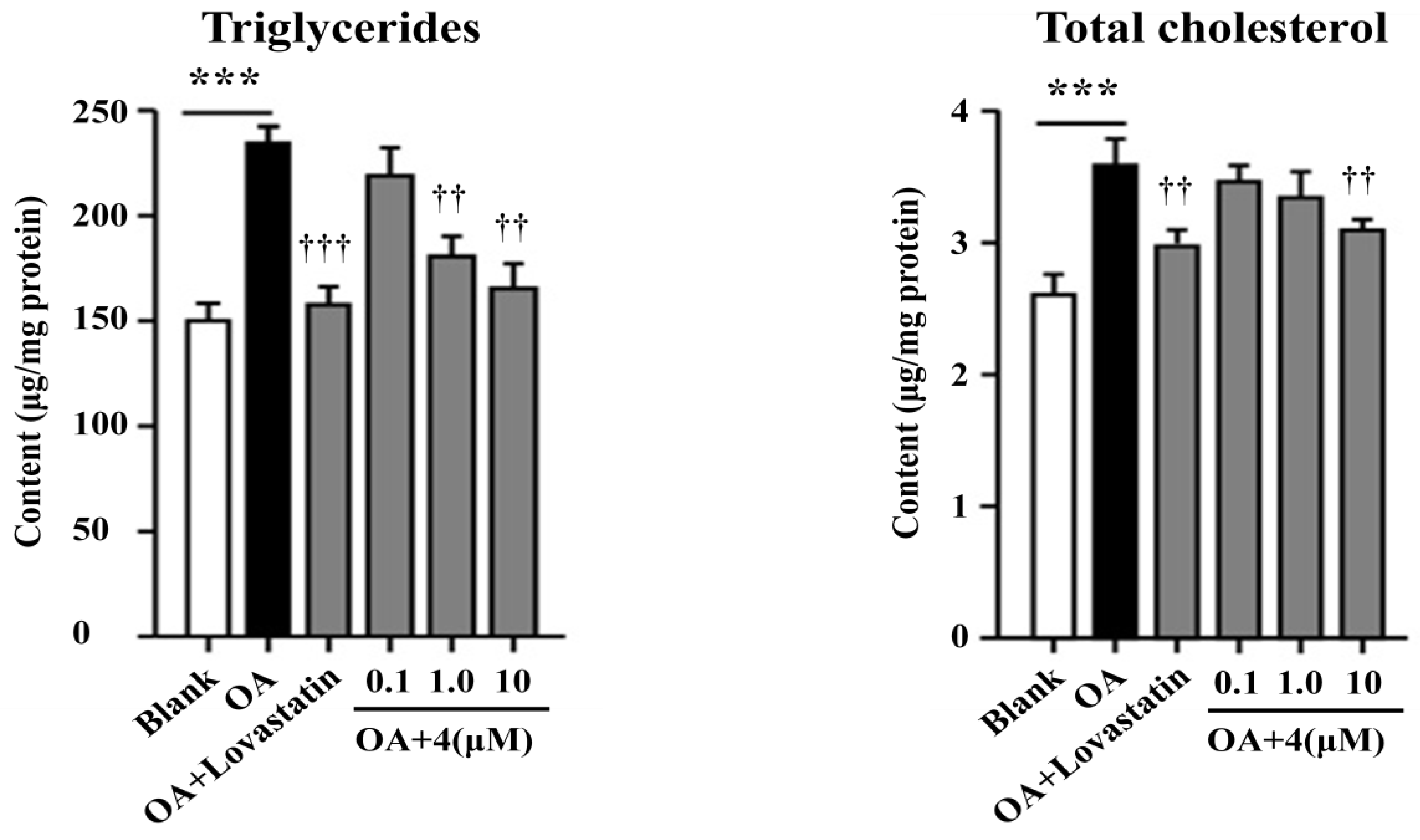
2.2. Bioassay Results
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material and Fermentation
3.3. Extraction and Isolation
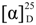 −83 (c 0.3, CH3OH). UV (CH3OH) λmax 208, 198 nm; IR (KBr) νmax 3350, 2961, 2934, 2870, 1708, 1530, 1383, 1263, 1122, 1024 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1675 [M + Na]+ (calcd for C17H26NaO5, 333.1672). −139 (c 0.22, CH3OH). UV (CH3OH) λmax 216, 198 nm; IR (KBr) νmax 3343, 2932, 2870, 1714, 1648, 1620, 1463, 1373, 1256, 1122 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1669 [M + Na]+ (calcd for C17H26NaO5, 333.1672). −37 (c 0.65, CH3OH). UV (CH3OH) λmax 221.1 196.5 nm; IR (KBr) νmax 3326, 2939, 2873, 1715, 1664, 1633, 1537, 1438, 1373, 1260, 1047, 1026 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 438.2481 [M + H]+ (calcd for C23H36NO7, 438.2486). −64 (c 0.58, CH3OH). UV (CH3OH) λmax 222, 195 nm; IR (KBr) νmax 3295 (br), 2936, 2870, 1723, 1663, 1629, 1533, 1439, 1365, 1202, 1060 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 380.2429 [M + H]+ (calcd for C21H34NO5, 380.2431).
−83 (c 0.3, CH3OH). UV (CH3OH) λmax 208, 198 nm; IR (KBr) νmax 3350, 2961, 2934, 2870, 1708, 1530, 1383, 1263, 1122, 1024 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1675 [M + Na]+ (calcd for C17H26NaO5, 333.1672). −139 (c 0.22, CH3OH). UV (CH3OH) λmax 216, 198 nm; IR (KBr) νmax 3343, 2932, 2870, 1714, 1648, 1620, 1463, 1373, 1256, 1122 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1669 [M + Na]+ (calcd for C17H26NaO5, 333.1672). −37 (c 0.65, CH3OH). UV (CH3OH) λmax 221.1 196.5 nm; IR (KBr) νmax 3326, 2939, 2873, 1715, 1664, 1633, 1537, 1438, 1373, 1260, 1047, 1026 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 438.2481 [M + H]+ (calcd for C23H36NO7, 438.2486). −64 (c 0.58, CH3OH). UV (CH3OH) λmax 222, 195 nm; IR (KBr) νmax 3295 (br), 2936, 2870, 1723, 1663, 1629, 1533, 1439, 1365, 1202, 1060 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 380.2429 [M + H]+ (calcd for C21H34NO5, 380.2431).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | 1 | 2 | 3 | 4 | 5 | 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 1 | 80.9 | 81.2 | 83.4 | 82.7 | 77.8 | 77.5 | ||||||
| 2 | 74.6 | 4.90, brd (3.6) | 76.1 | 5.11, dd (10.9, 4.0) | 77.4 | 5.08, dd (9.6, 5.2) | 32.9 | 1.57, m; 1.94, m | 71.4 | 4.53, dd (11.2, 4.8) | 70.8 | 4.94, dd (10.8, 5.0) |
| 3 | 44.9 | 1.36, d (15.8)
2.75, dd (15.8, 3.6) | 40.3 | 2.13, dd (10.8, 4.0)
2.42, dd (10.9, 10.8) | 35.1 | 2.19, dd (11.0, 9.6)
2.22, dd (11.0, 5.2) | 28.7 | 1.95, brd (12.2)
2.05, m | 39.0 | 1.66, dd (12.3, 4.8)
1.80, dd (12.3, 11.2) | 36.5 | 1.62, dd (13.0, 10.8)
1.81, dd (13.0, 5.0) |
| 4 | 72.1 | 127.4 | 143.5 | 148.3 | 52.6 | 54.4 | ||||||
| 5 | 143.2 | 5.75, d (13.2) | 133.4 | 5.17, d (9.8) | 62.5 | 4.10, t (8.0) | 62.3 | 4.24, t (8.5) | 145.6 | 5.71, d (5.6) | 80.0 | 4.77, brs |
| 6 | 124.3 | 5.63, brd (13.2) | 66.1 | 4.70, dd, (9.8, 4.2) | 69.5 | 4.36, dd (9.0, 8.0) | 69.6 | 4.43, dd (8.5,9.2) | 130.1 | 5.61, dd (5.6, 2.6) | 136.1 | 5.52, brd (6.0) |
| 7 | 123.6 | 5.81, brs | 141.8 | 5.83, s | 133.3 | 5.38, d (9.0) | 131.5 | 5.36, d (9.2) | 80.3 | 3.94, d (2.6) | 132.2 | 5.48, d (6.0) |
| 8 | 139.0 | 136.1 | 136.1 | 137.2 | 49.4 | 53.5 | ||||||
| 9 | 39.0 | 3.26, dd (11.0, 8.4) | 40.6 | 3.04, dd (10.2, 7.9) | 40.8 | 3.26, t (10.0) | 43.9 | 3.16, dd (9.0, 8.7) | 42.7 | 2.00, m | 45.3 | 1.98, m |
| 10 | 33.5 | 1.42, dd (9.1, 8.4)
2.02, dd (11.0, 9.1) | 33.3 | 1.57, dd (12.0, 10.2)
1.92, dd (12.0, 7.9) | 33.8 | 1.43, t (10.0)
2.04, t (10.0) | 33.7 | 1.46, dd (9.0, 8.7)
2.02, t (9.0) | 35.4 | 1.50, m
2.00, m | 35.4 | 1.39, dd (5.0, 4.5)
2.00, m |
| 11 | 40.6 | 41.6 | 41.0 | 40.9 | 41.6 | 42.5 | ||||||
| 12 | 24.5 | 1.03, s | 24.5 | 0.94, s | 24.7 | 1.08, s | 24.3 | 1.07, s | 23.6 | 1.02, s | 23.7 | 1.08, s |
| 13 | 24.1 | 0.98, s | 25.5 | 1.02, s | 24.0 | 0.98, s | 22.5 | 0.86, s | 24.3 | 0.98, s | 24.3 | 0.97, s |
| 14 | 63.4 | 3.87, d (13.2)
4.13, d (13.2) | 65.4 | 3.78, d (11.2)
4.11, d (11.2) | 64.3 | 3.76, d (11.0)
3.94, d (11.0) | 64.3 | 3.74, d (12.4)
3.90, d (12.4) | 60.3 | 3.50, d (11.0)
3.93, d (11.0) | 61.9 | 3.46, d (11.0)
3.53, d (11.0) |
| 15 | 31.9 | 1.08, s | 17.5 | 1.83, s | 119.6 | 4.79, s; 5.02, s | 117.9 | 4.66, s; 4.86, s | 27.3 | 1.15, s | 17.8 | 0.85, s |
| 1′ | 166.4 | 166.4 | ||||||||||
| 2′ | 120.6 | 5.76, s | 120.6 | 5.76, s | ||||||||
| 3′ | 149.5 | 149.4 | ||||||||||
| 4′ | 18.2 | 2.05, s | 18.2 | 2.07, s | ||||||||
| 5′ | 44.1 | 2.21, t (6.2) | 44.1 | 2.20, t (6.2) | ||||||||
| 6′ | 59.6 | 3.54, t (6.5) | 59.5 | 3.54, t (6.5) | ||||||||
| Ac | 170.8 | 170.1 | 169.8 | 170.2 | 170.1 | |||||||
| 21.6 | 1.99, s | 21.7 | 1.96, s | 21.5 | 1.97, s | 21.4 | 1.94, s | 21.4 | 1.97, s | |||
| NH | 7.79, d (8.0) | 7.92, d (8.5) | ||||||||||
−48 (c 0.10, CH3OH); UV (CH3OH) λmax 199 nm; IR (KBr) νmax 3336, 2952, 2933, 2870, 1729, 1682, 1529, 1454, 1350, 1251, 1028 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1677 [M + Na]+ (calcd for C17H26O5Na, 333.1672). +49 (c 0.12, CH3OH). UV (CH3OH) λmax 199 nm; IR (KBr) νmax 3265, 2957, 2933, 2865, 1718 (br), 1463, 1377, 1249, 1017 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 333.1677 [M + Na]+ (calcd for C17H26O5Na, 333.1672).3.4. X-Ray Single Crystallographic Analyses
3.5. Cell-Based Lipid Accumulation Assay
3.6. Cytotoxic and Antibacterial Assays
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Collado, I.G.; Hanson, J.R.; Macías-Sánchez, A.J. Recent advances in the chemistry of caryophyllene. Nat. Prod. Rep. 1998, 15, 187–204. [Google Scholar] [CrossRef]
- Ghalib, R.M.; Hashim, R.; Sulaiman, O.; Mehdi, S.H.; Valkonen, A.; Rissanen, K.; Trifunovi, S.R.; Ahamed, M.B.K.; Majid, A.M.S.A. A novel caryophyllene type sesquiterpene lactone from Asparagus falcatus (Linn.); Structure elucidation and anti-angiogenic activity on HUVECs. Eur. J. Med. Chem. 2012, 47, 601–607. [Google Scholar] [CrossRef]
- Orav, A.; Stulova, I.; Kailas, T.; Muurisepp, M. Effect of storage on the essential oil composition of Piper nigrum L. fruits of different ripening states. J. Agric. Food Chem. 2004, 52, 2582–2586. [Google Scholar] [CrossRef]
- Mockute, D.; Bernotiene, G.; Judzentiene, A. The essential oil of Origanum vulgare L. ssp. vulgare growing wild in Vilnius district (Lithuania). Phytochemistry 2001, 57, 65–69. [Google Scholar] [CrossRef]
- Li, Y.; Cui, C.; Liu, X.; Che, Y. Cytosporinols A–C, new caryophyllene sesquiterpenoids from Cytospora sp. Nat. Prod. Bioprospect. 2012, 2, 70–75. [Google Scholar]
- Deyrup, S.T.; Swenson, D.C.; Gloer, J.B.; Wicklow, D.T. Caryophyllene sesquiterpenoids from a fungicolous isolate of Pestalotiopsis disseminate. J. Nat. Prod. 2006, 69, 608–611. [Google Scholar] [CrossRef]
- Yang, S.; Chan, T.M.; Terracciano, J.; Boehm, E.; Patel, R.; Chen, G.; Loebenberg, D.; Patel, M.; Gullo, V.; Pramanik, B.; et al. Caryophyllenes from a fungal culture of Chrysosporium pilosum. J. Nat. Prod. 2009, 72, 484–487. [Google Scholar] [CrossRef]
- Wichlacz, M.; Ayer, W.A.; Trifonov, L.S.; Chakravarty, P.; Khasa, D. A caryophyllene-related sesquiterpene and two 6,7-seco-caryophyllenes from liquid cultures of Hebeloma longicaudum. J. Nat. Prod. 1999, 62, 484–486. [Google Scholar] [CrossRef]
- Pulici, M.; Sugawara, F.; Koshino, H.; Uzawa, J.; Yoshida, S. Pestalotiopsins A and B: New caryophyllenes from an endophytic fungus of Taxus brevifolia. J. Org. Chem. 1996, 61, 2122–2124. [Google Scholar] [CrossRef]
- Pulici, M.; Sugawara, F.; Koshino, H.; Okada, G.; Esumi, Y.; Uzawa, J.; Yoshida, S. Metabolites of Pestalotiopsis spp., endophytic fungi of Taxus brevifolia. Phytochemistry 1997, 46, 313–319. [Google Scholar] [CrossRef]
- Anderson, J.R.; Edwards, R.L.; Poyser, J.P.; Whalley, A.J.S. Metabolites of the higher fungi 23. the punctaporonins novel bi-cyclic, tri-cyclic, and tetra-cyclic sesquiterpenes related to caryophyliene, from the fungus Poronia punctata (Linnaeus, Fries) Fries. J. Chem. Soc. Perkin Trans. 1 1988, 4, 823–831. [Google Scholar]
- Anderson, J.R.; Edwards, R.L.; Freer, A.A.; Mabelis, R.P.; Poyser, J.P.; Spencer, H.; Whalley, A.J.S. Punctatins B and C (antibiotics M95154 and M95155): Further sesquiterpene alcohols from the fungus Poronia punctata. J. Chem. Soc. Chem. Commun. 1984, 14, 917–919. [Google Scholar]
- Ahmed, A.F.; Su, J.H.; Shiue, R.T.; Pan, X.J.; Dai, C.F.; Kuo, Y.H.; Sheu, J.H. New β-caryophyllene-derived terpenoids from the soft coral Sinularia nanolobata. J. Nat. Prod. 2004, 67, 592–597. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Guo, Y. Terpenoids of Sinularia soft corals: Chemistry and bioactivity. Acta Pharm. Sin. B 2012, 2, 227–237. [Google Scholar] [CrossRef]
- Wang, G.H.; Ahmed, A.F.; Sheu, J.H.; Duh, C.Y.; Shen, Y.C.; Wang, L.T. Suberosols A–D, four new sesquiterpenes with β-caryophyllene skeletons from a Taiwanese gorgonian coral Subergorgia suberosa. J. Nat. Prod. 2002, 65, 887–891. [Google Scholar] [CrossRef]
- Smetanina, O.F.; Kuznetsova, T.A.; Gerasimenko, A.V.; Kalinovsky, A.I.; Pivkin, M.V.; Dmitrenok, P.C.; Elyakov, G.B. Metabolites of the marine fungus Humicola fuscoatra KMM 4629. Russ. Chem. Bull. 2004, 53, 2643–2646. [Google Scholar] [CrossRef]
- Collado, I.G.; Aleu, J.; Macias-Sanchez, A.J.; Hernandez-Galan, R. Inhibition of Botrytzas cznerea by new sesquiterpenoid compounds obtained from the rearrangement of isocaryophyllene. J. Nat. Prod. 1994, 57, 738–746. [Google Scholar] [CrossRef]
- Poyser, J.P.; Edwards, R.L.; Hursthouse, M.B.; Walker, N.P.C.; Sheldrick, G.M.; Whalley, A.J.S. Punctaporonins A, D, E, and F (antibiotics M95464, M167906, M171950, and M189122), isomeric allylic alcohols from the fungus Poronia punctata: X-ray crystal structures of D and of E acetonide. J. Antibiot. 1986, 39, 167–169. [Google Scholar] [CrossRef]
- Flack, H.D. On enantiomorph-polatity estimation. Acta Cryst. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, C.; Wu, H.; Sheng, L.; Su, Y.; Zhang, X.; Luan, H.; Sun, G.; Sun, X.; Tian, Y.; et al. Anti-hyperlipidemic effects and potential mechanisms of action of the caffeoylquinic acid-rich Pandanus tectorius fruit extract in hamsters fed a high fat-diet. PLoS One 2013, 8, e61922. [Google Scholar]
- Ghuge, G.D.; Zine, R.; Mogrekar, M. Lipid profile of patients with ischaemic heart disease from rural area of Marthawada region, India. Int. J. Biomed. Adv. Res. 2012, 3, 767–769. [Google Scholar]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef]
- D’Adamo, E.; Cali, A.M.; Weiss, R.; Santoro, N.; Pierpont, B.; Northrup, V.; Caprio, S. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 2010, 33, 1817–1822. [Google Scholar]
- Wan, C.W.; Wong, C.N.; Pin, W.K.; Wong, M.H.; Kwok, C.Y.; Chan, R.Y.; Yu, P.H.; Chan, S.W. Chlorogenic acid exhibits cholesterol lowering and fatty liver attenuating properties by up-regulating the gene expression of PPAR-alpha in hypercholesterolemic rats induced with a high-cholesterol diet. Phytother. Res. 2013, 27, 545–551. [Google Scholar] [CrossRef]
- Poulsen, M.M.; Larsen, J.O.; Hamilton-Dutoit, S.; Clasen, B.F.; Jessen, N.; Paulsen, S.K.; Kjaer, T.N.; Richelsen, B.; Pedersen, S.B. Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet. Nutr. Res. 2012, 32, 701–708. [Google Scholar] [CrossRef]
- Guo, P.; Kai, Q.; Gao, J.; Lian, Z.Q.; Wu, C.M.; Wu, C.A.; Zhu, H.B. Cordycepin prevents hyperlipidemia in hamsters fed a high-fat diet via activation of AMP-activated protein kinase. J. Pharmacol. Sci. 2010, 113, 395–403. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre (CCDC). Available online: http://www.ccdc.cam.ac.uk (accessed on 20 June 2014).
- Kobayashi, M.; Nakano, E. Stereochemical course of the transannular cyclization, in chloroform, of epoxycembranoids derived from the geometrical isomers of (14S)-14-hydroxy-1,3,7,1l-cembratetraen. J. Org. Chem. 1990, 55, 1947–1951. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, Z.; Liu, D.; Proksch, P.; Guo, P.; Lin, W. Punctaporonins H–M: Caryophyllene-Type Sesquiterpenoids from the Sponge-Associated Fungus Hansfordia sinuosae. Mar. Drugs 2014, 12, 3904-3916. https://doi.org/10.3390/md12073904
Wu Z, Liu D, Proksch P, Guo P, Lin W. Punctaporonins H–M: Caryophyllene-Type Sesquiterpenoids from the Sponge-Associated Fungus Hansfordia sinuosae. Marine Drugs. 2014; 12(7):3904-3916. https://doi.org/10.3390/md12073904
Chicago/Turabian StyleWu, Zehong, Dong Liu, Peter Proksch, Peng Guo, and Wenhan Lin. 2014. "Punctaporonins H–M: Caryophyllene-Type Sesquiterpenoids from the Sponge-Associated Fungus Hansfordia sinuosae" Marine Drugs 12, no. 7: 3904-3916. https://doi.org/10.3390/md12073904
APA StyleWu, Z., Liu, D., Proksch, P., Guo, P., & Lin, W. (2014). Punctaporonins H–M: Caryophyllene-Type Sesquiterpenoids from the Sponge-Associated Fungus Hansfordia sinuosae. Marine Drugs, 12(7), 3904-3916. https://doi.org/10.3390/md12073904